

Background: Development of new therapeutic approaches for Alzheimer disease (AD) is crucial.
Results: A recombinant enzyme engineered to cross the blood-brain barrier treats the accumulation of Aβ in a mouse model.
Conclusion: This study shows a therapy can improve the symptoms of AD while also increasing the population of new neurons.
Significance: The enzyme could be delivered weekly or even monthly to patients.
Keywords: Alzheimer Disease, Lipoprotein Receptor, Neurogenesis, Protein Targeting, Transcytosis, Aβ, Blood Brain Barrier, Brain Penetrating Peptides, NPY, Neprilysin
Abstract
Alzheimer disease (AD) is characterized by widespread neurodegeneration throughout the association cortex and limbic system, deposition of amyloid-β peptide (Aβ) in the neuropil and around the blood vessels, and formation of neurofibrillary tangles. The endopeptidase neprilysin has been successfully used to reduce the accumulation of Aβ following intracranial viral vector delivery or ex vivo manipulated intracranial delivery. These therapies have relied on direct injections into the brain, whereas a clinically desirable therapy would involve i.v. infusion of a recombinant enzyme. We previously characterized a recombinant neprilysin that contained a 38-amino acid brain-targeting domain. Recombinant cell lines have been generated expressing this brain-targeted enzyme (ASN12). In this report, we characterize the ASN12 recombinant protein for pharmacology in a mouse as well as efficacy in two APPtg mouse models of AD. The recombinant ASN12 transited to the brain with a t½ of 24 h and accumulated to 1.7% of injected dose at 24 h following i.v. delivery. We examined pharmacodynamics in the tg2576 APPtg mouse with the prion promoter APP695 SWE mutation and in the Line41 mThy1 APP751 mutation mouse. Treatment of either APPtg mouse resulted in reduced Aβ, increased neuronal synapses, and improved learning and memory. In addition, the Line41 APPtg mice showed increased levels of C-terminal neuropeptide Y fragments and increased neurogenesis. These results suggest that the recombinant brain-targeted neprilysin, ASN12, may be an effective treatment for AD and warrant further investigation in clinical trials.
Introduction
Alzheimer disease (AD)2 is an incurable progressive neurodegenerative disorder affecting over 10 million people in the United States alone (1). This neurological disorder is characterized by widespread neurodegeneration throughout the association cortex and limbic system, deposition of Aβ in the neuropil and around the blood vessels, and formation of neurofibrillary tangles (2). Despite the considerable progress toward better understanding of the pathogenesis of AD, no effective therapeutic approaches are currently available.
Recent therapeutic approaches for the treatment of Alzheimer's disease have focused on immunotherapy to decrease the accumulation of Aβ and small molecules to block the formation of Aβ. Another approach that we and others have investigated has been the delivery of proteases for the targeted degradation of Aβ (3). One such protease, neprilysin (NEP), a zinc metalloendopeptidase, has been identified as a critical Aβ-degrading enzyme (4–6). Neprilysin has been shown to degrade Aβ monomers; however, the ability of NEP to degrade Aβ oligomers is controversial, and although some groups have reported that this endopeptidase breaks down oligomers (7, 8), others have not seen such effects (9, 10).
In addition to Aβ, NEP is capable of cleaving a wide range of neuropeptides with neurotrophic activity, including somatostatin, substance P, enkephalin, and neuropeptide Y (NPY) (11), and might regulate the activity of growth factors. Studies by Saito et al. (12) have shown that somatostatin and NEP co-regulate their expression, and we have recently shown that NPY fragments resulting from NEP proteolysis might be neuroprotective (13). Thus, NEP represents a unique example of a proteolytic enzyme with dual action, degradation (Aβ) and processing (NPY); and both actions are neuroprotective.
Peripheral delivery of neprilysin with viral vector-mediated gene delivery (14, 15), ex vivo cellular manipulation (16, 17), or intravenous recombinant protein delivery (18) failed to deliver neprilysin enzyme to the CNS. We recently developed a neprilysin containing a brain-transport peptide that was able to reduce CNS Aβ and improve learning and memory in the APPtg mouse (14). This therapy was delivered by viral vector-mediated gene delivery to the liver such that the liver acted as depot organ for production and secretion of the brain-targeted neprilysin protein. For this report, we developed cell lines to produce the recombinant brain-targeted neprilysin, allowing us to examine the pharmacokinetics and pharmacodynamics of i.v. delivery of recombinant brain-targeted neprilysin protein.
The recombinant brain-targeted neprilysin allowed us to examine the pharmacokinetics of intravenous delivery in a mouse. We found the brain-targeted protein was transported across the blood-brain barrier and accumulated in the brain with a t½ of 24 h. Treatment of two different APPtg mouse models of AD by repeated injections resulted in reduced intraneuronal Aβ, and ameliorating Aβ induced neuropathology. In addition, mice treated with the brain-targeted neprilysin showed increased levels of C-terminal NPY fragments as well as increased neurogenesis. Furthermore, the brain-targeted neprilysin-treated APPtg mice showed improvements in learning and memory in the water maze. We believe these results are consistent with a potentially viable new therapeutic for AD.
EXPERIMENTAL PROCEDURES
Protein Expression in Human Fibroblasts
To construct clonal recombinant cell lines, adherent human fibroblast 293T cells were transfected with a vector expressing either ApoBSecNep or the control SecNep cDNA described previously (14). Cells were selected for transfection by continuous application of Zeocin (400 μg/ml) for 2 weeks. Individual clones were picked and grown in selection media before screening for protein expression, secretion, and enzyme activity. ASN12 represented the best clone expressing ApoBSecNep, and SN5 represented the best clone expressing SecNep. Clones were propagated in DMEM supplemented with 10% FBS and 100 μg/ml Zeocin.
Purification of Recombinant Protein
Large scale production of recombinant ASN12 and SN5 was conducted by Biologics Process Development (Poway, CA). Briefly, clonal cell lines were grown in batch culture roller bottles containing DMEM + 10% FBS for 96 h. Cultured media were handled with low endotoxin procedure. Media were filtered with a 0.45 μm cellulose acetate filter and then dialyzed with 20 mm Tris, pH 7.5, overnight. The dialyzed media were applied to a 5-ml HiTrap Q FF column (GE Healthcare), washed, and eluted with 1 m NaCl, 20 mm Tris, pH 7.5.
Pooled fractions of SN5 were then applied to an 8-ml FLAG affinity column (Sigma) in 150 mm NaCl, 50 mm Tris, pH 7.5. Recombinant SN5 was eluted with 0.1 m glycine, pH 3.5, and immediately neutralized with 1 m Tris, pH 9.0.
Pooled fractions of ASN12 were applied to a 1-ml HiTrap Blue HP column (GE Healthcare), which bound residual serum. The flow-through containing ASN12 was applied to a 1-ml HiTrap Q HP column (GE Healthcare) in 20 mm Tris, pH 7.5. Protein was eluted with 1 m NaCl, 20 mm Tris, pH 7.5.
Pooled fractions containing ASN12 or SN5 were pooled, dialyzed with 20 mm Tris, pH 7.5, overnight, and filtered with a 0.2-μm filter. Endotoxin was measured (Pierce) at <20 endotoxin units/mg for both proteins.
NEP ELISA
An electrochemiluminescence ELISA-based assay for human neprilysin was generated from the MSD ELISA conversion kit (MesoScale Discovery). Briefly, goat anti-human neprilysin (R&D Systems, 100 ng/well) was coated onto 96-well MSD plates overnight at 4 °C. Prior to testing, blood was diluted 1:5 with PBS. Tissue samples were tested at 100 μg/well. Samples were added to the wells and incubated at room temperature for 2 h with shaking, and then a biotinylated anti-human neprilysin (R&D Systems, 1 μg/ml) antibody was used for detection of the recombinant protein. Following this, samples were incubated with streptavidin linked Sulfo-Tag (MSD), and plates were read on an MSD-7500 plate reader. Standards included recombinant human neprilysin ranging from 1 pg to 1 μg.
Neprilysin Activity Assay
The proteolytic activity of NEP was measured as described previously (19) using the substrate 3-dansyl-d-Ala-Gly-p-(nitro)-Phe-Gly (DAGNPG; Sigma). Cell lysate was incubated with 50 μm DAGNPG and 1 μm captopril (to inhibit ACE cleavage of DAGNPG) in a volume of 200 μl at 37 °C. Reactions were stopped by heating samples to 100 °C for 5 min and then centrifuging. The supernatant was diluted into 50 mm Tris, pH 7.4, and fluorescence was determined using a Victor2 multilabel plate reader (excitation 342 nm; emission 562 nm).
In Vitro Assays
Uptake of recombinant protein was tested in adherent HepG2 human hepatocellular cells. Cells were plated in 12-well tissue culture dishes for 48 h in complete media and then switched to serum-reduced media consisting of DMEM + 0.5% FBS. 24 h after incubation in serum-starved media, cells were washed once with PBS and then treated with ASN12, SN5, or saline control for 4 h at 37 °C. Cells were then washed three times with ice-cold PBS and lysed with RIPA buffer (14) containing protease inhibitors (Roche Applied Science). Protein concentration was assessed by Bradford colorimetric assay, and recombinant protein was measured by the MSD ELISA described above.
