

Abstract
Purpose
Determine whether Compound 49b treatment ameliorates retinal changes due to the lack of β2-adrenergic receptor signaling.
Methods
Using retinas from 3-month-old β2-adrenergic receptor-deficient mice, we treated mice with our novel β1-/β2-adrenergic receptor agonist, Compound 49b, to assess the effects of adrenergic agonists acting only on β1-adrenergic receptors due to the absence of β2-adrenergic receptors. Western blotting or enzyme-linked immunosorbent assay (ELISA) analyses were performed for β1- and β2-adrenergic receptors, as well as key insulin resistance proteins, including TNF-α, SOCS3, IRS-1Ser307, and IRTyr960. Analyses were also performed on key anti- and proapoptotic proteins: Akt, Bcl-xL, Bax, and caspase 3. Electroretinogram analyses were conducted to assess functional changes, while histological assessment was conducted for changes in retinal thickness.
Results
A 2-month treatment of β2-adrenergic receptor-deficient mice with daily eye drops of 1 mM Compound 49b, a novel β1- and β2-adrenergic receptor agonist, reversed the changes in insulin resistance markers (TNF-α and SOCS3) observed in untreated β2-adrenergic receptor-deficient mice, and concomitantly increased morphological integrity (retinal thickness) and functional responses (electroretinogram amplitude). These results suggest that stimulating β1-adrenergic receptors on retinal endothelial cells or Müller cells can compensate for the loss of β2-adrenergic receptor signaling on Müller cells, restore insulin signal transduction, reduce retinal apoptosis, and enhance retinal function.
Conclusions
Since our previous studies with β1-adrenergic receptor knockout mice confirmed that the reverse also occurs (β2-adrenergic receptor stimulation can compensate for the loss of β1-adrenergic receptor activity), it appears that increased activity in either of these pathways alone is sufficient to block insulin resistance–based retinal cell apoptosis.
Introduction
Diabetic retinopathy is associated with dysfunction in retinal endothelial cells, Müller cells, pericytes, as well as neuronal cells [1]. We previously reported that β-adrenergic receptor stimulation can prevent many of the deleterious changes associated with diabetic retinopathy in retinal endothelial cells [2,3] and retinal Müller cells [4,5]. Recently, we showed that β2-adrenergic receptor knockout mice have a phenotype similar to that noted in other rodent models of diabetic retinopathy, including increased tumor necrosis factor alpha (TNF-α) levels, Müller cell activation, and decreased amplitude of electroretinograms (ERGs) [6]. Additionally, we demonstrated that β2-adrenergic receptor stimulation is critical for inhibiting apoptosis in retinal Müller cells cultured in high glucose (diabetic-like) conditions [5]. It appears likely that the observed changes in TNF-α levels, insulin receptor phosphorylation, and apoptotic protein levels in β2-adrenergic receptor knockout mice are due to increased Müller cell activation in these mice, as evidenced by the attendant increase in glial fibrillary acidic protein levels in knockout mice [6]. Taken together, our in vitro and in vivo data suggest that β2-adrenergic receptors are present on retinal Müller cells and are critical for maintaining insulin signaling in these cell types.
In addition to these β2-adrenergic receptor actions associated with Müller cells, it appears from our in vitro data that retinal endothelial cells have β1- and β3-adrenergic receptors [7], which may be important in regulating insulin signaling. We have demonstrated that TNF-α is increased in retinal endothelial cells cultured under diabetic-like conditions [2]. This was not surprising since it has been shown in several cell types that increased TNF-α levels can lead to insulin resistance [8,9] through phosphorylation of insulin-receptor substrate 1 (IRS-1) on serine 307 to inhibit insulin signal transduction. As a result of TNF-α-mediated insulin resistance, Akt is decreased while suppressor of cytokine signaling 3 (SOCS3) is increased, both of which lead to the promotion of apoptosis [8,10,11]. Thus, our current understanding is that retinal Müller cells regulate insulin signaling primarily through β2-adrenergic receptors [5], whereas retinal endothelial cells do likewise through either β1- and β3-adrenergic receptors [7]. The degree to which these two cell type/subunit type-specific pathways interact or mutually compensate in their control of retinal insulin signaling is currently unknown. In the experiments reported here, we treated β2-adrenergic receptor knockout mice with a novel β1-/β2-adrenergic receptor agonist, Compound 49b, which can be applied in an eye drop formulation in vivo [2]. Under these conditions, β2-adrenergic receptor-mediated Müller cell actions should be blunted, allowing Compound 49b to selectively activate β1-adrenergic receptors on retinal endothelial cells [7]. This allowed us to determine if restoring β1-adrenergic receptor activity in retinal endothelial cells—in the absence of β2-adrenergic receptors on Müller cells—was sufficient to restore insulin signaling and enhance retinal function.
