

Background: Corin is a protease that acts in the pregnant uterus to prevent pregnancy-induced hypertension.
Results: Corin mutations K317E and S472G from preeclamptic patients impaired corin zymogen activation and intracellular trafficking, respectively.
Conclusion: Mutations in the CORIN gene may impair corin function by different mechanisms.
Significance: Genetic variants and mutations disrupting corin function may contribute to pregnancy-induced hypertension in patients.
Keywords: genetic disease, genetic polymorphism, hypertension, membrane protein, natriuretic peptide, pregnancy, protease, proteolytic enzyme, corin, preeclampsia
Abstract
Corin is a membrane-bound serine protease that acts as the atrial natriuretic peptide (ANP) convertase in the heart. Recent studies show that corin also activates ANP in the pregnant uterus to promote spiral artery remodeling and prevent pregnancy-induced hypertension. Two CORIN gene mutations, K317E and S472G, were identified in preeclamptic patients and shown to have reduced activity in vitro. In this study, we carried out molecular modeling and biochemical experiments to understand how these mutations impair corin function. By molecular modeling, the mutation K317E was predicted to alter corin LDL receptor-2 module conformation. Western blot analysis of K317E mutant in HEK293 cells showed that the mutation did not block corin expression on the cell surface but inhibited corin zymogen activation. In contrast, the mutation S472G was predicted to abolish a β-sheet critical for corin frizzled-2 module structure. In Western blot analysis and flow cytometry, S472G mutant was not detected on the cell surface in transfected HEK293 cells. By immunostaining, the S472G mutant was found in the ER, indicating that the mutation S472G disrupted the β-sheet, causing corin misfolding and ER retention. Thus, these results show that mutations in the CORIN gene may impair corin function by entirely different mechanisms. Together, our data provide important insights into the molecular basis underlying corin mutations that may contribute to preeclampsia in patients.
Introduction
Preeclampsia is a serious disease in pregnancy. The disease, characterized by gestational hypertension and proteinuria, is a major risk factor for maternal and neonatal deaths (1, 2). To date, the disease mechanism remains unclear. Studies indicate that factors causing a compromised maternal-fetal interface play a critical role in the disease (3–8). Consistent with this hypothesis, delayed trophoblast invasion, and poorly remodeled uterine spiral arteries are common pathological findings in preeclamptic patients (5–9).
Corin is a modular protease that consists of an N-terminal cytoplasmic tail, a transmembrane segment and an extracellular region with two frizzled (Fz)2 domains, eight LDL receptor (LDLR) repeats, a scavenger receptor domain, and a C-terminal trypsin-like protease domain (10, 11). In cardiomyocytes, corin activates atrial natriuretic peptide (ANP), a cardiac hormone essential for regulating body fluid and electrolyte homeostasis (12). In recent years, variants and mutations in the NPPA and NPR1 genes, encoding ANP and its receptor NPR-A, have been found in patients with hypertension and heart disease (13–18). Similarly, variants and mutations in the CORIN gene also have been reported in hypertensive patients (19–21). These results indicate that defects in the corin-ANP pathway may play an important role in hypertension.
In addition to the heart, corin is expressed in the pregnant uterus (11, 22, 23). Similar uterine expression of ANP and NPR-A mRNA and protein also have been reported (24–28). Our recent studies indicate that locally produced ANP by corin in the pregnant uterus is critical for promoting trophoblast invasion and spiral artery remodeling (29). We show that pregnant corin- or ANP-knock-out mice develop late gestational hypertension and proteinuria, resembling the phenotype in preeclamptic patients (22). Moreover, we identified two CORIN gene mutations in patient families with histories of preeclampsia; one mutation changed Lys-317 to Glu (K317E) in the LDLR2 domain and the other changed Ser-472 to Gly (S472G) in the Fz2 domain (22). In pro-ANP processing assays, K317E and S472G mutants had markedly reduced activities (22). It was unclear, however, how these mutations, which were located outside the protease domain, impaired corin activity.
To understand how K317E and S472G mutations alter corin structure and function, we expressed K317E and S472G mutants in HEK293 cells and analyzed the mutant proteins in functional experiments. We found that the K317E and S472G mutations impaired corin activity by entirely different mechanisms. Our results provide new insights into the molecular basis underlying corin mutations that may contribute to preeclampsia in patients.
EXPERIMENTAL PROCEDURES
Plasmid Construction
Plasmids expressing human wild-type (WT) corin, activation cleavage site mutant R801A, and active site mutant S985A were described previously (30, 31). Plasmid expressing corin mutants K317A, K317D, K317E, K317Q, K317R, S472A, and S472G were made by site-directed mutagenesis using the WT corin plasmid as a template. The constructed plasmids were verified by DNA sequencing. The corin proteins expressed by these plasmids contained a C-terminal V5 tag to be detected in Western blot analysis using an anti-V5 antibody (Invitrogen).
Plasmid Transfection and Western Blot Analysis
Human embryonic kidney (HEK) 293 cells were grown in DMEM with 10% FBS at 37 °C in humidified incubators with 5% CO2. At ∼80% confluency, the cells were transfected with the plasmids using the FuGENE reagent (Roche Diagnostics). After 24 to 48 h, the conditioned medium from the cells was collected. The cells were washed once with PBS and lysed in a solution containing 150 mmol/liter NaCl, 50 mmol/liter Tris-HCl, pH 8.0, 1% (v/v) Triton X-100, and a protease inhibitor mixture (1:100 dilution, Sigma). Immunoprecipitation and Western blot analysis to examine corin proteins in the conditioned medium and cell lysates were described previously (30, 31).
Pro-ANP Processing Assay
Corin activity was examined in a cell-based pro-ANP processing assay. In this assay, stable HEK293 cells were established to express human pro-ANP. Plasmids expressing corin WT and mutants were transfected into the cells. The cells were cultured at 37 °C for 16 h, and the conditioned medium was collected. Immunoprecipitation and Western blot analysis were performed to analyze pro-ANP processing, as described previously (30, 31).