In vitro rat blood brain cultures were grown according to the manufacturer's directions (PharmaCo Cell). Prior to treatment with the test proteins, trans-endothelial electrical resistance (TEER) was measured to verify the integrity of the barrier. All wells had a TEER measurement of >250 megohms/cm2.
To determine the rate of transport, 1 μg of protein (ASN12, SN5, or GCmB6) was added to the blood side (insert) of the culture, and then media were sampled at various time points from the brain side (well). Samples were assayed with a human neprilysin-specific MSD ELISA along with a standard curve.
To test for autoantibodies that may have developed in the mouse against the recombinant protein, Western blots were run with recombinant ASN12 (1 μg) and then probed with whole blood from mice. Briefly, ASN12 recombinant protein was run on a 4–12% BisTris-PAGE and then blotted to PVDF. Blots were probed with whole blood samples from mice diluted 1:500 in PBS. Secondary antibody consisted of anti-mouse HRP, and blots were analyzed with Lumiphos on a Bio-Rad VersaDoc CCD camera. A positive control was performed with mouse monoclonal anti-neprilysin (R&D Systems).
Pharmacokinetics
To determine the pharmacokinetics of the ASN12 and SN5 proteins in vivo, 5–6-week-old female C57/Bl6 mice were purchased from The Jackson Laboratory and housed for a minimum of 48 h prior to injection. Mice received a single intravenous injection of 1 mg/kg ASN12, SN5, or saline in a volume of 100 μl. Three mice were used for each time point for ASN12, SN5, and saline control. At time points ranging from 15 min to 48 h after injection, heparinized blood was sampled from the mandibular joint. Mice were sacrificed by cervical dislocation, and brains and peripheral tissues were removed and divided sagitally. The right hemibrain was post-fixed in phosphate-buffered 4% PFA, pH 7.4, at 4 °C for 48 h for immunohistological analysis, whereas the left hemibrain was snap-frozen and stored at −70 °C for subsequent protein analysis. Tissues were homogenized as described previously in RIPA buffer containing protease inhibitors (Roche Applied Science) (14). Fixed brains were sectioned on a sliding microtome (40 μm) prior to staining as described previously (20).
Transgenic Animals
Two separate APPtg mouse lines were used in the course of this project. The APPtg tg2576 mouse was purchased from Taconic. These mice express the human APP Swedish mutation (APP695 gene containing the double mutation K670N/M671L) using the hamster prion promoter (21). 11–12-week-old female transgenic and nontransgenic littermate mice were intravenously injected in the tail vein with 1 mg/kg dose of ASN12, SN5, or saline in a 100-μl volume every 72 h for a total of 3 weeks. Five transgenic and nontransgenic mice each were used for ASN12, SN5, and saline injection. Mice were weighed prior to the start of the experiment and every week during the experiment. At the conclusion of the experiment, heparinized blood was sampled from the mandibular joint. Mice were sacrificed by cervical dislocation, and brains and peripheral tissues were removed and divided sagitally. The right hemibrain was post-fixed in phosphate-buffered 4% PFA, pH 7.4, at 4 °C for 48 h for immunohistological analysis, whereas the left hemibrain was snap-frozen and stored at −70 °C for subsequent protein analysis. Tissues were homogenized as described previously in RIPA buffer containing protease inhibitors (Roche Applied Science) (14). Fixed brains were sectioned on a sliding microtome (40 μm) prior to staining as described previously (20).
The second mouse line used for the project was the Line 41 APPtg mouse developed by Eliezer Masliah at the University of California (22, 23). The mice express the express the human APP751 under the control of the mThy-1 promoter. The 6-month-old male transgenic and nontransgenic littermate mice received intraperitoneal injections twice weekly for a total of 3 weeks. The five transgenic and nontransgenic mice each were used for ASN12, SN5, and saline injection. At the conclusion of the dosing, mice were examined by the Morris water maze, and then heparinized blood was sampled from mandibular joint. Mice were anesthetized with chloral hydrate and flush-perfused transcardially with 0.9% saline. Brains and peripheral tissues were removed and divided sagitally. The right hemibrain was post-fixed in phosphate-buffered 4% PFA, pH 7.4, at 4 °C for 48 h for immunohistological analysis, and the left hemibrain was snap-frozen and stored at −70 °C for subsequent protein analysis. Tissues were homogenized as described previously in RIPA buffer containing protease inhibitors (Roche Applied Science) (14). Fixed brains were sections on a vibratome (40 μm) prior to staining as described previously (14).
Analysis of Aβ by Immunocytochemistry
To evaluate neuronal Aβ, sections were immunolabeled with the mouse monoclonal antibody against Aβ (clone 4G8; Senetek or clone 82E1; IBL International) followed by incubation with secondary biotinylated anti-mouse IgG and then Avidin biotin complex and 3,3′-diaminobutyric acid. Alternatively, sections were immunolabeled with the mouse monoclonal antibody against the pyroglutamate modified Aβ (clone D129; Affiris AG) followed by incubation with secondary FITC-conjugated anti-mouse (24). Sections were transferred to SuperFrost slides (Fisher) and mounted under glass coverslips with anti-fading media (Vector Laboratories) (14). All sections were processed under the same standardized conditions. Three immunolabeled sections were analyzed per mouse, and the average of individual measurements was used to calculate group means.
Analysis of NEP Expression, Double Immunolabeling, and Neurodegeneration
To verify the expression levels of NEP, sections were immunolabeled with a monoclonal antibody against NEP (Abcam) and detected with Alexa-conjugated secondary antibodies (Invitrogen). To evaluate the co-localization of NEP, double immunocytochemical analysis was performed as described previously (25). For this purpose, sections were immunolabeled with a monoclonal antibody against NEP (Abcam), detected with the Tyramide Signal Amplification (Invitrogen), and the mouse monoclonal antibodies against MAP2 (Millipore), NeuN (Millipore), or glial fibrillary acidic protein (GFAP, astroglial marker, Invitrogen). All sections were processed simultaneously under the same conditions, and experiments were performed twice to assess reproducibility. Sections were imaged with a Zeiss 63× (N.A. 1.4) objective on an Axiovert 35 microscope (Zeiss) with an attached MRC1024 LSCM system (Bio-Rad) (25). To confirm the specificity of primary antibodies, control experiments were performed where sections were incubated overnight in the absence of primary antibody (deleted) or preimmune serum and primary antibody alone.
To determine whether the neprilysin protein delivery ameliorated the neurodegenerative alterations in the APPtg mice, blind-coded sections from mouse brains were immunolabeled with the mouse monoclonal antibodies against microtubule-associated protein-2 (MAP2, dendritic marker, Invitrogen) or GFAP (astroglial marker, Invitrogen) (26). After overnight incubation, labeled sections were incubated with FITC-conjugated horse anti-mouse IgG secondary antibody (Vector Laboratories), transferred to SuperFrost slides (Fisher), and mounted under glass coverslips with anti-fading media (Vector Laboratories). GFAP-labeled sections were incubated with biotinylated secondary antibody and reacted with diaminobenzidine.
The integrity of the neuronal structure was evaluated as described previously (23, 27); briefly, blind-coded sections from mouse brains were immunolabeled with the mouse monoclonal antibodies against synaptophysin (Millipore) and detected with the tyramide signal amplification (Invitrogen). After staining, sections were transferred to SuperFrost slides (Fisher) and mounted under glass coverslips with anti-fading media (Vector Laboratories). All sections were processed under the same standardized conditions. The immunolabeled blind-coded sections were serially imaged with Zeiss and analyzed with the Image 1.43 program (National Institutes of Health), as described previously (26). For each mouse, a total of three sections were analyzed, and for each section, four fields in the frontal cortex and hippocampus were examined.
Additional immunohistochemistry was performed with antibodies against DCX (Santa Cruz Biotechnology, goat polyclonal), PCNA (Santa Cruz Biotechnology, mouse monoclonal), full-length NPY (Bachem, rabbit polyclonal), C-terminal NPY (CT-NPY, Invitrogen, rabbit polyclonal (13)), and NPY Y2 receptor (Y2-R, Novus Biologics, rabbit polyclonal). Following incubation with primary antibodies, sections were incubated with appropriate biotinylated conjugated secondary antibodies, followed by ABC and DAB. Sections were transferred to SuperFrost slides (Fisher) and mounted under glass coverslips with anti-fading media (Vector Laboratories). All sections were processed under the same standardized conditions. For purposes of cell quantitation, morphometric optical dissector analysis was performed as described previously (28).
Water Maze Testing
As described previously (27) to evaluate the functional effects of treatment in mice, groups of APPtg and non-tg animals were tested in the water maze. For this purpose, a pool (diameter 180 cm) was filled with opaque water (24 °C), and mice were first trained to locate a visible platform (days 1–3) and then a submerged hidden platform (days 4–7) in three daily trials 2–3 min apart. Mice that failed to find the hidden platform within 90 s were placed on it for 30 s. The same platform location was used for all sessions and all mice. The starting point at which each mouse was placed into the water was changed randomly between two alternative entry points located at a similar distance from the platform. Time to reach the platform (latency) and percentage of time in the target quadrant were recorded with a Noldus Instruments EthoVision video tracking system (San Diego Instruments) set to analyze two samples/s.
Statistical Analyses
Analyses were carried out with the Excel program (Microsoft Corp.). Differences among means were assessed by one-way ANOVA. All values in the figures are expressed as means ± S.D. Comparisons between two groups were done with the unpaired two-tailed Student's t test, and the null hypothesis was rejected at the 0.05 level.