Methods
Mice
All mice experiments, including those for dark-adaptation and tail electrodes for ERG analyses, were approved by the Institutional Animal Care and Use Committee at the University of Tennessee Health Science Center (Protocol #1992). β1-/β2-adrenergic receptor knockout mice (Adrb1tm1Bkk Adrb2tm1Bkk/J) were purchased from Jackson laboratories (Bar Harbor, ME). C57BL6 wild-type mice were purchased from Charles River Laboratories (Durham, NC). β1-/β2-adrenergic receptor knockout mice were generated on a mixed background (129S1/Sv * 129X1/SvJ * C57BL/6J * DBA/2 * FVB/N). From the β1-/β2-adrenergic receptor knockout mice, we bred mice to generate only β2-adrenergic receptor homozygous knockout mice. We appreciate that C57BL6 may not be the ideal wild-type control, but the original β1-/β2-adrenergic receptor knockout mice were from a mixed background containing C57BL6, therefore we chose the C57BL6 for wild-type. Since we use these mice at 3 months of age, other issues from the C57BL6 background should be minimized. We have previously published use of this animal model and genotyping [6] to demonstrate that neuronal markers of diabetic retinopathy are present, as well as increased apoptosis in the retina. Five mice in each group (control, β2-adrenergic receptor knockout (KO)+49b, β2-adrenergic receptor KO) of both genders were used for these studies.
Compound 49b treatment
A subset of the β2-adrenergic receptor knockout mice were administered Compound 49b (1 mM administered in one daily treatment of 4 eye drops) as we have done previously [2,12]. Eye drop administration was initiated when the animals were 1 month old. The animals were treated each day for 2 months. After the final treatment, C57BL6 control, β2-adrenergic receptor knockout mice+49b, and β2-adrenergic knockout mice were dark-adapted for electroretinogram analyses before they were euthanized by ketamine/xylazine overdose.
Electroretinography
Before the animals were euthanized for morphological and biochemical analyses, the animals were subjected to ERG analyses to evaluate the changes in the electrical activity of the retina as we have done previously [2,6]. Briefly, mice were dark-adapted overnight. ERG responses were recorded from both eyes together using platinum wire corneal electrodes, a forehead reference electrode, and a ground electrode in the tail. Pupils were fully dilated using 1% tropicamide solution (Alcon, Ft Worth, TX). Methylcellulose (Celluvise; Allergan, Irvine, CA) drops were applied as well to maintain a good electrical connection, and body temperature was maintained at 37 °C with a water-based heating pad. All ERG experiments were approved by the University of Tennessee Institutional Animal Care and Use Committee on Protocol #1992. ERG waveforms were recorded with a bandwidth of 0.3–500 Hz and samples at 2 kHz by a digital acquisition system and were analyzed a custom-built program (MatLab software, Mathworks, Natick, MA). Statistics was done on the mean ± standard deviation (SD) amplitudes of the a- and b-waves of each treatment group. Comparisons were made of ERG amplitudes, but implicit times were not measured.
Neuronal analyses
Formalin-fixed paraffin sections were stained with hematoxylin and eosin for light microscopy and morphometry of retinal thickness as previously described [13]. Photomicrographs were assessed for retinal thickness using methods previously described. The thickness of the retina and the cell count were measured using OpenLab software (Improvision, Lexington, MA) [2].
Western blot analysis
Equal amounts of protein from the tissue extracts were separated on the precast Tris-glycine gel (Invitrogen, Carlsbad, CA) and blotted onto a nitrocellulose membrane. After blocking in TBST (10 mM Tris-HCl buffer, pH 8.0, 150 mM NaCl, 0.1% Tween-20) and 5% (w/v) BSA, the membrane was treated with appropriate primary antibodies followed by incubation with secondary antibodies labeled with horseradish peroxidase. Antigen-antibody complexes were detected with a chemiluminescence reagent kit (Thermo Scientific, Pittsburgh, PA). The primary antibodies used were phosphorylated Akt (serine 473), Akt, Bax, Bcl-xL, SOCS3, phosphorylated insulin receptor (tyrosine 1150/1151), insulin receptor (all purchased from Cell Signaling, Danvers, MA), insulin receptor phosphorylated on Tyr960 (Cell Applications, San Diego, CA), and β1-adrenergic receptor, β2-adrenergic receptor, and β actin (Santa Cruz, Santa Cruz, CA).