Cell Surface Protein Labeling
HEK293 cells expressing corin proteins were labeled with sulfo-NHS-biotin (1 mmol/liter) (Pierce) in PBS (pH 8.0) at 4 °C for 5 min. The reaction was stopped by glycine (100 mmol/liter) in PBS. The cells were washed and lysed in the lysis solution described above. Streptavidin-Sepharose beads (30 μl) were added to the cell lysate and the mixture was rotated at 4 °C for 2 h. After washing, the beads were boiled in a sample-loading buffer. Proteins were examined by SDS-PAGE followed by Western blot analysis with an anti-V5 antibody.
Flow Cytometry
Flow cytometry was used to analyze corin expression on the cell surface (30). HEK293 cells expressing corin were detached from culture plates with an EDTA solution and incubated with an anti-V5 antibody, followed with a fluorescein isothiocyanate (FITC)-labeled secondary antibody (BD Biosciences). The cells were analyzed using FACSCalibur (BD Biosciences). The data analysis was done using the FlowJo software (Tree Star).
Immunostaining
HEK293 cells were cultured on coverslips in 6-well plates and transfected with plasmids expressing corin WT and mutants. After 48 h at 37 °C, the cells were fixed with 4% paraformaldehyde or ice-cold acetone, and blocked with 5% bovine serum albumin in PBS. The fixed cells were incubated with primary antibodies against corin, TGN46 (Sigma) or protein-disulfide isomerase (PDI) (BD Biosciences). Secondary antibodies used were conjugated with Alexa Fluor 488 (green) or 594 (red) (Invitrogen). The coverslips were mounted with a solution containing 6-diamidino-2-phenylindole dihydrochloride (DAPI) to stain the nuclei (32). Immunostaining was examined and images were taken with a confocal microscope (Olympus).
Molecular Modeling
Computer-based modeling of corin domains was performed, as described previously (19). Molecular models of corin LDLR2 and Fz2 domains were constructed based on the crystal structures of human LDLR ligand-binding repeat 5 (LDLR-LR5) (33) and mouse Fz8 (34), respectively. The amino acid sequences of LDLR-LR5 and corin LDLR2 were aligned using the Clustal W program (35). Similar alignments were made between the mouse Fz8 and human corin Fz2 sequences. Three-dimensional (3-D) models were created using the iterative threading assembly refinement (I-TASSER) server (36). The model inspection and image generation were done using the PyMOL program (www.pymol.org).
Statistical Analysis
Analysis was done using the SPSS 12 software (SPSS, Chicago, IL). The data are presented as means ± S.D. Student's t test was used to compare the data from two groups. ANOVA was used to compare the data from multi-groups. Differences were considered statistically significant when p values were less than 0.05.
RESULTS
Model of Corin LDLR2 Domain
Previously, we identified two CORIN gene mutations in preeclamptic patients (22). One mutation, K317E, was in the LDLR2 domain and the other, S472G, was in the Fz2 domain (Fig. 1A). Both mutations reduced the pro-ANP processing activity of corin (22). To understand the potential impact of K317E mutation, we generated a corin LDLR2 model based on the crystal structure of human LDLR-LR5 (33). The structure of human LDLR-LR5 is maintained by calcium-binding, disulfide bonds, and the hydrogen-bonding network through backbone amides (Fig. 1B). While the overall folding of corin LDLR2 is similar to that of LDLR-LR5 (Fig. 1C), our model suggests a distinct local structural environment at Lys-317 and adjacent loop containing Gly-332 (Lys-202 in LDLR-LR5) and Asp-333. As shown in Fig. 1C, Lys-317 appears to be solvent-exposed. The substituted Glu-317 may assume a similar side-chain orientation to that of the corresponding Glu-187 in LDLR-LR5, leading to significant charge repulsion of Glu-317 with Asp-333 and possible steric clash of Glu-317 with Thr-315 (Fig. 1C). This may in turn alter the conformation of corin LDLR2.
FIGURE 1.

Corin domain structure and LDLR2 molecular model. A, corin domains. The transmembrane (TM), frizzled (Fz), LDL receptor (LDLR), scavenger receptor (SR), and protease domains of corin are shown. K317E and S472G mutations are indicated. An arrow indicates the corin activation cleavage site between Arg-801 and Ile-802. Catalytic active sites His (H), Asp (D), and Ser (S) in the protease domain are indicated. B, ribbon model of human LDLR-LR5 module structure. Calcium (yellow ball)-binding site is indicated. Hydrogen bonds involving residues Glu-187, Lys-202, and Ser-185 are shown. C, computer-aided homology model of the corin LDLR2 domain. Close-up views highlight a hypothetical side-chain conformation of Lys-317 in stick model (left panel) and its simulation model upon disease-related mutation into Glu-317 (right panel).
Corin Zymogen Activation and Activity
To test these predictions, we expressed corin mutants K317E, K317A, K317D, K317Q, and K317R and tested their zymogen activation and activity in HEK293 cells. Corin WT, activation cleavage site mutant R801A and active site mutant S985A were used as controls. By Western blot analysis, corin zymogen activation, as indicated by an ∼40-kDa band representing the corin protease domain (corin-p) fragment, was detected in the lysate from the HEK293 cells (Fig. 2A). As predicted, this band was absent in the lysate of R801A mutant, in which the activation cleavage site was abolished. In samples of K317E and K317D mutants, the intensity of the ∼40-kDa band was less than that in WT or mutants K317R, K317A, and K317Q (Fig. 2A). When corin zymogen (∼170 kDa) and corin-p bands were quantified, the cleaved corin-p fragment represented 20.2 ± 3.2% of total corin protein in WT (Fig. 2B). Similar percentages were found in mutants K317R (20.1 ± 7.3%), K317A (19.9 ± 6.0%) and K317Q (17.4 ± 5.6%) (n = 5, all p values >0.05 versus WT). In contrast, the percentage was significantly lower in mutants K317E (13.1 ± 4.2%) (n = 5, p < 0.05) and K317D (10.0 ± 2.6%) (n = 5, p < 0.01) compared with that in WT (Fig. 2B).