RESULTS
ASN12 Protein Purification
Previously, we showed that delivery of the ApoBSecNep gene by lentivirus vector injection was able to reduce Aβ accumulation and restore synaptic density in APPtg mice. To develop a more clinically amenable approach, we constructed recombinant cell lines to express and secrete the previously described ApoBSecNep protein or the control SecNep protein (14). Human fibroblast 293T cells were transfected with plasmids expressing either ApoBSecNep or SecNep, and positive clones were selected by addition of the selection agent Zeocin. Individual clones were examined for expression of active neprilysin enzyme. The clone ASN12 expressed an active ApoBSecNep, and clone SN5 expressed an active SecNep protein. Both clones secreted ∼10 mg/liter of recombinant protein. Protein was purified as described under “Experimental Procedures” and analyzed by Coomassie-stained gel for impurities (Fig. 1A). A similar gel was run alongside with a recombinant human neprilysin and was probed with the anti-human neprilysin antibody resulting in a single band at the appropriate ∼100-kDa size (Fig. 1A). Functional assay measurement of the purified recombinant protein showed SN5 had activity similar to commercially purchased recombinant neprilysin enzyme activity/μg (Fig. 1B). In contrast, the ASN12 protein showed ∼75% of the recombinant neprilysin activity (Fig. 1B). This is similar to results observed with protein activity from the original lentivirus vector (14) and probably represents some loss of activity with the addition of the 38-amino acid apoB LDLR binding domain to the N terminus of the protein. Addition of thiorphan, a specific inhibitor of neprilysin, to the reaction blocked activity of both SN5 and ASN12 enzymatic activity. The purified SN5 and ASN12 proteins were used for all future in vitro and in vivo analyses.
FIGURE 1.

Purified ASN12 is taken up and transported by the LDL receptor. A, ASN12 and SN5 were affinity column-purified. Recombinant protein was analyzed by Coomassie-stained polyacrylamide gel. M, marker; RFU, relative fluorescence units. B, recombinant protein was assayed for enzymatic activity with the DAGNP substrate. C, dose response of ASN12 and SN5 protein uptake by serum-starved HepG2 cells. D, diagram of the blood-brain barrier, and E, in vitro system used to examine the rate of protein transport at the blood-brain barrier. F, rates of blood-brain barrier transcytosis of ASN12 and SN5 were calculated with the in vitro blood-brain barrier system. Competition for transport was determined with the LDLR binding GCmB6 recombinant protein.
ASN12 Receptor-mediated Uptake and Transport
To examine the selective uptake of the ASN12 protein, serum-starved HepG2 cells were used as a surrogate of LDLR-expressing cells. Serum/lipid starvation up-regulates the cell surface expression of LDLR thus increasing the uptake of apolipoprotein-binding proteins (29). HepG2 cells were incubated with increasing concentrations of ASN12 or SN5 for 4 h and then were assayed for internalized neprilysin (Fig. 1C). The ASN12 protein was specifically taken up by HepG2 cells following a 4-h incubation in a dose-dependent manner. In contrast, the control SN5 protein was not taken up by the cells.
To examine the kinetics of transport of the ASN12 protein, an in vitro cell culture system designed to approximate the blood-brain barrier was used (Fig. 1, D and E). Cells were grown until TEER measurements reached >250 megohms/cm2. Recombinant ASN12 (1 μg) or SN5 (1 μg) was added to the “blood” side of the culture system. At various times, the “brain” side of the culture was sampled and analyzed with the neprilysin ELISA. ASN12 displayed a linear first-order kinetics transport rate across the in vitro blood-brain barrier at 3.5 ng/min (Fig. 1F). This contrasted to the control neprilysin lacking the brain targeting peptide (SN5), which did not cross the blood-brain barrier at any appreciable rate. Competition with equal molar amounts of another brain-targeted protein, GCmB6 (glucocerebrosidase fused to the same 38-amino acid LDLR binding domain of apoB (20)), reduced the rate of transport of the ASN12 protein to 1.9 ng/min as would be expected if they were competing for the same receptor/transport mechanism (Fig. 1F).
ASN12 Protein Pharmacokinetics in a Mouse
To determine the pharmacokinetics of the ASN12 and SN5 proteins in vivo, 5–6-week-old female C57/Bl6 mice received a single intravenous injection of 1 mg/kg ASN12 or SN5. At time points ranging from 15 min to 48 h after injection, whole blood, brain, and liver were examined for accumulation of the recombinant human neprilysin protein.
Analysis of whole heparinized blood with a human neprilysin-specific ELISA showed an initial peak of 1.2 to 1.5 μg/ml recombinant protein in the blood. This concentration diminished over time with a t½ of ∼7.5 h (Fig. 2A and Table 1). The concentration at 24 h was 131 ng/ml of SN5 and 41 ng/ml of ASN12. Similarly, an initial peak of recombinant protein was observed in the whole liver homogenates at the earliest time point analyzed (15 min) (Fig. 2B and Table 1). Accumulation of neprilysin peaked with ∼470 ng/mg tissue in the liver with a rapid reduction over time. The t½ for SN5 was 7.0 h; however, the t½ for ASN12 was only 3.0 h.
FIGURE 2.

Pharmacokinetics of ASN12 and SN5 in C57/Bl6 mice. C57/Bl6 mice received 1 mg/kg recombinant ASN12 or SN5 in a 100-μl volume or 100 μl of saline as a control by intravenous delivery to the tail vein. A, at various times after injection, mice were sacrificed and blood (A), liver (B), and brain (C) were sampled for recombinant human neprilysin with an ELISA. n = 3 for each group. D, fixed brains (frontal cortex) were sectioned and stained for recombinant human neprilysin followed by anti-mouse Alexa488 (green) and counterstained with DAPI (nuclei, blue). Scale bars = 25 μm.
TABLE 1.
Pharmacokinetic parameters of ASN12 intravenous delivery to mice
Recombinant ASN12 was delivered by intravenous tail vein injection at 1 mg/kg to C57/Bl6, female, 6-month-old mice. PK calculations were made from the concentration versus time curves in Fig. 2. n = 3 per time point. ND means not determined. AUC mean area under the curve.
| Plasma | Brain | Liver | |
|---|---|---|---|
| AUC(0-inf) (ng/ml (or mg)·h) | 9438.8 | 29.28 | 398.12 |
| Clearance ng/h | 2.12 ng/h | ND | 50.23 ng/h |
| t½ | 7.51 h | 22.54 h | 7.00 h |
| Vd | 22.97 ml | ND | ND |
| % ID at 24 h | 0.29 | 1.70 | 37.23 |
Analysis of whole brain homogenates by ELISA showed an initial peak of recombinant protein at the earliest time point examined (15 min) (Fig. 2C and Table 1). This may reflect circulating recombinant protein, as the brain was not perfused prior to analysis. By 6 h, we observed a significant reduction in SN5 in the brain with protein detected at 24 h post-injection at the lower limit of our assay. In contrast, ASN12 was detectable at 24 h (210 pg/mg) and at 48 h (140 pg/mg). At 24 h ∼1.7% of the injected dose had accumulated in the brain (Table 1).
To determine whether these levels of recombinant protein were relevant, we analyzed human neprilysin levels in human cortical brain homogenates. Levels of neprilysin were measured at 200 pg/mg in human tissues indicating that we were obtaining endogenous levels of neprilysin 24 and 48 h after intravenous injection in the mouse (data not shown).
The ELISA results for recombinant neprilysin were confirmed by immunohistochemistry of sectioned mouse brains. SN5 was observed in the frontal cortex of mice 30 min after injection; however, this protein was confined to vessel-like structures, and protein was not observed in the neuropil beyond 4 h (Fig. 2C). In contrast, at 30 min post-injection and extending to 15 h, ASN12 was observed in the neuropil in the frontal cortex of injected mice. Similar results were observed in the hippocampus (data not shown). These results suggest that following intravenous injection with the ASN12 protein; we are able to obtain physiological levels of human neprilysin in the CNS and the protein accumulated in the cortex and hippocampus of the brain, two regions primarily affected in Alzheimer disease.
ASN12 Efficacy in an APPtg Mouse
To determine whether delivery of the ASN12 recombinant protein is effective at reducing the accumulation of Aβ, we utilized the tg2576 mouse model of Alzheimer disease purchased from Taconic, which has the APP Swedish mutation (APP695 gene containing the double mutation K670N and M671L) using the hamster prion promoter (21). Based on data from the PK experiments, recombinant protein was administered every 72 h to retain physiological levels in the CNS. The 11–12-week-old female transgenic and nontransgenic littermate mice were intravenously injected by the tail vein with 1 mg/kg dose of either ASN12 or SN5 in a 100-μl volume of saline. Control mice were injected with 100 μl of saline. Blood was sampled weekly during the experiment. At the conclusion of the experiment, blood and CSF were sampled, mice were sacrificed, and whole brain was removed for analysis.
ELISA analysis of human neprilysin in the whole blood of treated mice showed significant levels of SN5 in both APPtg and non-tg mice across the study (Fig. 3A). Levels of SN5 appeared to decline slowly over the course of the study with a 25% decline over the 3-week injection period. In contrast, low levels of ASN12 were detected in the whole blood of either APPtg or non-tg treated mice across the whole study (Fig. 3A). These levels also appeared to decline slightly over the course of the treatments.
FIGURE 3.