Enzyme-linked immunosorbent assay analysis
A cleaved caspase 3 enzyme-linked immunosorbent assay (ELISA; Cell Signaling, Danvers, MA) was used to measure the levels of the active apoptotic marker in the whole retinal lysates. TNF-α protein concentrations were measured using a TNF-α ELISA (ThermoFisher, Pittsburgh, PA). For cleaved caspase 3 ELISA analyses, equal protein was loaded (50 μg) in all wells to allow for comparisons based on optical density (OD). For the TNF-α ELISA, 50 μl protein was loaded into all wells, with analyses based on a standard curve.
Statistical analysis
Data are presented as mean ± standard error of the mean (SEM), with statistical analyses using Kruskal–Wallis non-parametric testing, followed by Dunn’s test. For western blot analyses, data was normalized to β actin levels.
Results
Compound 49b increases β1-adrenergic receptor levels with no action on β2-adrenergic receptor
To ensure that Compound 49b was effective in activating the β1-adrenergic receptor, we tested retinal lysates from control, β2-adrenergic receptor knockout+1 mM Compound 49b for 2 months, and 3-month old β2-adrenergic receptor knockout mice. Figure 1A demonstrates that Compound 49b increased β1-adrenegic receptor protein levels in the β2-adrenergic receptor knockout mice. Figure 1B shows the control to demonstrate that Compound 49b can only slightly increase β2-adrenergic receptor protein levels in β2-adrenergic receptor KO mice, while limited β2-adrenergic receptor levels were observed in the β2-adrenergic receptor KO mice. Taken together, these data demonstrate that Compound 49b is acting on β1-adrenergic receptors in these studies.
Figure 1.
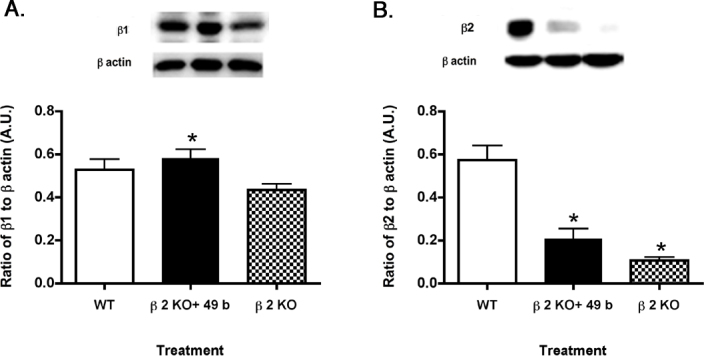
β-adrenergic receptor protein levels in control, β2-adrenergic receptor knockout mice+49b, and β2-adrenergic receptor knockout mice. A: Compound 49b significantly increased β1-adrenergic receptor levels compared to untreated β2-adrenegic receptor knockout (KO) mice. B: β2-adrenergic receptor protein levels are minimally restored in β2-adrenergic receptor KO mice treated with Compound 49b. *p<0.05 versus wild-type (WT); #p<0.05 versus β2-adrenergic receptor KO mice. n = 5 for all groups. Data are mean ± standard error of the mean (SEM).
Tumor necrosis factor α and insulin-receptor substrate 1Ser307 are increased in β2-adrenergic receptor knockout mice but not in knockout mice treated with Compound 49b
Figure 2A validates our previous work showing that Compound 49b significantly reduces TNF-α levels (p<0.05 versus β2KO, Figure 2A), which we reported previously for retinal endothelial cells [2,3], likely the key retinal cell type involved in signaling in β2-adrenergic receptor knockout mice. Additionally, Figure 2A supported published data that TNF-α is significantly increased in β2-adrenergic receptor knockout mice [6]. Since TNF-α preferentially phosphorylates IRS-1Ser307 to initiate insulin resistance [9,11], we wanted to measure the phosphorylation of IRS-1 on serine 307 in the β2-adrenergic receptor knockout mice and knockout mice treated with Compound 49b. Figure 2B demonstrates that Compound 49b significantly decreased IRS-1Ser307 in β2-adrenegic receptor KO mice. IRS-1Ser307 is increased in β2-adrenergic receptor KO mice with no treatment (p<0.05 versus β2KO, Figure 2B).
