

Abstract
The fast advancing RNA-seq technology has unveiled an unexpected diversity and expression specificity of 3′ untranslated regions (3′UTRs) of mRNAs. In particular, neural mRNAs seem to express significantly longer 3′UTRs, some of which are over 10 kb in length. The extensive elongation of 3′UTRs in neural tissues provides intriguing possibilities for cell type-specific regulations that are governed by miRNAs, RNA-binding proteins and ribonucleoprotein aggregates. In this article, we review recent progress in the characterization of mRNA 3′UTRs and discuss their implications in the understanding of 3′UTR-mediated gene regulation.
Keywords: ceRNAs, polyadenylation, RNP aggregates, transcriptome, 3′UTR
Introduction
One of the most important discoveries in the genomic era is the realization that the coding regions only constitute a tiny fraction (~ 2%) of the human genome. In contrast, a large portion of the genome is expressed as non-coding, potentially regulatory, RNAs. Although many of these non-coding transcripts have been discovered recently as discrete RNA molecules, the functional importance of non-coding portions of protein coding mRNAs, 3′UTRs in particular, has long been recognized. A large number of regulatory elements including binding sites for miRNAs and RNA-binding proteins (RBPs) have been identified in the 3′UTRs. In turn, the recruitment of these RNA binding complexes modulates the production of proteins from mRNAs. In recent years, transcriptome profiling has revealed that a large number of coding mRNAs harbor different 3′UTR isoforms in mammals [1-3], zebrafish [1-3], flies [4], worms [5], yeast [3, 6, 7] as well as plants [6, 7]. In general, these 3′UTR isoforms transcribed from a single coding region are produced by a molecular event called alternative polyadenylation (APA) during mRNA biogenesis. Both genome-wide studies and characterization of individual APA events have helped to provide evidence for regulatory roles of different 3′UTR lengths in many biological processes. Recent reports have begun to examine differential 3′UTR expression in a tissue-specific manner. In particular, neural cell-specific 3′UTR extension has been observed in flies and mammals [4, 8, 38]. These findings lay the foundation for understanding the functional significance of APA in both normal development and neurodegenerative diseases. In this article, we examine recent progress in accurately defining 3′UTR isoforms and discuss experimental approaches to characterize the global, interactive behavior of 3′UTRs as a means to study the role of APA in development, tissue homeostasis, and disease.
Extremely long 3′UTRs are broadly expressed in mammalian neural transcriptome
A recent study by Miura et al. [8] reported a group of mRNA transcripts with extremely long 3′UTRs. In their study, Miura et al. reanalyzed published RNA-seq data and discovered that some previously annotated long intergenic non-coding RNAs (lincRNAs) downstream of coding mRNAs are not discrete transcripts. Instead, they are extended 3′UTRs of the upstream mRNAs. The authors further designed an effective bioinformatic pipeline and examined genome-wide 3′UTR extensions in a total of 20 tissues using mouse and human RNA-seq data. Surprisingly, they revealed more than 3,850 unannotated long 3′UTRs. Some of them are extended more than 10 kb in length compared with current Ensembl database entries. By Northern blotting and in situ hybridization, the authors experimentally validated the existence of these long transcripts. Furthermore, the authors examined tissue specificity for these long transcripts. A total of 15 pairwise comparisons of 3′UTR profiles from six mouse tissues were performed. Interestingly, hippocampus emerged as the only tissue showing a strong preference for the long 3′UTRs. The same results were also observed in human data, in which a total of 16 tissues were examined. Although up-regulation of distal 3′UTRs in neural cells has been reported previously, Miura et al. directly visualized the in vivo expression pattern of extended 3′UTRs using in situ hybridization. These data depicted even more restricted expression patterns of extended 3′UTRs specifically in the central nervous system.