Distribution of ASN12 and SN5 following repeat i.v. administration to tg2576 APPtg mice. tg2576 APPtg and non-tg mice received i.v. injections of ASN12 or SN5 (1 mg/kg) or saline every 3 days for 3 weeks. A, 3 days after the last injection, whole blood was sampled and assayed for human neprilysin with an ELISA. Mice were sacrificed, and whole brains were removed half-frozen for protein analysis and half-fixed for tissue section immunohistochemistry. B, brain homogenates were assayed for human neprilysin with an ELISA. C, brain sections were stained for human neprilysin (green), the neuronal marker MAP2 (red), and counterstained for nuclei (DAPI, blue). * indicates ANOVA p < 0.05 compared with mice that received SN5. Arrows indicate co-localization of neprilysin with the neuronal marker MAP2. Scale bars = 7 μm. Arrows highlight cells staining for neprilysin. n = 5 for each group.
Brain homogenates of APPtg- and non-tg-treated mice only showed accumulated human neprilysin in those mice that received the ASN12 recombinant protein (Fig. 3B). The level of ASN12 was higher in APPtg mice similar to previous results observed following lentiviral vector delivery of the gene for ASN in APPtg mice (14). At the conclusion of the 3-week dosing regimen and 3 days after the last injection, ASN12 levels in APPtg mice were ∼200 pg/mg of brain homogenate, similar to levels observed 24 and 48 h after i.v. delivery of the protein in the PK study. Immunohistochemical analysis of mice that received the ASN12 protein showed recombinant protein accumulated in the brain and co-localizing with the neuronal marker, MAP2 (Fig. 3C).
Intraneuronal Aβ may play a role in neuronal dysfunction and degeneration by disturbing ERK-cAMP-response element-binding protein signaling and increasing intracellular calcium levels (30). The tg2576 APPtg mouse showed elevated levels of intraneuronal Aβ that was ameliorated by treatment with the ASN12 protein but not with saline or SN5 protein treatment (Fig. 4, A and B). Three weeks of treatment were sufficient to reduce the intracellular levels of Aβ by 60% as measured by both 4G8 and 82E1 antibodies against Aβ. The pyroglutamate modified Aβ fragments play a significant role in the pathogenesis of Alzheimer's disease, and these too were significantly reduced following treatment with ASN12 as measured with the D129 antibody (Fig. 4, A and B).
FIGURE 4.

ASN12 reduces intraneuronal Aβ and ameliorates Aβ-induced neuropathology in the tg2576 APPtg mice. tg2576 APPtg and non-tg mice received ASN12, SN5 (1 mg/kg), or saline i.v. every 3 days for 3 weeks. Following protein delivery, mice were sacrificed, and brains were removed and divided sagitally with one-half fixed in paraformaldehyde. A, representative brain sections were stained for Aβ (4G8, 82E1, or D129), neuronal dendritic marker (MAP2), post-synaptic marker (synaptophysin), or astrocytic marker (GFAP). B, image analysis of the % area of the neuropil covered or corrected optical density for images analyzed. * indicates statistical significance p < 0.05 when compared with non-tg controls. # indicates statistical significance p < 0.05 when ASN12 or SN5 are compared with saline-treated APPtg mice. Scale bars = 7 μm. n = 5 for each group.
Aβ accumulation has been reported to reduce synaptic density in neurons prior to neuronal death. Similar to other APPtg mouse lines, the tg2576 line shows decreased neuronal dendritic MAP2 staining (31) and synaptic synaptophysin staining (Fig. 4, A and B) (32). Treatment with ASN12 prevented the loss of the MAP2 and synaptophysin staining although neither the SN5 nor saline treatment showed significant improvement (Fig. 4, A and B). In fact, treatment with ASN12 was sufficient to restore MAP2 and synaptophysin to wild-type mouse levels. The tg2576 also displays increased astrocytosis as shown by increased GFAP staining. Treatment with either SN5 or ASN12 prevented or reduced the astrocytosis observed in this mouse model of AD (Fig. 4, A and B).
Toxicity
Neprilysin has been shown to cleave NPY (13), which is a known regulator of appetite (33). To determine whether increased soluble neprilysin would affect appetite of treated mice, animals were weighed prior to treatment and weekly during treatment. There was no significant change in weight between groups of treated or untreated mice suggesting neprilysin had no effect on appetite (Fig. 5A). Additionally, during the treatment, three mice died during injection of recombinant protein (Fig. 5B). Two mice died receiving ASN12 injection with one mouse an APPtg and the other a non-tg. One additional mouse died following SN5 treatment, a non-tg mouse. All three mice died shortly after injection, and it is believed the cause was the repeat i.v. injection in the tail vein possibly releasing a previous clot that led to cardiac failure. Necropsy of the mice that died following treatment did not reveal any obvious signs of internal trauma, hemorrhaging, or necrosis of internal organs.
FIGURE 5.

Neither recombinant protein, ASN12 or SN5, appears to show toxicity in APP tg2576 or non-tg mice. A, mice were weighed prior to treatment and weekly afterward during treatment. B, survival curve of mice during the course of the experiment. C, representative Western blot of autoantibodies to human neprilysin or the human 38-amino acid LDLR domain from apoB detected at the conclusion of the 3-week experiment. Recombinant ASN12 was run on a gel, blotted to PVDF, and probed with whole blood from mice following the conclusion of the 3-week dosing. A positive control consisted of probing with an anti-human neprilysin antibody. n = 5 for each group.
The recombinant proteins (ASN12 and SN5) are based on the human neprilysin gene with the addition of the human 38-amino acid apoB LDLR binding domain to the ASN12 protein. To determine whether mice generated neutralizing antibodies to the recombinant protein, whole blood was used to detect recombinant ASN12 following a Western blot protocol. A total of two of the 20 mice that received intravenous injections generated an antibody response to the recombinant protein (Fig. 5C). The two mice both received the SN5 recombinant protein with one mouse an APPtg and the other a non-tg.
ASN12 Reverses Memory Deficits in the APPtg Mouse
To corroborate the results observed in the APPtg tg2576 mouse line, similar experiments were completed with a separate APPtg mouse model. APPtg mice used in these studies express human APP751 under the control of the mThy-1 promoter (mThy1-human APP751; Line41) (22, 23).
The Line41 APPtg and non-tg mice used in this study were injected twice weekly by intraperitoneal injection at 1 mg/kg for 3 weeks. Because these mice were subjected to the Morris water maze test for learning and memory after the final injection, the analysis of tissues did not occur until 10 days after the final injection. Immunohistochemical analysis of the hippocampus showed recombinant protein taken up by MAP2-positive neuronal cells in both APPtg and non-tg mice (Fig. 6A and B). Although a few APPtg mice showed some SN5 protein uptake, this was only slightly above background and is consistent with the previous result observed with viral vector delivery to the periphery (14). In contrast, intraperitoneal delivery of the ASN12 protein results in significant co-localization of recombinant human neprilysin with neurons in the hippocampus (Fig. 6, A and B) in both APPtg and non-tg mice. Similar results were observed in the cortex.
FIGURE 6.

ASN12 transits to the CNS following intraperitoneal delivery to the Line41 APPtg mouse. Line41 APPtg and non-tg mice received intraperitoneal injections of ASN12 or SN5 (1 mg/kg) or saline twice weekly for 3 weeks. A, 10 days after the last injection, mice were sacrificed, and whole brains were removed half-frozen for protein analysis and half-fixed for tissue section immunohistochemistry. B, brain sections were stained for human neprilysin (green), the neuronal marker MAP2 (red), and counterstained for nuclei (DAPI, blue). * indicates ANOVA p < 0.05 compared with mice that received SN5. # indicates ANOVA p < 0.05 compared with non-tg mice that received ASN12. Arrows indicate co-localization of neprilysin with the neuronal marker MAP2. Scale bars = 7 μm. Arrows highlight cells staining for neprilysin. n = 5 for each group.
The ASN12 protein is targeted to the LDLR on endothelial cells of the blood-brain barrier for transport to the CNS; however, LDLR are also expressed on astrocytes and neurons (34) in the brain. To determine whether these cells in the brain take up the brain-targeted neprilysin, we performed immunohistochemical co-localization analysis. ASN12 protein co-localized with the neuronal marker NeuN in the Line41 mice consistently showing a distinct cytoplasmic staining pattern (Fig. 7A). Approximately 15% of NeuN-positive neurons stained positive for the ASN12 protein (Fig. 7B). In contrast, very little recombinant ASN12 protein was observed co-staining with astrocytes (Fig. 7, C and D).
FIGURE 7.

ASN12 is taken up by neurons in the CNS of the Line41 APPtg mouse. Line41 APPtg and non-tg mice received intraperitoneal injections of ASN12 or SN5 (1 mg/kg) or saline twice weekly for 3 weeks. A, brain sections were stained for human neprilysin (red), the neuronal marker NeuN (green), and counterstained for nuclei (DAPI, blue). B, quantitation of % co-localization of neprilysin and NeuN staining. C, brain sections stained for neprilysin (red), the astrocytic marker GFAP (green), and counterstained for nuclei (DAPI, blue). D, quantitation of % co-localization of neprilysin and GFAP staining. * indicates ANOVA p < 0.05 compared with mice that received SN5. Arrows highlight cells staining for neprilysin. Scale bars = 7 μm. n = 5 for each group.