Figure 2.
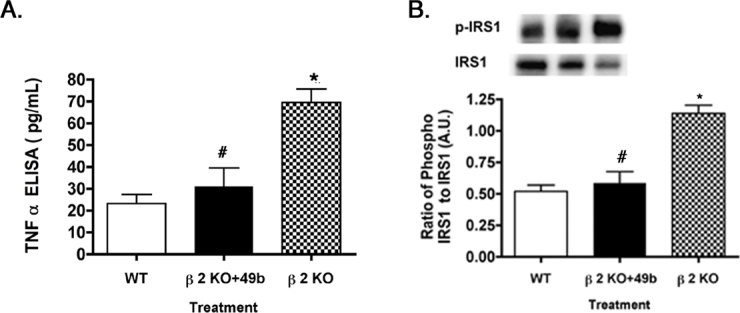
Tumor necrosis factor α and insulin-receptor substrate 1Ser307 phosphorylation in control, β2-adrenergic receptor knockout mice+49b and β2-adrenergic receptor knockout mice. A: Tumor necrosis factor α (TNF-α) levels are returned to near control levels in β2-adrenergic receptor knockout (KO) mice treated with Compound 49b. B: Similar results for the phosphorylation of serine 307 on insulin-receptor substrate 1 (IRS-1). *p<0.05 versus wild-type (WT); #p<0.05 versus β2-adrenergic receptor KO mice. n = 5 for all groups. Data are mean ± standard error of the mean (SEM).
Compound 49b restores suppressor of cytokine signaling 3 and insulin receptorTyr960 to wild-type levels in β2-adrenergic receptor knockout mice
We have reported previously that TNF-α activates SOCS3 and initiates phosphorylation of IRTyr960 in retinal endothelial cells cultured in high glucose [11]. To study whether a similar response occurred in vivo, we measured SOCS3 and IRTyr960 in β2-adrenergic receptor knockout mice treated with Compound 49b and untreated β2-adrenergic receptor knockout mice. Because Compound 49b actions on β2-adrenergic receptor knockout mice primarily target retinal endothelial cells, we assessed whether Compound 49b could reduce SOCS3 and IRTyr960. As in retinal endothelial cells in diabetic-like conditions, Compound 49b reduced SOCS3 levels (Figure 3A) and phosphorylation of IRTyr960 (Figure 3B), suggesting that Compound 49b can restore normal insulin signal transduction. Since β2-adrenergic receptor knockout mice have a phenotype similar to diabetes, we expected that SOCS3 and IRTyr960 would be increased in these mice. Indeed, loss of β2-adrenergic receptor signaling produced increased SOCS3 (Figure 3A) and phosphorylation of the insulin receptor on tyrosine 960 (Figure 3B), which inhibits proper signal transduction by blocking the insulin receptor/IRS-1 interaction [14].
Figure 3.
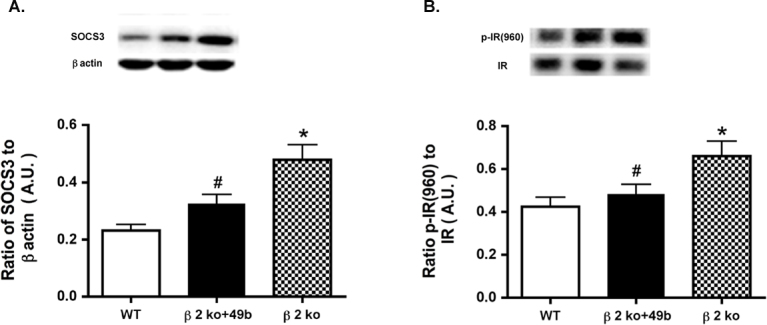
Compound 49b treatment reduces suppressor of cytokine signaling 3 and insulin receptorTyr960 in β2-adrenergic receptor knockout mice. A: Suppressor of cytokine signaling 3 (SOCS3) levels in control, β2-adrenergic receptor knockout mice+49b, and β2-adrenergic receptor knockout (KO) mice are shown. B: Phosphorylation of tyrosine 960 on the insulin receptor in the same animal groups. *p<0.05 versus wild-type (WT); #p<0.05 versus β2-adrenergic receptor KO mice. n = 5 for all groups. Data are mean ± standard error of the mean (SEM).