Alternative polyadenylation plays regulatory roles in mammalian gene expression
Many high-throughput methods – especially the ones based on next-generation sequencing – have been developed to annotate APA. Comparative studies using RNAs obtained from different cell lines or organs usually detect a large number of differentially expressed APAs [1-5]. Although they provide the evidence for dynamic 3′UTR formation in different cells, these results also reflect distinct biological states of these cells. Examination of whether APA plays a causative role in cell fate determination through the regulation of gene expression requires the knowledge of spatiotemporal patterns of APA during dynamic processes, such as developmental transition and disease progress. To achieve this goal, it is critical to delineate the heterogeneity of mRNA expression. The first level is associated with different cell lineages. Comparisons between closely related cell lineages during development, such as stem cell lineages, could provide more insights to the regulatory role of 3′UTR usage in cell fate determination. For example, in muscle stem cells, Pax7, a critical myogenic transcription factor, maintains high levels of expression by preferentially expressing an isoform with a short 3′UTR. The short 3′UTR lacks binding sites for miR-206, a highly expressed miRNA that promotes muscle differentiation, thus escaping repression by this miRNA [9]. Although this study offers an interesting example, a global analysis of 3′UTR length during the activation of muscle stem cells has not been determined. It is unclear whether the shortening of 3′UTRs represents a general mechanism for the maintenance of “stemness”. On the other hand, studies in T cell development and tumor cells suggest that 3′UTR shortening is generally associated with enhanced proliferation in these cells [10-12]. However, 3′UTR profiling with closely related stem cell lineages in the skin did not detect any noticeable trend of 3′UTR shortening between the highly proliferative progenitors and terminally differentiated daughter cells that exit the cell cycle [13]. Collectively, these studies suggest that APA may be differentially utilized in different cell lineages. The second level is the heterogeneity in mRNA expression within a cell population. In one scenario, a single cell could differentially express multiple 3′UTRs for a single coding mRNA. Alternatively, a subset of cells could dominantly express one isoform of 3′UTR whereas the other expresses a different 3′UTR isoform. If total RNAs are isolated from the cell population to examine the 3′UTR heterogeneity, no technique could distinguish these possibilities, as reviewed by [14]. In contrast, individual APA events can be examined in situ, as reported by Miura et al. They clearly showed cell-type specificity of 3′UTR isoforms. We note that recent developments in RNA in situ detection [15] and live cell imaging [16] are beginning to achieve single-molecule level resolution. Aided by these technical advances, more insights can be obtained to further understand the regulation of APA in complex cell populations. Ultimately, single-cell APA profiling could reveal global dynamics of 3′UTRs in a high-throughput manner.
Multiple approaches are required to map poly(A) signals to primary transcripts
In the Miura paper, the authors noted the incompleteness of transcriptome annotation in RefSeq and Ensembl database. In order to generate a comprehensive atlas for global APA of the transcriptome, one challenging task is to assign numerous 3′ end-sequencing tags to nearby protein coding mRNAs or to annotate them as novel transcripts. Current protocols for APA profiling only detect the 3′ end of polyadenylated transcripts. As a result, linking the 3′ end signals to their primary transcripts is implemented bioinformatically. This usually depends upon existing transcript annotation. Most of the published APA profiling utilizes RefSeq, Ensembl, or equivalent gene annotation databases and assigns novel polyadenylation sites based on the localization of annotated 3′UTRs and stop codons. This strategy becomes problematic when dealing with splicing-dependent 3′UTRs and extremely long 3′UTRs. In the former case, a novel polyadenylation site could be a shortened 3′UTR for a distal stop codon, or an extended 3′UTR for a proximal stop codon (Fig. 1A). In the latter case, extremely extended 3′UTRs could be mis-annotated as a discrete lincRNA as revealed by Miura’s work. To solve this issue, it is necessary to incorporate full-length RNA-seq data or to utilize methodologies that define a transcription unit either at RNA or DNA level. Combination of full-length and 3′end RNA-seq data to annotate novel 3′UTRs is equivalent to the reassembly of a transcriptome. As noted by Miura et al. and reviewed in [17], existing software such as Cufflink and Miso often generate truncated 3′UTRs, because of their highly uneven RNA-seq coverage. To alleviate these problems, Miura et al. designed a straightforward “sliding window plus filtering” strategy. By leveraging the distribution of RNA-Seq coverage within a defined window, they successfully identified thousands of 3′UTR extensions by using full-length RNA-seq data. However, full-length RNA-seq data usually show strongly diminished coverage towards the 3′ end. And this makes it more difficult to accurately define the 3′ end position.