The Line41 APPtg mice show increased intraneuronal Aβ (35), decreased dendritic neuronal staining, MAP2 (23), and synaptic staining (synaptophysin) (22), as well as increased astrocytosis (GFAP) (Fig. 8, A and B) (23). Treatment of the Line41 APPtg mice with ASN12 significantly reduced intracellular Aβ and pyroglutamate-modified Aβ to wild-type levels and restored levels of MAP2 and GFAP (Fig. 8, A and B). In contrast to the tg2576 line, SN5 was unable to reduce astrocytosis (GFAP) in the Line41 APPtg mouse model (Fig. 8, A and B). Also in contrast to tg2576 where treatment with ASN12, but not SN5, restored synaptophysin levels, treatment of the Line41 APPtg with either ASN12 or SN5 was able to restore the levels of synaptophysin levels of staining in the APPtg mouse (Fig. 8, A and B). This may reflect differences in physiology between the two strains or differences in the stress of the expression patterns the APP transgene puts on the system.
FIGURE 8.

ASN12 reduces intraneuronal Aβ and ameliorates Aβ-induced neuropathology in the Line41 APPtg mouse. Line41 APPtg and non-tg mice received ASN12, SN5 (1 mg/kg), or saline intraperitoneally twice weekly for 3 weeks. Following protein delivery, mice were sacrificed, brains removed, divided sagitally with one-half fixed in paraformaldehyde. A, representative brain sections were stained for Aβ (4G8, 82E1, or D129), neuronal dendritic marker (MAP2), post-synaptic marker (synaptophysin), or astrocytic marker (GFAP). B, image analysis of the % area of the neuropil covered or corrected optical density for images analyzed. * indicates statistical significance p < 0.05 when compared with non-tg controls. # indicates statistical significance p < 0.05 when ASN12 or SN5 are compared with saline-treated APPtg mice. Scale bars = 7 μm. n = 5 for each group.
We have previously shown that neprilysin can cleave NPY at Tyr-20 and Leu-30 to produce the C-terminal fragment of 21–36 and 30–36 residues (13) and that these fragments are neuroprotective. To determine whether the peripherally delivered ASN12 would cleave full-length NPY in the brain, we examined the subgranular zone of the hippocampus by immunohistochemistry with an antibody that specifically recognizes the C-terminal fragment of neprilysin-cleaved NPY (CT-NPY). The Line41 APPtg mice showed a significant reduction in NPY C-terminal fragments compared with non-tg controls (Fig. 9, A and B). Treatment of the Line41 mice with ASN12 for 3 weeks restored levels of the C-terminal NPY fragment to wild-type levels, whereas treatment with SN5 or saline did not (Fig. 9, A and B). In contrast, full-length NPY was not altered in the Line41 APPtg mouse nor was expression of the NPY Y2 receptor (Fig. 9, A and B).
FIGURE 9.

ASN12 restores levels of C-terminal NPY fragment in the Line41 APPtg mouse. Line41 APPtg and non-tg mice received ASN12, SN5 (1 mg/kg) or saline intraperitoneally twice weekly for 3 weeks. A, brains were fixed in paraformaldehyde and representative sections were stained for full-length NPY (FL-NPY), C-terminal NPY (CT-NPY), or NPY Y2 receptor (Y2-R). B, analysis of tissue staining was completed by examining cell counts by stereology or total optical density. ML, medial layer; GC, granular cells; SG, subgranular layer. * indicates statistical significance p < 0.05 compared with non-tg controls. # indicates statistical significance p < 0.05 compared with vehicle or SN5-treated Line41 APPtg mice. Scale bars = 20 μm. n = 5 per group.
We recently showed a significant reduction in neurogenesis in the Line41 APPtg mouse (28, 36). To determine whether delivery of the recombinant human neprilysin to the brain altered the neurogenesis in the subgranular zone of the dentate gyrus of the hippocampus, Line41 APPtg and non-tg mice were examined by immunohistochemistry for the neurogenesis marker, DCX, and PCNA. Cells staining for either or both of these markers would indicate proliferating neurons in the area known for neurogenesis. Line41 APPtg mice contained significantly fewer DCX- (50%) and PCNA (25%)-stained cells in the subgranular zone of the dentate gyrus (Fig. 10, A and B). In contrast, treatment with the ASN12 human neprilysin protein restored levels of DCX and PCNA staining to levels observed in wild-type non-tg mice, whereas treatment with SN5 did not (Fig. 10, A and B). This indicates that treatment of APPtg mice with the ASN12 protein therapeutic restored the neurogenic environment to allow neurogenesis to occur at a rate similar to that observed in non-transgenic wild-type animals.
FIGURE 10.
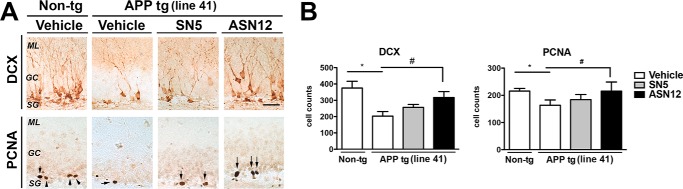
ASN12 restores neurogenesis in the Line41 APPtg mouse. Line41 APPtg and non-tg mice received ASN12, SN5 (1 mg/kg), or saline intraperitoneally twice weekly for 3 weeks. A, brains were fixed in paraformaldehyde, and representative sections were stained for DCX or PCNA. B, analysis of tissue staining was completed by examining cell counts by stereology. ML, medial layer; GC, granular cells; SG- subgranular layer. * indicates statistical significance p < 0.05 compared with Non-tg controls. # indicates statistical significance p < 0.05 compared with vehicle or SN5-treated Line41 APPtg mice. Arrows indicate cells positive for CT-NPY or Y2-R staining. Arrows indicate cells positive for PCNA staining. Scale bars = 20 μm. n = 5 per group.
The alterations in synaptic markers in APPtg mice have been associated with memory and learning deficits beginning at 3–6 months of age as measured in the Morris water maze (23). To determine whether treatment with ASN12 could ameliorate these deficits, APPtg mice were tested 1 week after the final intraperitoneal injection of either ASN12, SN5, or control saline. During the training period of the test, all groups of mice performed similarly at locating the visible platform after 3 days of testing. In the subsequent days of testing (days 4–7) with the platform submerged, non-tg mice located the platform in the water pool, with progressively shorter path distances over time (Fig. 11A). Compared with non-tg controls, APPtg mice that received saline injections did not show improvements in the spatial learning with the submerged platform (Fig. 11A). Similarly, the APPtg treated with SN5 protein displayed poor performance locating the hidden platform (days 4–7) (Fig. 11A). In contrast, APPtg mice that received the ASN12 protein were able to learn the location of the hidden platform significantly faster and at a rate similar to non-tg mice (Fig. 11A). Similarly, during the final probe test, APPtg mice that received the ASN12 protein spent significantly more time in the target quadrant than APPtg that received saline SN5 protein or saline injections (Fig. 11B).
FIGURE 11.
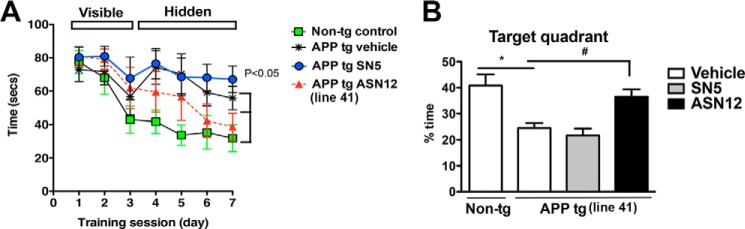
ASN12 improves water maze performance in Line41 APPtg mice. A, mice were trained on the cued platform on days 1–3 and then tested for spatial learning on days 4–7. B, probe test was performed at day 8, and the number of entrances of the mouse in the target quadrant containing the hidden platform was quantified. * indicates statistical significance p < 0.05 when compared with non-tg controls. One-way ANOVA with post hoc Dunnett's. n = 5 per group.
DISCUSSION
Development of new and novel therapeutic approaches for AD is of fundamental importance. Recent clinical failures of several high profile therapeutics highlight the need for new approaches (37). Although the reason for these recent failures is not clear, one possibility is the poor transport of molecules and proteins to the CNS, the site of action of Aβ accumulation in AD (38). We have developed an approach for delivery of protein therapeutics to the CNS by targeted transport across the blood-brain barrier (20). In this study, we examined this approach for delivery of the Aβ-degrading enzyme, neprilysin. We found that the brain-targeted neprilysin (ASN12) accumulated in the brain with a half-life of ∼24 h. When delivered by intravenous injection to APPtg mice, the ASN12 protein was able to increase levels of the synaptic protein, synaptophysin, decrease intracellular Aβ, and improve learning and memory. This is the first report to show this brain-targeted approach is effective for delivering recombinant proteins to the CNS while also increasing neurogenesis.