Antiapoptotic proteins are increased after Compound 49b treatment to β2-adrenergic receptor knockout mice
Since one of the primary actions of insulin signaling is to prevent apoptosis, we wanted to determine whether Compound 49b treatment of β2-adrenergic receptor knockout mice could decrease proapoptotic markers. As expected based on the decreased TNF-α and SOCS3 levels, Compound 49b significantly increased the protein levels of the antiapoptotic proteins (Figure 4C–D), while decreasing levels of the proapoptotic proteins (Figure 4A–B) in the β2-adrenergic receptor knockout mice, thus suggesting that Compound 49b restores normal insulin signal transduction to prevent apoptosis. Data demonstrate the expected finding that the proapoptotic factors Bax (Figure 4A) and cleaved caspase 3 (Figure 4B) are increased in β2-adrenergic receptor knockout mice, while the antiapoptotic proteins Bcl-xL (Figure 4C) and Akt (Figure 4D) are decreased.
Figure 4.

Pro- and anti-apoptotic proteins in control, β2KO+49b and β2KO lysates. Proapoptotic (Panels A–B) and antiapoptotic (Panels C–D) in control, β2-adrenergic receptor knockout mice+49b, and β2-adrenergic receptor knockout mice. A–D: Compound 49b reduces proapoptotic protein levels of Bax (A) and cleaved caspase 3 (B), while increasing the antiapoptotic proteins Bcl-xL (C) and phosphorylated Akt (D). *p<0.05 versus wild-type (WT); #p<0.05 versus β2-adrenergic receptor knockout (KO) mice. n = 5 for all groups. Data are mean ± standard error of the mean (SEM).
Compound 49b improves amplitudes of electroretinographic waves in β2-adrenergic receptor knockout mice
We reported that β2-adrenergic receptor knockout mice have reduced amplitudes of the a-wave, b-wave, and oscillatory potentials [6]. Although retinal endothelial cells likely do not directly alter ERG waveforms, restoration of insulin signaling in these cells appeared to improve retinal health, since β2-adrenergic receptor knockout mice treated with Compound 49b had significantly higher amplitudes than β2-adrenergic receptor knockout mice (Figure 5). Compound 49b appeared to increase the b-wave and oscillatory potentials much more than the a-wave (Figure 5). Since the b-wave and oscillatory potentials are believed to originate from the inner retina, while the a-wave is electrical signals from the photoreceptors, the data suggest that Compound 49b works primarily on the inner retina in β2-adrenergic receptor knockout mice.
Figure 5.
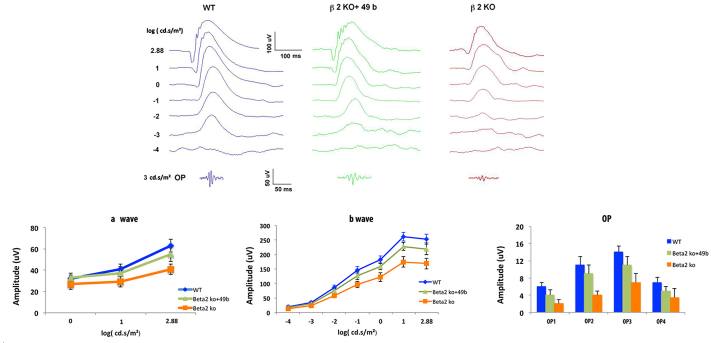
Electroretinogram analyses on control, β2-adrenergic receptor knockout mice+49b and β2-adrenergic receptor knockout mice. Compound 49b significantly increased the a-wave, b-wave, and oscillatory potentials in the β2-adrenergic receptor knockout (KO) mice. #p<0.05 versus β2-adrenergic receptor KO mice. n = 5 for all groups. Data are mean ± standard deviation (SD).
Retinal thickness is increased in β2-adrenergic receptor knockout mice treated with Compound 49b
Similar to the ERG analyses, we wanted to ascertain whether maintaining normal insulin signaling in β2-adrenergic receptor knockout mice would prevent the decreased retinal thickness noted in β2-adrenergic receptor knockout mice (Figure 6) [6]. Treatment with 1 mM Compound 49b for 2 months significantly increased the thickness of the retina of β2-adrenergic receptor knockout mice when compared to untreated β2-adrenergic receptor knockout mice (p<0.05 versus β2KO mice).