FIGURE 2.
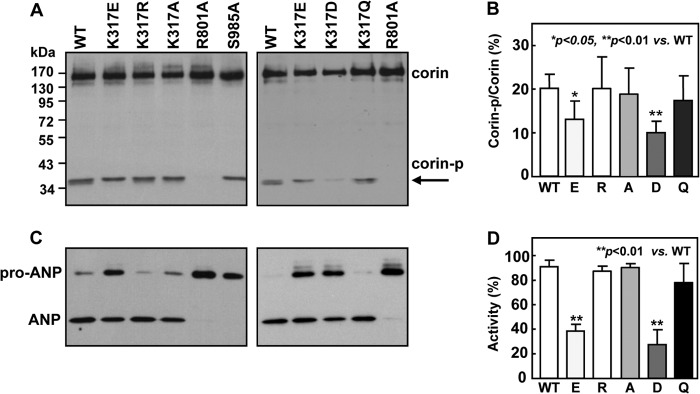
Corin zymogen activation and pro-ANP processing activity. A, Western blot analysis of corin expression and activation in transfected HEK293 cell lysate. The corin protease domain fragment (corin-p) from activation cleavage is indicated by an arrow. B, percentage of the activated corin protease domain fragment versus zymogen protein was estimated by densitometry. Data are means ± S.D. from five independent experiments. C, Western blot analysis of pro-ANP processing activity in corin WT and mutants. D, pro-ANP processing activity in corin WT and mutants, as estimated by densitometry. Data are means ± S.D. from five independent experiments.
In pro-ANP processing assays, corin WT and mutants K317R, K317A, and K317Q had similar activities, whereas mutants K317E and K317D had significantly reduced activities (Fig. 2C). In these studies, negative control mutants R801A and S985A had little activity. By densitometric analysis, mutants K317E and K317D had 40.5 ± 4.9 and 27.5 ± 12.1% activity, respectively, compared with that of WT (n = 5, p values <0.01) (Fig. 2D). The data indicate that changing Lys-317 to an acidic residue (Glu or Asp), but not basic (Arg) or neutral (Ala or Gln) residues, impaired corin zymogen activation and pro-ANP processing activity.
Cell Surface Expression of Corin Mutants
Corin is a transmembrane protein. We examined if the low activity of K317E and K317D mutants was due to low levels of cell surface expression. By Western blot analysis of biotin-labeled cell surface proteins, corin WT and all mutants appeared to be expressed at similar levels (Fig. 3A). To verify this result, we examined corin expression on the surface of intact cells by flow cytometry. As shown in Fig. 3B, the percentage and fluorescent intensity of corin-positive cells were similar in HEK293 cells expressing corin WT and mutants K317E, K317A, K317R, K317D, and K317Q (Fig. 3B). The results indicate that K317E and K317D mutations did not prevent corin expression on the cell surface.
FIGURE 3.
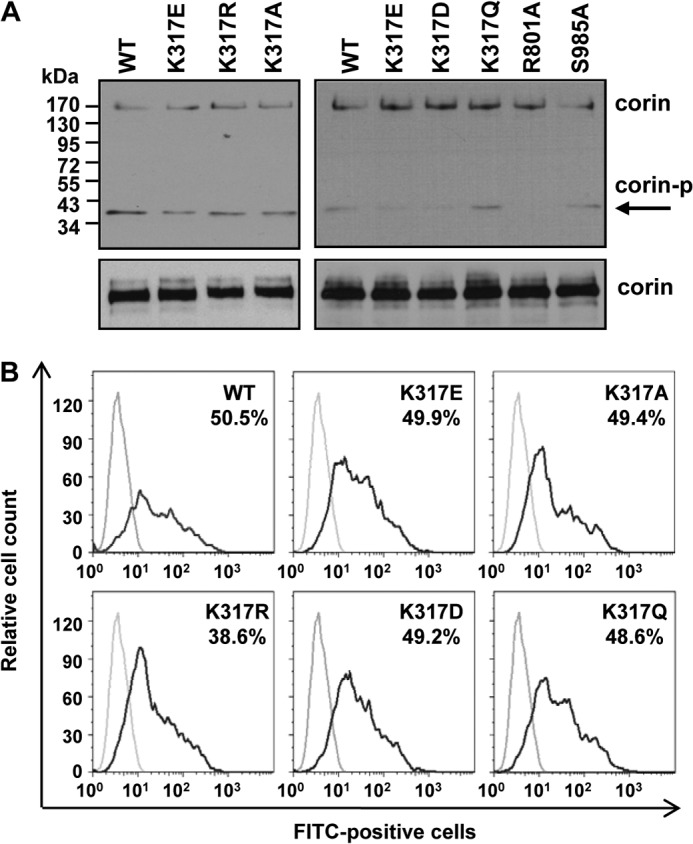
Cell surface expression of corin. A, Western blot analysis of biotin-labeled cell surface corin proteins in transfected HEK293 cells (top panels). As a control, total corin proteins in cell lysates were verified (bottom panels). B, flow cytometric analysis of corin expression on the surface of transfected HEK293 cells. Percentages of corin-positive cells are indicated. Data are representative of five independent experiments.
Soluble Corin Fragments in Cultured Cells
Previous studies show that corin is shed from the cell surface (37). We examined corin shedding in cultured HEK293 cells expressing corin WT and the mutants. Three corin fragments of ∼180, ∼160, and ∼100 kDa, respectively, were found in the conditioned medium from the cells expressing WT corin (Fig. 4). The ∼180-kDa band represented the fragment cleaved near the cell membrane by ADAM proteases, whereas the ∼160- and ∼100-kDa bands represented the fragments from corin autocleavage in the Fz1 and LDLR5 domains, respectively (37). In samples from cells expressing mutants K317R, K317A, and K317Q, three similar bands were detected (Fig. 4). In contrast, only the ∼180-kDa band was detected in samples from K317E and K317D mutants. Similarly, only the ∼180-kDa band was detected in samples from inactive mutants R801A and S985A (Fig. 4). The results indicate that K317E and K317D mutants were expressed on the cell surface but had little activity for autocleavage.