Neprilysin is a membrane-bound endo-peptidase that is found in the CNS at the plasma membrane, axonal processes, and synapses of neurons (39). The neprilysin used in this study has had the trans-membrane domain removed. The protein has been further modified with the addition of a secretory signal and the apolipoprotein B LDLR binding sequence. The apoB sequence facilitates binding to and transport with the LDL receptor at the blood-brain barrier (20). The LDLR receptor is also highly expressed in the liver and the spleen as well as blood- circulating macrophages (40). Single-dose administration of the apoB-neprilysin protein resulted in significant accumulation in the liver as would be expected (20). Previous investigation of LDLR targeted recombinant proteins has shown uptake in spleen and muscle (20). These organs were not examined in this study; however, ASN12 protein would be expected to accumulate in spleen and muscle. Neprilysin is expressed in numerous tissues, including kidney, lung, and brain (41), and is involved in the degradation of many immune and neuronal signaling molecules (4). Increased accumulation of neprilysin is not expected to cause pathology in these tissues as increased expression by transgenesis (42), viral vector gene delivery (14, 35, 43–45), or ex vivo cellular gene delivery (16, 17) has not shown overt pathology in the mouse nor has increased degradation of other known neprilysin substrates been reported. Thus, based on previous reports (13, 15) and the limited toxicity reported here, it appears that the increased soluble neuronal neprilysin does not exhibit any overt gross pathology.
We and others have reported that delivery of neprilysin to the brain can reduce the accumulation of Aβ in the APPtg mouse model (43–45). Although this report relies on the fact that neprilysin is acting as a proteolytic enzyme to degrade Aβ in the brain, it cannot be ruled out that at least part of the beneficial effect of the LDLR targeted neprilysin is on the LDLR. Up-regulation of the LDLR has been shown to decrease CNS accumulation of Aβ in an APPtg mouse model (46); therefore, it is possible that binding of the ASN12 protein containing the apoB LDLR binding domain could effect an up-regulation of the LDLR at the blood-brain barrier, thus having the effect of transporting more Aβ out of the CNS. This potentially beneficial aspect of the recombinant protein requires more investigation.
Delivery of the ASN12 protein to the CNS was able to reduced intracellular Aβ as determined by several anti-Aβ antibodies. Immunohistochemical analysis of the recombinant protein in the brain co-localized the protease with neurons and only to a minor degree with astrocytes. Both neurons and astrocytes have been shown to express the LDLR (34), so it is not clear why there would be such a drastic difference in the observed co-localization. One possible explanation is differences in intracellular trafficking. The LDLR binds proteins on the cell surface for endocytosis and targeting to the autophagy pathway (47). It may be that this pathway is different or proteins are degraded faster in astrocytes, so little is left to observe. In a previous study we showed that microglia take up ApoBSecNep (14). Further investigation is needed to determine the extent of the contribution of protein uptake and site of proteolytic cleavage.
Examining the distribution of ASN12 24 h after i.v. injection is crucial in this study. The control protein SN5 is detected in whole brain homogenates up to 24 h after injection. This is probably due to protein circulating in the blood vessels of the brain. Immunohistochemical examination of the brains of mice that received the SN5 protein showed recombinant neprilysin in structures that resemble vessels but not in neuronal cell bodies. The t½ of SN5 in the blood was 7.5 h, and at 24 h less than 0.3% of the injected dose was in circulation. Indeed, recombinant neprilysin detected in whole brain homogenates from mice treated with SN5 at 24 h shows 3.6 pg/mg brain tissue. Thus, the measurement of neprilysin in whole brain homogenates following ASN12 injection of 840 pg/mg brain tissue is a more reliable measure of protein that has accumulated in the brain. In fact, 24 h after ASN12 i.v. injection, the percentage of injected dose in the brain remains at 1.70%, although in the blood it is only 0.29%. A similar analysis of recombinant iduronate 2-sulfotase fused to the transferrin receptor found ∼1.5% uptake in the brain 1 h after i.v. delivery (48). This study included a PBS perfusion of the mice prior to examination of the brain protein, so this figure is likely reliable at this early time point. A thorough analysis of the ASN12 protein at 60 min after injection would probably find a much higher percentage accumulation in the brain.
Treatment of APPtg mice with ASN12 resulted in a significant reduction in intracellular Aβ. Several studies have shown that intracellular Aβ could be deleterious (30, 35, 49–51) by triggering an aberrant signaling via cAMP-response element-binding protein (52). Supporting this possibility, previous studies have shown that neprilysin ameliorates the deficits in the fly (53) and rodent models (35) by diminishing Aβ from the intracellular compartment. Although ASN12 is primarily extracellular, it is not clear if this is capable of degrading Aβ, and this shifts the equilibrium of Aβ from the intracellular space to the extracellular space, thus reducing the intracellular accumulation of Aβ indirectly. Alternatively, the 38-amino acid apoB LDLR binding domain can bind to the LDLR and trigger endocytosis through the ESCRT-mediated pathway for delivery to the multivesicular body for transport to the autophagosome (data not shown). This compartment and pathway may be another mechanism for intracellular Aβ degradation by ASN12.
Delivery of neprilysin to the CNS and in particular the hippocampus appeared to increase the C-terminal fragment of NPY and increase neurogenesis. The increase in C-terminal NPY fragment is probably directly attributable to increased neprilysin as we have previously shown that NPY can be cleaved by neprilysin to form these 21–36 and 30–36 C-terminal fragments (13). NPY C-terminal fragments bind preferentially to NPY Y2 receptors, which are important for learning and memory in the hippocampus and are expressed specifically in the dentate gyrus (54). This may help to account for the improvements in learning and memory observed in the Line41 APPtg mice. Another effect of increased NPY 21–36 and 30–36 C-terminal fragments may be to increase learning and memory through increased synaptic protection. Addition of NPY C-terminal fragments to primary neurons in the presence of Aβ prevents the loss of synapses in culture (13). We found that delivery of the ASN12 protein to the CNS ameliorated the loss of synaptic staining in the hippocampus of APPtg mice. The increased synaptic staining could be due increased NPY 21–36 and 30–36, decreased Aβ, or more likely a combination of the two.
In contrast, the improvement in neurogenesis probably cannot be directly attributed to the increase in neprilysin in the subgranular zone of the dentate gyrus. The NPY C-terminal fragment binds specifically to the NPY Y2 receptor and not the NPY Y1 receptor that is expressed preferentially on the neural progenitor cells (55). Dysregulation in neurogenesis in Alzheimer disease has been controversial with some reporting an increase (56–58) and others reporting a decrease (28, 36, 59–63) in neural progenitor cells in humans and APPtg mice. We have shown specifically in the Line41 APPtg mouse a decrease in neurogenesis (28, 36), and we linked this to an increase in BMP6 protein levels in the hippocampus (36). It is not clear how neprilysin is increasing neurogenesis in the subgranular zone; however, we cannot rule out the fact that neprilysin may be decreasing the expression of BMP6 in the hippocampus or may be activating some neurogenic growth factor. Alternatively, the increase in neurogenesis may be due to a decrease in Aβ in the subgranular zone; which provides an improved environment for neurogenesis. Thus, delivery of neprilysin to the hippocampus in general may provide an indirect mechanism for increased neurogenesis by improving the environment.
The results presented here are favorable for continued exploration of the brain-targeted neprilysin for clinical trials with repeated infusions of the recombinant protein. The only limit to repeat injections in this study was the potential for the generation of antibodies to recombinant protein. The human neprilysin is 91% identical to the mouse cDNA. Antibodies were only detected in mice that received the SN5 protein suggesting that the apoB LDLR binding domain was not sufficiently immunogenic to generate an antibody response on its own. Thus, although human antibody generation against the recombinant protein remains possible, these results suggest the risk is small. Extrapolation of the mouse data using allometric scaling suggests the ASN12 brain t½ of 24 h in the mouse brain would equate to a t½ of 8 days in the human (64). Therefore, a repeat i.v. treatment could be performed at a minimum of once weekly and maybe even once monthly. In summary, a recombinant anti-Aβ protein therapeutic that is transported across the blood-brain barrier may be a feasible, noninvasive therapeutic option for treatment for Alzheimer disease.
This work was supported, in whole or in part, by National Institutes of Health Grants AG5131, AG022074, AG188440, and AG010435. B. S. was the principal investigator at NeuroTransit, Inc., during the course of these studies. The pharmacokinetics and the pharmacodynamics in the APPtg2576 line were completed at NeuroTransit.
- AD
- Alzheimer disease
- Aβ
- amyloid-β peptide
- NEP
- neprilysin
- ANOVA
- analysis of variance
- GFAP
- glial fibrillary acidic protein
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- PFA
- paraformaldehyde
- PCNA
- proliferating cell nuclear antigen
- DAGNPG
- 3-dansyl-d-Ala-Gly-p-(nitro)-Phe-Gly
- dansyl
- 5-dimethylaminonaphthalene-1-sulfonyl
- DCX
- doublecortin
- NPY
- neuropeptide Y
- NEP
- neprilysin
- non-tg
- nontransgenic
- TEER
- trans-endothelial electrical resistance
- LDLR
- LDL receptor
- APP
- amyloid precursor protein.