Figure 6.
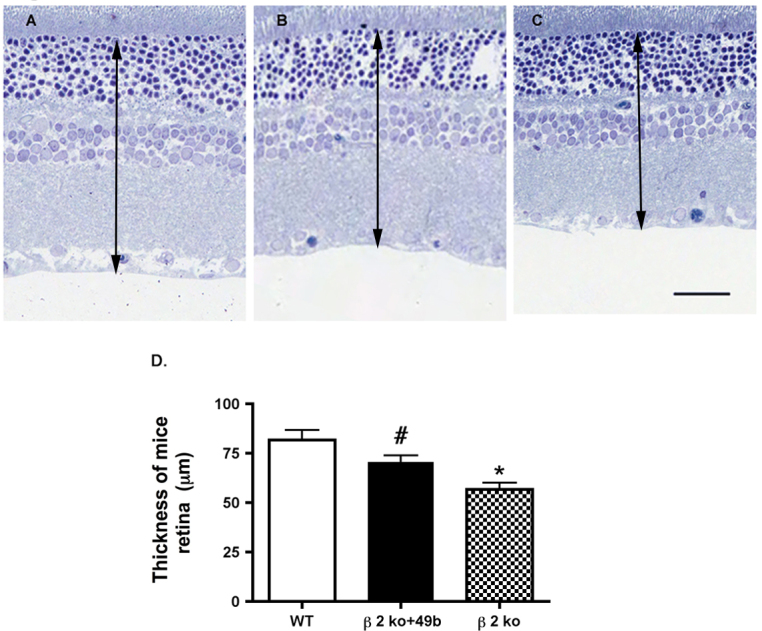
Images of the retina of the control, β2-adrenergic receptor knockout mice+Compound 49b, and β2-adrenergic receptor knockout mice. A–C: Compound 49b increased retinal thickness compared to untreated β2-adrenergic receptor knockout mice. A: Control. B: β2-adrenergic receptor knockout (KO) mice+49b. C: β2KO mice only. D: Graph of the mean ± standard error of the mean (SEM) of the retinal thickness of five mice in each group. Scale bar = 50 μm. The black arrow demarcates the thickness area measured. *p<0.05 versus wild-type (WT); #p<0.05 versus β2-adrenergic receptor KO mice. n = 5 for all groups. Data are mean ± SEM.
Discussion
The symptoms of diabetes and diabetic retinopathy are triggered in part by the loss of normal β-adrenergic receptor signaling [15]. It has been shown experimentally that a decrease in β-adrenergic receptor signaling through the loss of dopamine beta hydroxylase [16], β1-adrenergic receptor knockout [17], or β2-adrenergic receptor knockout [6] can all produce a diabetic-like phenotype. Studies also show that the effect is preventable since treating diabetic rats with β-adrenergic agonists such as isoproterenol [13] or a novel β1-/β2-adrenergic receptor agonist, Compound 49b, blocks diabetic-like changes [2].
The downstream pathways regulated by insulin and β-adrenergic receptor signaling have been well characterized in Müller cells and retinal endothelial cells in the retina [5,11] as well as several other cell types, including liver, muscle, and fat cells [8,18]. The TNF-α pathway is involved in these cell types. For example, we recently showed that TNF-α activates SOCS3 in retinal endothelial cells, leading to increased apoptosis due to inhibition of normal insulin signal transduction [11]. Similarly, we found that Müller cells cultured in diabetic-like conditions respond rapidly to high glucose, resulting in high levels of TNF-α, which can be decreased by treatment with salmeterol, a β2-adrenergic receptor agonist [5]. The decrease in levels of TNF-α led to increased survival of Müller cells [5]. Elimination of the key insulin signaling protein, IRS-1, also protects retinal Müller cells from apoptosis [19], suggesting that functional insulin signal transduction may be key to Müller cell survival. Although insulin receptor signaling appears to be regulated by different adrenergic receptor subclasses in these two cell types (β1-adrenergic receptors in retinal endothelial cells and β2-adrenergic receptors in Müller cells), the overall protective effects in the two systems appear to be similar.