FIGURE 4.
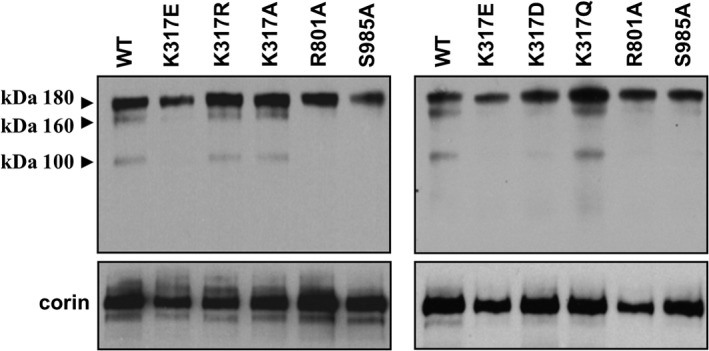
Soluble corin fragments in cell culture medium. Western blot analysis of corin fragments in the conditioned medium from HEK293 cells expressing corin WT and mutants (top panels). As a control, corin proteins in cell lysate were verified by Western blot analysis (bottom panels). Data are representative of four independent experiments.
Model of Corin Fz2 Domain
We also made a three-dimensional model of the corin Fz2 domain, based on the mouse Fz8 crystal structure (34), to understand the potential impact of S472G mutation on corin structure. As shown in Fig. 5 (top circle), there are two antiparallel β-sheets in the Fz2 domain. Hydrogen bonds are predicted between the β-sheets that stabilize the structure. S472G mutation is likely to abolish the β-sheet, in which the mutation resides, and destabilize the overall domain structure (Fig. 5, lower left circle). The model also suggests that changing S472 to Ala (A) would not disrupt the β-sheet and thus have a minimal impact on the overall domain structure (Fig. 5, lower right circle).
FIGURE 5.

Molecular model of corin Fz2 domain. Ribbon model of the corin Fz2 domain was based on the mouse Fz8 crystal structure. Antiparallel β-sheets (green) are shown in zoomed areas for corin WT (top circle) and S472G (lower left circle) or S472A (lower right circle) mutants. N- (N-term) and C- (C-term) termini are indicated.
Functional Analysis of Corin S472G Mutant
We expressed S472G and S472A mutants in HEK293 cells. By Western blot analysis, corin WT and the mutants were found at similar levels in the transfected cells. However, the ∼40-kDa protease domain fragment was not detected in samples from S472G mutant (Fig. 6A). This fragment was present in samples from WT and S472A mutant, but not R801A mutant (Fig. 6A). The results indicate that S472G, but not S472A, mutation prevented corin activation in these cells. Consistent with these results, S472G mutant had little activity in processing pro-ANP, whereas S472A mutant converted pro-ANP to ANP as efficiently as WT corin (Fig. 6B).
FIGURE 6.
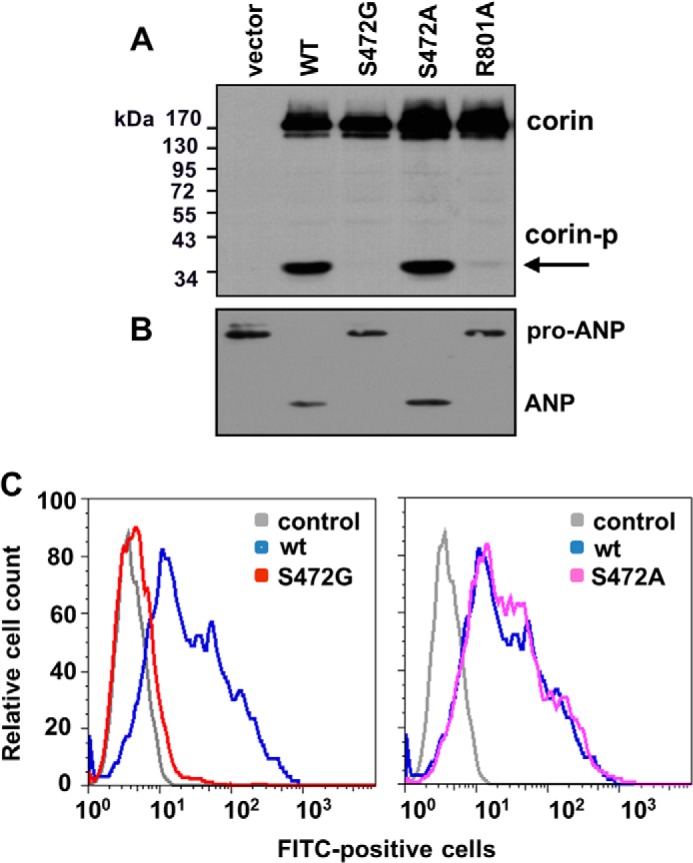
Functional analysis of corin S472G mutant. A, Western blot analysis of corin expression and activation in transfected HEK293 cells. The cleaved corin protease domain fragment (corin-p) is indicated by an arrow. B, Western blot analysis of pro-ANP processing activity in corin WT and mutants. C, flow cytometric analysis of corin expression on the surface of transfected HEK293 cells. Data are representative of four independent experiments.
Cell Surface Expression of S472G Mutant
We examined the surface expression of S472G and S472A mutants in transfected HEK293 cells. In flow cytometry, there appeared minimal S472G mutant protein on the cell surface (Fig. 6C, left panel), whereas the expression levels for S472A mutant and WT were similar (Fig. 6C, right panel). The data indicate that S472G, but not S472A, mutation prevented corin expression on the cell surface.