REFERENCES
- 1. Ashford J. W. (2004) APOE genotype effects on Alzheimer's disease onset and epidemiology. J. Mol. Neurosci. 23, 157–165 [DOI] [PubMed] [Google Scholar]
- 2. Terry R., Hansen L., Masliah E. (1994) in Alzheimer disease (Terry R., Katzman R., eds) pp. 179–196, Raven Press, New York [Google Scholar]
- 3. Marr R. A., Spencer B. J. (2010) Diabetes NEP-like endopeptidases and Alzheimer's disease. Curr. Alzheimer. Res. 7, 223–229 [DOI] [PubMed] [Google Scholar]
- 4. Howell S., Nalbantoglu J., Crine P. (1995) Neutral endopeptidase can hydrolyze β-amyloid(1–40) but shows no effect on β-amyloid precursor protein metabolism. Peptides 16, 647–652 [DOI] [PubMed] [Google Scholar]
- 5. Iwata N., Tsubuki S., Takaki Y., Shirotani K., Lu B., Gerard N. P., Gerard C., Hama E., Lee H. J., Saido T. C. (2001) Metabolic regulation of brain Aβ by neprilysin. Science 292, 1550–1552 [DOI] [PubMed] [Google Scholar]
- 6. Iwata N., Tsubuki S., Takaki Y., Watanabe K., Sekiguchi M., Hosoki E., Kawashima-Morishima M., Lee H. J., Hama E., Sekine-Aizawa Y., Saido T. C. (2000) Identification of the major Aβ1–42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat. Med. 6, 143–150 [DOI] [PubMed] [Google Scholar]
- 7. Huang S. M., Mouri A., Kokubo H., Nakajima R., Suemoto T., Higuchi M., Staufenbiel M., Noda Y., Yamaguchi H., Nabeshima T., Saido T. C., Iwata N. (2006) Neprilysin-sensitive synapse-associated amyloid-β peptide oligomers impair neuronal plasticity and cognitive function. J. Biol. Chem. 281, 17941–17951 [DOI] [PubMed] [Google Scholar]
- 8. Kanemitsu H., Tomiyama T., Mori H. (2003) Human neprilysin is capable of degrading amyloid β peptide not only in the monomeric form but also the pathological oligomeric form. Neurosci. Lett. 350, 113–116 [DOI] [PubMed] [Google Scholar]
- 9. Meilandt W. J., Cisse M., Ho K., Wu T., Esposito L. A., Scearce-Levie K., Cheng I. H., Yu G. Q., Mucke L. (2009) Neprilysin overexpression inhibits plaque formation but fails to reduce pathogenic Aβ oligomers and associated cognitive deficits in human amyloid precursor protein transgenic mice. J. Neurosci. 29, 1977–1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Betts V., Leissring M. A., Dolios G., Wang R., Selkoe D. J., Walsh D. M. (2008) Aggregation and catabolism of disease-associated intra-Aβ mutations: reduced proteolysis of AβA21G by neprilysin. Neurobiol. Dis. 31, 442–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Skidgel R. A., Erdös E. G. (2004) Angiotensin converting enzyme (ACE) and neprilysin hydrolyze neuropeptides: a brief history, the beginning and follow-ups to early studies. Peptides 25, 521–525 [DOI] [PubMed] [Google Scholar]
- 12. Saito T., Iwata N., Tsubuki S., Takaki Y., Takano J., Huang S. M., Suemoto T., Higuchi M., Saido T. C. (2005) Somatostatin regulates brain amyloid β peptide Aβ42 through modulation of proteolytic degradation. Nat. Med. 11, 434–439 [DOI] [PubMed] [Google Scholar]
- 13. Rose J. B., Crews L., Rockenstein E., Adame A., Mante M., Hersh L. B., Gage F. H., Spencer B., Potkar R., Marr R. A., Masliah E. (2009) Neuropeptide Y fragments derived from neprilysin processing are neuroprotective in a transgenic model of Alzheimer's disease. J. Neurosci. 29, 1115–1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Spencer B., Marr R. A., Gindi R., Potkar R., Michael S., Adame A., Rockenstein E., Verma I. M., Masliah E. (2011) Peripheral delivery of a CNS targeted, metalloprotease reduces Aβ toxicity in a mouse model of Alzheimer's disease. PLoS One 6, e16575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu Y., Studzinski C., Beckett T., Guan H., Hersh M. A., Murphy M. P., Klein R., Hersh L. B. (2009) Expression of neprilysin in skeletal muscle reduces amyloid burden in a transgenic mouse model of Alzheimer disease. Mol. Ther. 17, 1381–1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu Y., Guan H., Beckett T. L., Juliano M. A., Juliano L., Song E. S., Chow K. M., Murphy M. P., Hersh L. B. (2007) In vitro and in vivo degradation of Aβ peptide by peptidases coupled to erythrocytes. Peptides 28, 2348–2355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Guan H., Liu Y., Daily A., Police S., Kim M. H., Oddo S., LaFerla F. M., Pauly J. R., Murphy M. P., Hersh L. B. (2009) Peripherally expressed neprilysin reduces brain amyloid burden: a novel approach for treating Alzheimer's disease. J. Neurosci. Res. 87, 1462–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Walker J. R., Pacoma R., Watson J., Ou W., Alves J., Mason D. E., Peters E. C., Urbina H. D., Welzel G., Althage A., Liu B., Tuntland T., Jacobson L. H., Harris J. L., Schumacher A. M. (2013) Enhanced proteolytic clearance of plasma Aβ by peripherally administered neprilysin does not result in reduced levels of brain Aβ in mice. J. Neurosci. 33, 2457–2464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hemming M. L., Patterson M., Reske-Nielsen C., Lin L., Isacson O., Selkoe D. J. (2007) Reducing amyloid plaque burden via ex vivo gene delivery of an Aβ-degrading protease: a novel therapeutic approach to Alzheimer disease. PLoS Med. 4, e262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Spencer B. J., Verma I. M. (2007) Targeted delivery of proteins across the blood-brain barrier. Proc. Natl. Acad. Sci. U.S.A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hsiao K., Chapman P., Nilsen S., Eckman C., Harigaya Y., Younkin S., Yang F., Cole G. (1996) Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 274, 99–102 [DOI] [PubMed] [Google Scholar]
- 22. Rockenstein E., Mallory M., Mante M., Sisk A., Masliaha E. (2001) Early formation of mature amyloid-β protein deposits in a mutant APP transgenic model depends on levels of Aβ(1–42). J. Neurosci. Res. 66, 573–582 [DOI] [PubMed] [Google Scholar]
- 23. Rockenstein E., Mante M., Alford M., Adame A., Crews L., Hashimoto M., Esposito L., Mucke L., Masliah E. (2005) High β-secretase activity elicits neurodegeneration in transgenic mice despite reductions in amyloid-β levels: implications for the treatment of Alzheimer disease. J. Biol. Chem. 280, 32957–32967 [DOI] [PubMed] [Google Scholar]
- 24. Mandler M., Rockenstein E., Ubhi K., Hansen L., Adame A., Michael S., Galasko D., Santic R., Mattner F., Masliah E. (2012) Detection of peri-synaptic amyloid-β pyroglutamate aggregates in early stages of Alzheimer's disease and in AβPP transgenic mice using a novel monoclonal antibody. J. Alzheimers Dis. 28, 783–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Masliah E., Rockenstein E., Veinbergs I., Mallory M., Hashimoto M., Takeda A., Sagara Y., Sisk A., Mucke L. (2000) Dopaminergic loss and inclusion body formation in α-synuclein mice: implications for neurodegenerative disorders. Science 287, 1265–1269 [DOI] [PubMed] [Google Scholar]
- 26. Spencer B., Potkar R., Trejo M., Rockenstein E., Patrick C., Gindi R., Adame A., Wyss-Coray T., Masliah E. (2009) Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson's and Lewy body diseases. J. Neurosci. 29, 13578–13588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rockenstein E., Schwach G., Ingolic E., Adame A., Crews L., Mante M., Pfragner R., Schreiner E., Windisch M., Masliah E. (2005) Lysosomal pathology associated with α-synuclein accumulation in transgenic models using an eGFP fusion protein. J. Neurosci. Res. 80, 247–259 [DOI] [PubMed] [Google Scholar]
- 28. Rockenstein E., Mante M., Adame A., Crews L., Moessler H., Masliah E. (2007) Effects of cerebrolysin on neurogenesis in an APP transgenic model of Alzheimer's disease. Acta Neuropathol. 113, 265–275 [DOI] [PubMed] [Google Scholar]
- 29. Srivastava R. A., Ito H., Hess M., Srivastava N., Schonfeld G. (1995) Regulation of low density lipoprotein receptor gene expression in HepG2 and Caco2 cells by palmitate, oleate, and 25-hydroxycholesterol. J. Lipid Res. 36, 1434–1446 [PubMed] [Google Scholar]
- 30. LaFerla F. M., Green K. N., Oddo S. (2007) Intracellular amyloid-β in Alzheimer's disease. Nat. Rev. Neurosci. 8, 499–509 [DOI] [PubMed] [Google Scholar]
- 31. Gerenu G., Dobarro M., Ramirez M. J., Gil-Bea F. J. (2013) Early cognitive stimulation compensates for memory and pathological changes in Tg2576 mice. Biochim. Biophys. Acta 1832, 837–847 [DOI] [PubMed] [Google Scholar]
- 32. Chapman P. F., White G. L., Jones M. W., Cooper-Blacketer D., Marshall V. J., Irizarry M., Younkin L., Good M. A., Bliss T. V., Hyman B. T., Younkin S. G., Hsiao K. K. (1999) Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat. Neurosci. 2, 271–276 [DOI] [PubMed] [Google Scholar]