Figure 1.
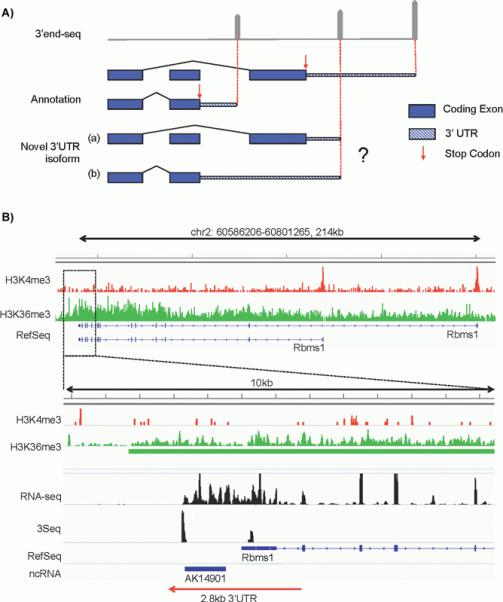
Strategies to map novel polyadenylation sites to primary transcripts. A: Example of potential difficulties in mapping a 3′ end-seq peak to primary transcripts when splicing-dependent isoforms are involved. Gene annotation indicates two 3′UTR isoforms with different stop codon (proximal and distal, red arrows) on different exons. In the top panel, gray pointed bars indicate 3′ end-sequencing peaks. The most proximal and distal peaks match the annotation, however the novel peak that resides in the middle can be interpreted as a shortened 3′UTR using the distal stop codon as shown in (a) or an extended 3′UTR using the proximal stop codon as shown in (b). B: Example of utilizing Histone ChIP-seq data to facilitate 3′UTR annotation. Top panel shows gene structure of Rbms1, which is an example with an extended 3′UTR reported by Miura and colleagues. ChIP-seq of H3K4me3 (red track) demarcates transcription start site (TSS). The two prominent peaks match the two annotated transcription start sites. H3K36me3 (green track) demarcates active Pol II elongation with preferable signal towards the 3′ end. A zoom in of a 10 kb region of the 3′ end is shown in the bottom panel, where a long non-coding RNA (AK14901) was annotated previously [19]. No H3K4me3 peaks are detected for the novel transcript whereas strong H3K36me3 signals extend from the RefSeq annotated Rbms1 3′UTR region and cover the entire region of AK14901, indicating that this non-coding RNA is likely an extended 2.8 kb 3′UTR. This conclusion is supported by 3′ end-seq profiling data (3Seq, data from [13]) and full-length RNA-seq (unpublished data).
An alternative strategy takes advantage of distinct histone modifications that are characteristic of actively transcribed genomic regions. For example, H3K4me3 generally marks the transcription start site and H3K36me3 or H3K79me3 marks transcription elongation. By performing ChIP-seq for these marks, one can define an actively transcribed genomic unit. Indeed, combination of H3K4me3 and H3K36me3 marks has been successfully utilized to identify novel lincRNAs [18]. In the case where a distal 3′ end-sequencing signal indicates a putative long extension of 3′UTR, the existence of continuous H3K36me3 coverage and the lack of H3K4me3 peaks in the extended region can serve as a strong indicator for an extended 3′UTR isoform, rather than an independent transcription event. As an example, we examined Rbms1, whose extended 3′UTR was originally identified as an ncRNA by Ponjavic et al. [19] and then re-annotated as a 3′UTR extension by Miura and colleagues. Rbms1 is also expressed in the skin lineages, hence our H3K4me3-H3K36me3 ChIP-Seq data clearly mark the ~3 kb region downstream of the previously annotated 3′ end as a 3′UTR extension. We judge this to be a 3′UTR extension rather than a discrete transcript, because it lacks H3K4me3 signals and contains continuous coverage of H3K36me3 from the upstream regions (Fig. 1B). In addition, our full-length RNA-Seq and 3′ end sequencing data also support this conclusion (Fig. 1B). Thus, this example highlights the potential advantage of combinatorial approaches with ChIP-Seq and RNA-Seq to accurately annotate the transcriptome.