Intracellular Localization of S472G Mutant
To examine intracellular distribution of S472G mutant, we did co-immunostaining with markers for the ER (PDI) and the Golgi (TGN46) in transfected HEK293 cells. As shown in Fig. 7A, the staining of S472G mutant and PDI overlapped. In contrast, there was little overlapping staining of S472G mutant and TGN46 (Fig. 7B). In controls, the staining of WT corin had minimal overlaps with that of PDI or TGN46 (Fig. 7, A and B). The results indicate that S472G mutant was retained in the ER in these cells.
FIGURE 7.
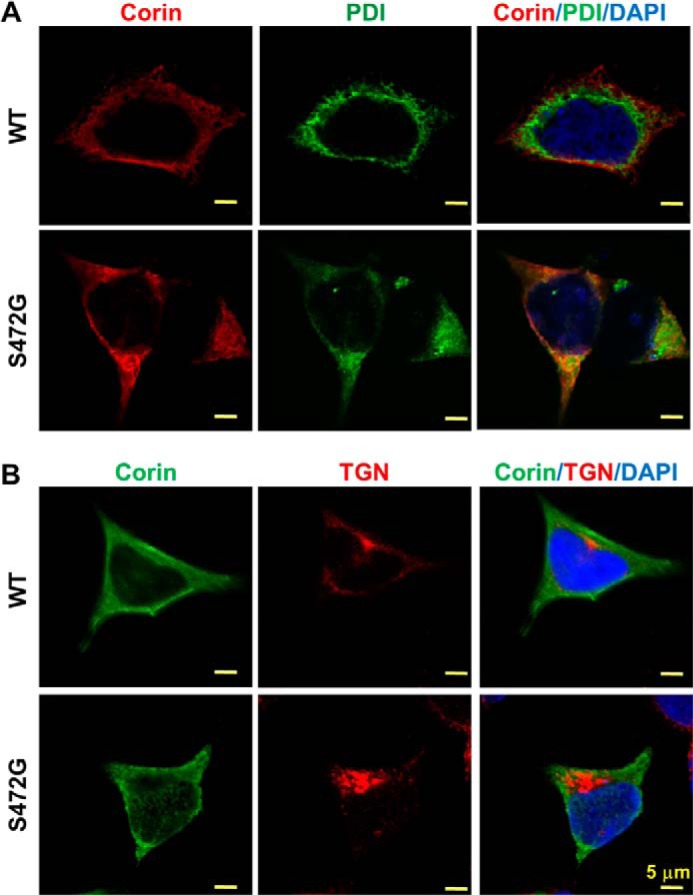
Intracellular distribution of corin WT and S472G mutant. HEK293 cells expressing corin WT and S472G mutant were co-immunostained for corin and PDI (A) or TGN46 (B) under cell membrane-permeable conditions. The images were obtained with a confocal microscope. Scale bars: 5 μm. Data are representative of four independent experiments.
DISCUSSION
In this study, we characterized corin mutations, K317E and S472G, identified in preeclamptic patients (22). The mutation K317E is in the LDLR2 repeat of the corin propeptide. In a three-dimensional model based on the crystal structure of human LDLR-LR5 (Fig. 1B), replacing the positively charged Lys-317 in corin with an acidic residue, Glu or Asp, is expected to induce charge-charge repulsion with Asp-333 and possible steric clash with Thr-315, thereby disturbing the module conformation (Fig. 1C). Consistent with this prediction, both K317E and K317D mutants had reduced pro-ANP processing activity, whereas K317R, K317A, and K317Q mutants had similar activities to that of WT corin (Fig. 2C). Apparently, either a positively charged or neutral residue may occupy the position 317 without compromising corin activity. The results suggest that the damage was caused mainly by the charge-charge repulsion with the neighboring Asp-333, when the negatively charged side-chain of Glu-317 or Asp-317 was introduced.
The position corresponding to corin Lys-317 in the human LDLR-LR5 is Glu-187 (Fig. 1B). In the LDLR-LR5 crystal structure, the side-chain of Glu-187 was shown to form hydrogen bonds with the backbone amides of Lys-202 and the side-chain hydroxyl group of Ser-185, which stabilizes the module structure (33). The local structural environments of corin Lys-317 and LDLR-LR5 Glu-187 appear different due to the unique Gly-332 position in corin LDLR2 versus K202 in LDLR-LR5. K317E substitution in corin LDLR2 and E187K substitution in LDLR-LR5 may disturb the local conformations in different ways. To date, more than 250 LDLR mutations have been reported in patients with familial hypercholesterolemia (38, 39). Among these mutations, many are located in the LDLR-LR5 module, including a particular mutation E187K (39). In a biophysical study, the mutation E187K was shown to alter the folding of the LDLR-LR5 module in a calcium binding-independent manner (40). Our results also suggest that the mutation K317E in corin may cause conformational changes in the LDLR2 module, thereby impairing the corin function.
By analyzing the cleaved corin protease domain fragment, we found that K317E and K317D mutants had reduced zymogen activation, which likely accounted for the reduced pro-ANP processing activity (Fig. 2). To date, it is unclear how corin is activated, as the physiological corin activator remains elusive. It is known, however, that cell surface expression is required for corin activation (30, 41, 42). Previously, mutations in the human LDLR-LR5 module that cause familial hypercholesterolemia were shown to impair the trafficking from the ER to the Golgi, thereby preventing LDLR expression on the cell surface (43). Surprisingly, the cell surface expression of corin mutants K317E and K317D appeared normal in both Western blot analysis and flow cytometric studies (Fig. 3). The results indicate that the mutations K317E and K317D did not disrupt corin intracellular trafficking, even though they altered the LDLR2 module structure.