- 33. Naveilhan P., Hassani H., Canals J. M., Ekstrand A. J., Larefalk A., Chhajlani V., Arenas E., Gedda K., Svensson L., Thoren P., Ernfors P. (1999) Normal feeding behavior, body weight and leptin response require the neuropeptide Y Y2 receptor. Nat. Med. 5, 1188–1193 [DOI] [PubMed] [Google Scholar]
- 34. Fan Q. W., Iosbe I., Asou H., Yanagisawa K., Michikawa M. (2001) Expression and regulation of apolipoprotein E receptors in the cells of the central nervous system in culture: a review. J. Am. Aging Assoc. 24, 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Spencer B., Marr R. A., Rockenstein E., Crews L., Adame A., Potkar R., Patrick C., Gage F. H., Verma I. M., Masliah E. (2008) Long-term neprilysin gene transfer is associated with reduced levels of intracellular Aβ and behavioral improvement in APP transgenic mice. BMC Neurosci. 9, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Crews L., Adame A., Patrick C., Delaney A., Pham E., Rockenstein E., Hansen L., Masliah E. (2010) Increased BMP6 levels in the brains of Alzheimer's disease patients and APP transgenic mice are accompanied by impaired neurogenesis. J. Neurosci. 30, 12252–12262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Misra S., Medhi B. (2013) Drug development status for Alzheimer's disease: present scenario. Neurol. Sci. 34, 831–839 [DOI] [PubMed] [Google Scholar]
- 38. Banks W. A., Terrell B., Farr S. A., Robinson S. M., Nonaka N., Morley J. E. (2002) Passage of amyloid β protein antibody across the blood-brain barrier in a mouse model of Alzheimer's disease. Peptides 23, 2223–2226 [DOI] [PubMed] [Google Scholar]
- 39. Fukami S., Watanabe K., Iwata N., Haraoka J., Lu B., Gerard N. P., Gerard C., Fraser P., Westaway D., St George-Hyslop P., Saido T. C. (2002) Aβ-degrading endopeptidase, neprilysin, in mouse brain: synaptic and axonal localization inversely correlating with Aβ pathology. Neurosci. Res. 43, 39–56 [DOI] [PubMed] [Google Scholar]
- 40. Hussain M. M., Strickland D. K., Bakillah A. (1999) The mammalian low-density lipoprotein receptor family. Annu. Rev. Nutr. 19, 141–172 [DOI] [PubMed] [Google Scholar]
- 41. Li C., Booze R. M., Hersh L. B. (1996) Tissue-specific expression of rat neutral endopeptidase mRNAs. Ann. N.Y. Acad. Sci. 780, 145–155 [DOI] [PubMed] [Google Scholar]
- 42. Leissring M. A., Farris W., Chang A. Y., Walsh D. M., Wu X., Sun X., Frosch M. P., Selkoe D. J. (2003) Enhanced proteolysis of β-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron 40, 1087–1093 [DOI] [PubMed] [Google Scholar]
- 43. Hong C. S., Goins W. F., Goss J. R., Burton E. A., Glorioso J. C. (2006) Herpes simplex virus RNAi and neprilysin gene transfer vectors reduce accumulation of Alzheimer's disease-related amyloid-β peptide in vivo. Gene Ther. 13, 1068–1079 [DOI] [PubMed] [Google Scholar]
- 44. Iwata N., Mizukami H., Shirotani K., Takaki Y., Muramatsu S., Lu B., Gerard N. P., Gerard C., Ozawa K., Saido T. C. (2004) Presynaptic localization of neprilysin contributes to efficient clearance of amyloid-β peptide in mouse brain. J. Neurosci. 24, 991–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Marr R. A., Rockenstein E., Mukherjee A., Kindy M. S., Hersh L. B., Gage F. H., Verma I. M., Masliah E. (2003) Neprilysin gene transfer reduces human amyloid pathology in transgenic mice. J. Neurosci. 23, 1992–1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Castellano J. M., Deane R., Gottesdiener A. J., Verghese P. B., Stewart F. R., West T., Paoletti A. C., Kasper T. R., DeMattos R. B., Zlokovic B. V., Holtzman D. M. (2012) Low-density lipoprotein receptor overexpression enhances the rate of brain-to-blood Aβ clearance in a mouse model of β-amyloidosis. Proc. Natl. Acad. Sci. U.S.A. 109, 15502–15507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Scotti E., Calamai M., Goulbourne C. N., Zhang L., Hong C., Lin R. R., Choi J., Pilch P. F., Fong L. G., Zou P., Ting A. Y., Pavone F. S., Young S. G., Tontonoz P. (2013) IDOL stimulates clathrin-independent endocytosis and MVB-mediated lysosomal degradation of the LDLR. Mol. Cell. Biol. 33, 1503–1514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhou Q. H., Boado R. J., Lu J. Z., Hui E. K., Pardridge W. M. (2012) Brain-penetrating IgG-iduronate 2-sulfatase fusion protein for the mouse. Drug Metab. Dispos. 40, 329–335 [DOI] [PubMed] [Google Scholar]
- 49. Cuello A. C. (2005) Intracellular and extracellular Aβ, a tale of two neuropathologies. Brain Pathol. 15, 66–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cuello A. C., Canneva F. (2008) Impact of intracellular β-amyloid in transgenic animals and cell models. Neurodegener. Dis. 5, 146–148 [DOI] [PubMed] [Google Scholar]
- 51. Gouras G. K., Tampellini D., Takahashi R. H., Capetillo-Zarate E. (2010) Intraneuronal β-amyloid accumulation and synapse pathology in Alzheimer's disease. Acta Neuropathol. 119, 523–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Arvanitis D. N., Ducatenzeiler A., Ou J. N., Grodstein E., Andrews S. D., Tendulkar S. R., Ribeiro-da-Silva A., Szyf M., Cuello A. C. (2007) High intracellular concentrations of amyloid-β block nuclear translocation of phosphorylated CREB. J. Neurochem. 103, 216–228 [DOI] [PubMed] [Google Scholar]
- 53. Iijima-Ando K., Hearn S. A., Granger L., Shenton C., Gatt A., Chiang H. C., Hakker I., Zhong Y., Iijima K. (2008) Overexpression of neprilysin reduces Alzheimer's amyloid-β 42 (Aβ42)-induced neuron loss and intraneuronal Aβ 42 deposits, but causes a reduction in CREB-mediated transcription, age-dependent axon pathology and premature death in Drosophila. J. Biol. Chem. 283, 19066–19076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Redrobe J. P., Dumont Y., Herzog H., Quirion R. (2004) Characterization of neuropeptide Y, Y(2) receptor knockout mice in two animal models of learning and memory processing. J. Mol. Neurosci. 22, 159–166 [DOI] [PubMed] [Google Scholar]
- 55. Decressac M., Wright B., David B., Tyers P., Jaber M., Barker R. A., Gaillard A. (2011) Exogenous neuropeptide Y promotes in vivo hippocampal neurogenesis. Hippocampus 21, 233–238 [DOI] [PubMed] [Google Scholar]
- 56. López-Toledano M. A., Shelanski M. L. (2007) Increased neurogenesis in young transgenic mice overexpressing human APP(Sw, Ind). J. Alzheimers Dis. 12, 229–240 [DOI] [PubMed] [Google Scholar]
- 57. Jin K., Galvan V., Xie L., Mao X. O., Gorostiza O. F., Bredesen D. E., Greenberg D. A. (2004) Enhanced neurogenesis in Alzheimer's disease transgenic (PDGF-APPSw, Ind) mice. Proc. Natl. Acad. Sci. U.S.A. 101, 13363–13367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Li B., Yamamori H., Tatebayashi Y., Shafit-Zagardo B., Tanimukai H., Chen S., Iqbal K., Grundke-Iqbal I. (2008) Failure of neuronal maturation in Alzheimer disease dentate gyrus. J. Neuropathol. Exp. Neurol. 67, 78–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Donovan M. H., Yazdani U., Norris R. D., Games D., German D. C., Eisch A. J. (2006) Decreased adult hippocampal neurogenesis in the PDAPP mouse model of Alzheimer's disease. J. Comp. Neurol. 495, 70–83 [DOI] [PubMed] [Google Scholar]
- 60. Haughey N. J., Liu D., Nath A., Borchard A. C., Mattson M. P. (2002) Disruption of neurogenesis in the subventricular zone of adult mice, and in human cortical neuronal precursor cells in culture, by amyloid β-peptide: implications for the pathogenesis of Alzheimer's disease. Neuromolecular Med. 1, 125–135 [DOI] [PubMed] [Google Scholar]
- 61. Taniuchi N., Niidome T., Goto Y., Akaike A., Kihara T., Sugimoto H. (2007) Decreased proliferation of hippocampal progenitor cells in APPswe/PS1dE9 transgenic mice. Neuroreport 18, 1801–1805 [DOI] [PubMed] [Google Scholar]
- 62. Dong H., Goico B., Martin M., Csernansky C. A., Bertchume A., Csernansky J. G. (2004) Modulation of hippocampal cell proliferation, memory, and amyloid plaque deposition in APPsw (Tg2576) mutant mice by isolation stress. Neuroscience 127, 601–609 [DOI] [PubMed] [Google Scholar]
- 63. Jin K., Peel A. L., Mao X. O., Xie L., Cottrell B. A., Henshall D. C., Greenberg D. A. (2004) Increased hippocampal neurogenesis in Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 101, 343–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mordenti J., Osaka G., Garcia K., Thomsen K., Licko V., Meng G. (1996) Pharmacokinetics and interspecies scaling of recombinant human factor VIII. Toxicol. Appl. Pharmacol. 136, 75–78 [DOI] [PubMed] [Google Scholar]