To develop better treatment strategies for diabetic retinopathy based on our knowledge of β2- and β1-adrenergic receptor/insulin resistance pathways, it is important to determine the degree to which these two receptor subtypes, each located on a different cell type, act synergistically or through mutual compensation to block the development of insulin receptor resistance in diabetes. The overall conclusion suggested from our experiments is that there is significant compensation between receptor subtypes, since restoration of β1-adrenergic receptor activity with a receptor agonist can reduce the β2-deficient phenotype by 1) lowering TNF-α levels and blocking TNF-α-triggered pathways, 2) blocking concomitant SOCS3 activation, 3) decreasing levels of negative insulin receptor regulators, IRS-1Ser307 and IRTyr960, 4) decreasing production of proapoptotic factors, and 5) thus decreasing cell death. At the same time, morphological integrity (as measured by retinal thickness) and functional capacity (as measured by electroretinogram amplitude) are enhanced by the treatment. These responses occur in retinas that lack β2-adrenergic receptors suggesting that β1-adrenergic receptor activation alone can compensate for loss of β2-adrenergic receptor activity and is thus sufficient to prevent insulin receptor resistance in vivo.
Müller cells and retinal endothelial cells represent two major non-neuronal cell types within the mammalian retina. Both are involved in maintaining the flow of oxygen and nutrients to the neural retina. As the only cell that extends through the full thickness of the retina, Müller cells function as retinal glia, providing cellular scaffolding for neuronal structure, and facilitating the flow of oxygen and nutrients into the retinal and metabolic waste out of the retina through the maintenance of junctional complexes that constitute an outer limiting membrane and inner limiting membrane at inner and outer retinal surfaces. Retinal endothelial cells regulate nutrient and oxygen flow through blood vessels generally confined to the inner layers of the retina. We hypothesize that as gatekeepers of retinal metabolism, these two cell types are responsive to adrenergic and insulin signaling that helps to control TNF-α levels and limit apoptosis. Our new observations indicate a high degree of synergism and compensation in these adrenergic pathways such that the overall loss of β2-adrenergic receptor input to Müller cells can be compensated for by increased β1-adrenergic receptor input in retinal endothelial cells. Our previous studies with β1-adrenergic receptor knockout mice confirmed that the reverse also occurs, in that β2-adrenergic receptor stimulation of Müller cells can compensate for the loss of β1-adrenergic receptor activity in retinal endothelial cells. It appears that increased activity in either pathway alone is sufficient to block retinal cell apoptosis. From this perspective, treatment therapies for diabetic retinopathy that consist of a combined β1-/β2-adrenergic receptor agonist, such as Compound 49b, would be expected to provide maximal benefit by stimulating antiapoptotic pathways in Müller cells and retinal endothelial cells thus compensating for the diabetes-induced decrease of adrenergic receptor signaling.
Acknowledgments
This work was supported by the National Eye Institute (R01EY022330 to JJS); Juvenile Diabetes Research Foundation (2–2011–597 to JJS); Oxnard Foundation (JJS); Research to Prevent Blindness (James C. Fleming); National Eye Institute Vision Core Grant (PHS 3P30 EY013080 to Dianna Johnson).
References
- 1.Gardner TW, Antonetti DA, Barber AJ, LaNoue KF, Levison SW. Diabetic retinopathy: more than meets the eye. Surv Ophthalmol. 2002;47(Suppl 2):S253–62. doi: 10.1016/s0039-6257(02)00387-9. [DOI] [PubMed] [Google Scholar]