Like many membrane proteins, corin is shed from the cell surface. In cultured cells, the shedding was mediated by ADAM proteases to produce an ∼180-kDa fragment and by corin autocleavage to produce an ∼160-kDa and an ∼100-kDa fragments (37). Similar shedding may occur in vivo, as soluble corin was detected in human blood (44–47). In patients with hypertension and heart disease, reduced plasma corin levels were reported (48–50). In this study, we detected the three corin fragments in the conditioned medium from the cells expressing WT and K317R, K317A, and K317Q mutants. In the medium from the cells expressing K317E and K317D mutants, only the ∼180-kDa fragment was detected (Fig. 4). The results support the idea that K317E and K317D mutants were expressed on the cell surface. The lack of the ∼160- and ∼100-kDa autocleavage fragments indicates that the mutants had poor activities and thus failed to cleave themselves, consistent with the impaired zymogen activation observed in these mutants.
Apparently, the mutation S472G in the Fz2 domain had a greater impact on corin conformation than the mutation K317E did. In Western blot analysis, zymogen activation was completely inhibited in S472G mutant, as indicated by the lack of the ∼40-kDa band (Fig. 6A). In a three-dimensional model based on the crystal structure of mouse Fz8, a Wnt signaling molecule (34), the mutation S472G is predicted to abolish a β-sheet that is critical for the overall Fz2 module structure. The model also predicts that the β-sheet would be preserved if S472 is replaced by Ala (Fig. 5). Indeed, S472A mutant had normal zymogen activation and pro-ANP processing activity in our experiments (Fig. 6, A and B). In previous studies, alanine-scanning mutagenesis disrupting the corresponding β-sheet in mouse Fz8 abolished its binding to Wnt proteins (34). Thus, data from both mouse Fz8 and corin S472G mutant studies support the importance of the β-sheet in maintaining Fz module structure and function.
By flow cytometry, we did not detect S472G mutant on the cell surface (Fig. 6C), suggesting that impaired intracellular trafficking may block the mutant in the cells. Immunostaining showed that the mutant protein was retained in the ER (Fig. 7A), indicating that the mutation disrupted the β-sheet, resulting in a misfolded protein trapped in the ER. As cell surface expression is important for corin activation, these results help to explain the lack of zymogen activation and activity of S472G mutant in our experiments.
Previously, corin mutants T555I/Q568P and R539C, both located in the Fz2 domain, were reported in hypertensive patients (19, 20). In functional studies, both T555I/Q568P and R539C proteins were shown on the cell surface. However, corin T555I/Q568P remained in an inactive zymogen form, whereas corin R539C was activated but quickly inactivated by autocleavage (19, 51). In this study, corin K317E was found on the cell surface with impaired zymogen activation, whereas corin S472G was found retained in the ER. It is amazing that mutations S472G, R539C, and T555I/Q568P, all located in the Fz2 domain, impaired corin activity by entirely different mechanisms; from ER retention to autoinactivation to impairing zymogen activation. In human LDLR, genetic mutations have been shown to disrupt LDLR at different steps, including protein synthesis in the ER, transport to the Golgi, LDL binding, clustering on the cell membrane, and recycling in endosomes (43). Each class of defects may be caused by mutations in different regions of the LDLR gene, and mutations in the same region of the gene may result in different defects (43). Remarkably, here we show a similar genotype-phenotype relationship in corin. Together, our results provide important insights into the molecular basis underlying corin mutations identified in preeclamptic patients. Our findings should help to understand the potential role of corin defects in hypertensive disease.
This work was supported, in whole or in part, by grants from the National Institutes of Health (HL089298 and HD064634) and grants from the National Natural Science Foundation of China (81170247, 81370718, and 31161130356), the Key Project of Chinese Ministry of Education (213016A), the Danish-Chinese Center for Proteases and Cancer, and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
- Fz
- frizzled
- ANP
- atrial natriuretic peptide
- HEK
- human embryonic kidney
- LDLR
- LDL receptor
- LDLR-LR5
- LDL receptor ligand-binding repeat 5
- PDI
- protein-disulfide isomerase.
REFERENCES
- 1. Sibai B., Dekker G., Kupferminc M. (2005) Pre-eclampsia. Lancet 365, 785–799 [DOI] [PubMed] [Google Scholar]
- 2. Steegers E. A., von Dadelszen P., Duvekot J. J., Pijnenborg R. (2010) Pre-eclampsia. Lancet 376, 631–644 [DOI] [PubMed] [Google Scholar]
- 3. Dechend R., Luft F. C. (2008) Angiogenesis factors and preeclampsia. Nat. Med. 14, 1187–1188 [DOI] [PubMed] [Google Scholar]
- 4. Parikh S. M., Karumanchi S. A. (2008) Putting pressure on pre-eclampsia. Nat. Med. 14, 810–812 [DOI] [PubMed] [Google Scholar]
- 5. Pijnenborg R., Vercruysse L., Hanssens M. (2006) The uterine spiral arteries in human pregnancy: facts and controversies. Placenta 27, 939–958 [DOI] [PubMed] [Google Scholar]
- 6. Red-Horse K., Zhou Y., Genbacev O., Prakobphol A., Foulk R., McMaster M., Fisher S. J. (2004) Trophoblast differentiation during embryo implantation and formation of the maternal-fetal interface. J. Clin. Invest. 114, 744–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Redman C. W., Sargent I. L. (2005) Latest advances in understanding preeclampsia. Science 308, 1592–1594 [DOI] [PubMed] [Google Scholar]
- 8. Roberts J. M., Cooper D. W. (2001) Pathogenesis and genetics of pre-eclampsia. Lancet 357, 53–56 [DOI] [PubMed] [Google Scholar]
- 9. Kaufmann P., Black S., Huppertz B. (2003) Endovascular trophoblast invasion: implications for the pathogenesis of intrauterine growth retardation and preeclampsia. Biol. Reprod 69, 1–7 [DOI] [PubMed] [Google Scholar]
- 10. Hooper J. D., Scarman A. L., Clarke B. E., Normyle J. F., Antalis T. M. (2000) Localization of the mosaic transmembrane serine protease corin to heart myocytes. Eur. J. Biochem. 267, 6931–6937 [DOI] [PubMed] [Google Scholar]
- 11. Yan W., Sheng N., Seto M., Morser J., Wu Q. (1999) Corin, a mosaic transmembrane serine protease encoded by a novel cDNA from human heart. J. Biol. Chem. 274, 14926–14935 [DOI] [PubMed] [Google Scholar]
- 12. Wu Q., Xu-Cai Y. O., Chen S., Wang W. (2009) Corin: new insights into the natriuretic peptide system. Kidney Int. 75, 142–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Barbato E., Bartunek J., Mangiacapra F., Sciarretta S., Stanzione R., Delrue L., Cotugno M., Marchitti S., Iaccarino G., Sirico G., Di Castro S., Evangelista A., Lambrechts D., Sinnaeve P., De Bruyne B., Van De Werf F., Janssens S., Fox K. A., Wijns W., Volpe M., Rubattu S. (2012) Influence of rs5065 atrial natriuretic peptide gene variant on coronary artery disease. J. Am. Coll Cardiol. 59, 1763–1770 [DOI] [PubMed] [Google Scholar]
- 14. Cannone V., Huntley B. K., Olson T. M., Heublein D. M., Scott C. G., Bailey K. R., Redfield M. M., Rodeheffer R. J., Burnett J. C., Jr. (2013) Atrial natriuretic peptide genetic variant rs5065 and risk for cardiovascular disease in the general community: a 9-year follow-up study. Hypertension 62, 860–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hodgson-Zingman D. M., Karst M. L., Zingman L. V., Heublein D. M., Darbar D., Herron K. J., Ballew J. D., de Andrade M., Burnett J. C., Jr., Olson T. M. (2008) Atrial natriuretic peptide frameshift mutation in familial atrial fibrillation. N. Engl. J. Med. 359, 158–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lynch A. I., Claas S. A., Arnett D. K. (2009) A review of the role of atrial natriuretic peptide gene polymorphisms in hypertension and its sequelae. Curr. Hypertens Rep. 11, 35–42 [DOI] [PubMed] [Google Scholar]
- 17. Nakayama T., Soma M., Takahashi Y., Rehemudula D., Kanmatsuse K., Furuya K. (2000) Functional deletion mutation of the 5′-flanking region of type A human natriuretic peptide receptor gene and its association with essential hypertension and left ventricular hypertrophy in the Japanese. Circ. Res. 86, 841–845 [DOI] [PubMed] [Google Scholar]
- 18. Sciarretta S., Marchitti S., Bianchi F., Moyes A., Barbato E., Di Castro S., Stanzione R., Cotugno M., Castello L., Calvieri C., Eberini I., Sadoshima J., Hobbs A. J., Volpe M., Rubattu S. (2013) C2238 atrial natriuretic peptide molecular variant is associated with endothelial damage and dysfunction through natriuretic peptide receptor C signaling. Circ. Res. 112, 1355–1364 [DOI] [PubMed] [Google Scholar]
- 19. Dong N., Fang C., Jiang Y., Zhou T., Liu M., Zhou J., Shen J., Fukuda K., Qin J., Wu Q. (2013) Corin mutation R539C from hypertensive patients impairs zymogen activation and generates an inactive alternative ectodomain fragment. J. Biol. Chem. 288, 7867–7874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dries D. L., Victor R. G., Rame J. E., Cooper R. S., Wu X., Zhu X., Leonard D., Ho S. I., Wu Q., Post W., Drazner M. H. (2005) Corin gene minor allele defined by 2 missense mutations is common in blacks and associated with high blood pressure and hypertension. Circulation 112, 2403–2410 [DOI] [PubMed] [Google Scholar]
- 21. Rame J. E., Drazner M. H., Post W., Peshock R., Lima J., Cooper R. S., Dries D. L. (2007) Corin I555(P568) allele is associated with enhanced cardiac hypertrophic response to increased systemic afterload. Hypertension 49, 857–864 [DOI] [PubMed] [Google Scholar]
- 22. Cui Y., Wang W., Dong N., Lou J., Srinivasan D. K., Cheng W., Huang X., Liu M., Fang C., Peng J., Chen S., Wu S., Liu Z., Dong L., Zhou Y., Wu Q. (2012) Role of corin in trophoblast invasion and uterine spiral artery remodelling in pregnancy. Nature 484, 246–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kaitu'u-Lino T. J., Ye L., Tuohey L., Dimitriadis E., Bulmer J., Rogers P., Menkhorst E., Van Sinderen M., Girling J. E., Hannan N., Tong S. (2013) Corin, an enzyme with a putative role in spiral artery remodeling, is up-regulated in late secretory endometrium and first trimester decidua. Hum. Reprod 28, 1172–1180 [DOI] [PubMed] [Google Scholar]
- 24. Cootauco A. C., Murphy J. D., Maleski J., Blakemore K. J., Slodzinski M. K. (2008) Atrial natriuretic peptide production and natriuretic peptide receptors in the human uterus and their effect on myometrial relaxation. Am. J. Obstet. Gynecol. 199, 429.e1–e6 [DOI] [PubMed] [Google Scholar]
- 25. Gililland J. L., Tseng Y. C., Troche V., Lahiri S., Wartofsky L. (1992) Atrial natriuretic peptide receptors in human endometrial stromal cells. J. Clin. Endocrinol. Metab. 75, 547–551 [DOI] [PubMed] [Google Scholar]
- 26. Hatta K., Carter A. L., Chen Z., Leno-Durán E., Ruiz-Ruiz C., Olivares E. G., Tse M. Y., Pang S. C., Croy B. A. (2011) Expression of the vasoactive proteins AT1, AT2, and ANP by pregnancy-induced mouse uterine natural killer cells. Reprod Sci. 18, 383–390 [DOI] [PubMed] [Google Scholar]