- 2.Zhang Q, Guy K, Pagadala J, Jiang Y, Walker RJ, Liu L, Soderland C, Kern TS, Ferry R, Jr, He H, Yates CR, Miller DD, Steinle JJ. Compound 49b Prevents Diabetes-Induced Apoptosis through Increased IGFBP-3 Levels. Invest Ophthalmol Vis Sci. 2012;53:3004–13. doi: 10.1167/iovs.11-8779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang Q, Jiang Y, Miller MJ, Peng B, Liu L, Soderland C, Tang J, Kern TS, Pintar J, Steinle JJ. IGFBP-3 and TNF-alpha Regulate Retinal Endothelial Cell Apoptosis. Invest Ophthalmol Vis Sci. 2013;54:5376–84. doi: 10.1167/iovs.13-12497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walker RJ, Steinle JJ. Role of beta-adrenergic receptors in inflammatory marker expression in Muller cells. Invest Ophthalmol Vis Sci. 2007;48:5276–81. doi: 10.1167/iovs.07-0129. [DOI] [PubMed] [Google Scholar]
- 5.Walker RJ, Anderson NM, Jiang Y, Bahouth S, Steinle JJ. Role of beta-adrenergic receptors regulation of TNF-alpha and insulin signaling in retinal Muller cells. Mol Vis. 2012;18:271–9. [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang Y, Zhang Q, Liu L, Tang J, Kern TS, Steinle JJ. beta2-adrenergic receptor knockout mice exhibit A diabetic retinopathy phenotype. PLoS ONE. 2013;8:e70555. doi: 10.1371/journal.pone.0070555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Steinle JJ, Booz GW, Meininger CJ, Day JN, Granger HJ. Beta 3-adrenergic receptors regulate retinal endothelial cell migration and proliferation. J Biol Chem. 2003;278:20681–6. doi: 10.1074/jbc.M300368200. [DOI] [PubMed] [Google Scholar]
- 8.Hotamisligil GS. The role of TNFalpha and TNF receptors in obesity and insulin resistance. J Intern Med. 1999;245:621–5. doi: 10.1046/j.1365-2796.1999.00490.x. [DOI] [PubMed] [Google Scholar]
- 9.Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS-1-Mediated Inhibition of Insulin Receptor Tyrosine Kinase Activity in TNF-alpha- and Obesity-Induced Insulin Resistance. Science. 1996;271:668–70. doi: 10.1126/science.271.5249.665. [DOI] [PubMed] [Google Scholar]
- 10.Guo S. Insulin signaling, resistance, and the metabolic syndrome: insights from mouse models into disease mechanisms. J Endocrinol. 2014;220:T1–23. doi: 10.1530/JOE-13-0327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiang Y, Zhang Q, Soderland C, Steinle JJ. TNFalpha and SOCS3 regulate IRS-1 to increase retinal endothelial cell apoptosis. Cell Signal. 2012;24:1086–92. doi: 10.1016/j.cellsig.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang Y, Liu L, Pagadala J, Miller DD, Steinle JJ. Compound 49b protects against blast-induced retinal injury. J Neuroinflammation. 2013;10:96. doi: 10.1186/1742-2094-10-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang Y, Walker RJ, Kern TS, Steinle JJ. Application of isoproterenol inhibits diabetic-like changes in the rat retina. Exp Eye Res. 2010;91:171–9. doi: 10.1016/j.exer.2010.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Senn JJ, Klover PJ, Nowak IA, Zimmers TA, Koniaris LG, Furlanetto RW, Mooney RA. Suppressor of cytokine signaling-3 (SOCS-3), a potential mediator of interleukin-6-dependent insulin resistance in hepatocytes. J Biol Chem. 2003;278:13740–6. doi: 10.1074/jbc.M210689200. [DOI] [PubMed] [Google Scholar]
- 15.Burnstock G. Changes in expression of autonomic nerves in aging and disease. J Auton Nerv Syst. 1990;30(Suppl):S25–34. doi: 10.1016/0165-1838(90)90096-2. [DOI] [PubMed] [Google Scholar]
- 16.Steinle JJ, Kern TS, Thomas SA, McFadyen-Ketchum LS, Smith CP. Increased basement membrane thickness, pericyte ghosts, and loss of retinal thickness and cells in dopamine beta hydroxylase knockout mice. Exp Eye Res. 2009;88:1014–9. doi: 10.1016/j.exer.2008.12.015. [DOI] [PubMed] [Google Scholar]
- 17.Panjala SR, Jiang Y, Kern TS, Thomas SA, Steinle JJ. Increased tumor necrosis factor-alpha, cleaved caspase 3 levels and insulin receptor substrate-1 phosphorylation in the beta-adrenergic receptor knockout mouse. Mol Vis. 2011;17:1822–8. [PMC free article] [PubMed] [Google Scholar]
- 18.Emanuelli B, Peraldi P, Filloux C, Chavey C, Freidinger K, Hilton DJ, Hotamisligil GS, Van Obberghen E. SOCS-3 inhibits insulin signaling and is up-regulated in response to tumor necrosis factor-alpha in the adipose tissue of obese mice. J Biol Chem. 2001;276:47944–9. doi: 10.1074/jbc.M104602200. [DOI] [PubMed] [Google Scholar]
- 19.Walker RJ, Anderson NM, Bahouth S, Steinle JJ. Silencing of insulin receptor substrate-1 increases cell death in retinal Muller cells. Mol Vis. 2012;18:271–9. [PMC free article] [PubMed] [Google Scholar]