- 27. Itoh H., Sagawa N., Hasegawa M., Nanno H., Kobayashi F., Ihara Y., Mori T., Komatsu Y., Suga S., Yoshimasa T. (1994) Expression of biologically active receptors for natriuretic peptides in the human uterus during pregnancy. Biochem. Biophys. Res. Commun. 203, 602–607 [DOI] [PubMed] [Google Scholar]
- 28. Reis A. M., Jankowski M., Mukaddam-Daher S., Tremblay J., Dam T. V., Gutkowska J. (1997) Regulation of the natriuretic peptide system in rat uterus during the estrous cycle. J. Endocrinol. 153, 345–355 [DOI] [PubMed] [Google Scholar]
- 29. Zhou Y., Wu Q. (2013) Role of corin and atrial natriuretic peptide in preeclampsia. Placenta 34, 89–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Qi X., Jiang J., Zhu M., Wu Q. (2011) Human corin isoforms with different cytoplasmic tails that alter cell surface targeting. J. Biol. Chem. 286, 20963–20969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wu F., Yan W., Pan J., Morser J., Wu Q. (2002) Processing of pro-atrial natriuretic peptide by corin in cardiac myocytes. J. Biol. Chem. 277, 16900–16905 [DOI] [PubMed] [Google Scholar]
- 32. Dong J., Zhao X., Shi S., Ma Z., Liu M., Wu Q., Ruan C., Dong N. (2012) Identification and functional analysis of a novel von Willebrand factor mutation in a family with type 2A von Willebrand disease. PLoS One 7, e33263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fass D., Blacklow S., Kim P. S., Berger J. M. (1997) Molecular basis of familial hypercholesterolaemia from structure of LDL receptor module. Nature 388, 691–693 [DOI] [PubMed] [Google Scholar]
- 34. Dann C. E., Hsieh J. C., Rattner A., Sharma D., Nathans J., Leahy D. J. (2001) Insights into Wnt binding and signalling from the structures of two Frizzled cysteine-rich domains. Nature 412, 86–90 [DOI] [PubMed] [Google Scholar]
- 35. Chenna R., Sugawara H., Koike T., Lopez R., Gibson T. J., Higgins D. G., Thompson J. D. (2003) Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 31, 3497–3500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Roy A., Kucukural A., Zhang Y. (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jiang J., Wu S., Wang W., Chen S., Peng J., Zhang X., Wu Q. (2011) Ectodomain shedding and autocleavage of the cardiac membrane protease corin. J. Biol. Chem. 286, 10066–10072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brown M. S., Herz J., Goldstein J. L. (1997) LDL-receptor structure. Calcium cages, acid baths and recycling receptors. Nature 388, 629–630 [DOI] [PubMed] [Google Scholar]
- 39. Hobbs H. H., Brown M. S., Goldstein J. L. (1992) Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum. Mutat. 1, 445–466 [DOI] [PubMed] [Google Scholar]
- 40. Blacklow S. C., Kim P. S. (1996) Protein folding and calcium binding defects arising from familial hypercholesterolemia mutations of the LDL receptor. Nat. Struct. Biol. 3, 758–762 [DOI] [PubMed] [Google Scholar]
- 41. Gladysheva I. P., King S. M., Houng A. K. (2008) N-glycosylation modulates the cell-surface expression and catalytic activity of corin. Biochem. Biophys. Res. Commun. 373, 130–135 [DOI] [PubMed] [Google Scholar]
- 42. Liao X., Wang W., Chen S., Wu Q. (2007) Role of glycosylation in corin zymogen activation. J. Biol. Chem. 282, 27728–27735 [DOI] [PubMed] [Google Scholar]
- 43. Hobbs H. H., Russell D. W., Brown M. S., Goldstein J. L. (1990) The LDL receptor locus in familial hypercholesterolemia: mutational analysis of a membrane protein. Annu. Rev. Genet. 24, 133–170 [DOI] [PubMed] [Google Scholar]
- 44. Dong N., Chen S., Wang W., Zhou Y., Wu Q. (2012) Corin in clinical laboratory diagnostics. Clin. Chim. Acta 413, 378–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dong N., Dong J., Liu P., Xu L., Shi S., Wu Q. (2010) Effects of anticoagulants on human plasma soluble corin levels measured by ELISA. Clin. Chim. Acta 411, 1998–2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ichiki T., Huntley B. K., Heublein D. M., Sandberg S. M., McKie P. M., Martin F. L., Jougasaki M., Burnett J. C., Jr. (2011) Corin is present in the normal human heart, kidney, and blood, with pro-B-type natriuretic peptide processing in the circulation. Clin. Chem. 57, 40–47 [DOI] [PubMed] [Google Scholar]
- 47. Peleg A., Jaffe A. S., Hasin Y. (2009) Enzyme-linked immunoabsorbent assay for detection of human serine protease corin in blood. Clin. Chim. Acta 409, 85–89 [DOI] [PubMed] [Google Scholar]
- 48. Dong N., Chen S., Yang J., He L., Liu P., Zheng D., Li L., Zhou Y., Ruan C., Plow E., Wu Q. (2010) Plasma soluble corin in patients with heart failure. Circ. Heart Fail 3, 207–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ibebuogu U. N., Gladysheva I. P., Houng A. K., Reed G. L. (2011) Decompensated heart failure is associated with reduced corin levels and decreased cleavage of pro-atrial natriuretic peptide. Circ. Heart Fail 2011, 114–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Peleg A., Ghanim D., Vered S., Hasin Y. (2013) Serum corin is reduced and predicts adverse outcome in non-ST-elevation acute coronary syndrome. Eur. Heart J. Acute Cardiovasc. Care 2, 159–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang W., Liao X., Fukuda K., Knappe S., Wu F., Dries D. L., Qin J., Wu Q. (2008) Corin variant associated with hypertension and cardiac hypertrophy exhibits impaired zymogen activation and natriuretic peptide processing activity. Circ. Res. 103, 502–508 [DOI] [PMC free article] [PubMed] [Google Scholar]