Recently, a new technique has been developed to capture 5′ and 3′ ends of the same RNA transcript simultaneously [20]. It appears to be a promising strategy because the pairing of 5′ and 3′ end tags belongs to a single RNA molecule, and it leads to much enhanced reads alignment [20]. Consequently, annotations of distinct transcripts – especially for isoforms at the UTR regions – are drastically improved. Although this technique is currently developed in yeast, its application in multicellular eukaryotes, where multi-exon transcripts and alternative splicing are much more common, can be easily achieved. This will help to elucidate the dynamics and complexity of UTRs.
Extremely long 3′UTRs provide a platform to regulate gene expression
The 3′UTRs of mRNAs have been implicated in numerous roles in the regulation of gene expression, such as mRNA stability (reviewed in [21]) and localization [22, 23], stem cell differentiation (reviewed in [14]), cell proliferation, and tumorigenesis [10-12]. Generally, these regulatory roles are associated with cis elements located in the 3′UTRs that serve as a platform to recruit RNA binding complex. Thus, the discovery of exceptionally long 3′UTRs in neural cells indicates additional possibilities for cell-specific regulation. One such possibility is miRNA-mediated regulation. Because of the lack of coding ability, 3′UTRs are under much less pressure to maintain a conserved sequence, and that leads to more sequence diversity. However, genome-wide analysis focusing on conserved sequences in the 3′UTRs has revealed many potentially functional motifs. Among these motifs, nearly one-half are miRNA recognition sites [24]. Subsequent bioinformatic analysis and experimental identification of miRNA binding sites have further validated the enrichment of miRNA binding sites located in the 3′UTRs [25-27]. However, it is not clear whether all of these miRNA binding sites mediate functionally important gene silencing to their host mRNAs. In the hypothesis of competitive endogenous RNAs (ceRNAs), some of the miRNA binding sites are proposed to simply serve as a decoy to compete for the pool of miRNAs and their associated RNA-induced silencing complex. In turn, these ceRNAs are proposed to regulate the accessibility of miRNAs to other targets [26, 28]. This mechanism suggests a bidirectional regulation of miRNAs and their mRNA binding partners. Recent experimental evidence has provided support for the ceRNA hypothesis and highlighted the possibility of 3′UTRs serving as trans regulators. It adds a new dimension to 3′UTR-mediated function, in addition to the well appreciated cis effect of 3′UTRs for the regulation of stability and translational efficiency of host mRNA molecules [29-33].
The extremely long 3′UTRs identified by Miura et al. significantly increase the probability of miRNA binding sites serving as a decoy to sequester miRNAs from the functional pool in trans, and as ceRNAs to regulate other mRNAs. Interestingly, this mechanism may lead to a synchronized expression pattern of mRNA species that share the same miRNA binding sites (Fig. 2A and B, “functional pool”). To estimate the possible trans effect of these long 3′UTRs, an accurate quantitative model of miRNA:mRNA interaction is required. With the advance of efficient miRNA cloning, target identification and quantitative mRNA profiling techniques, it is now possible to more precisely dissect the regulatory network of miRNAs and their associated RNA species. Since one miRNA can target multiple mRNA species and one mRNA may contain multiple miRNA binding sites, it is essential to establish a stoichiometry between miRNAs and mRNA target sites, in order to reliably model miRNA-mRNA regulatory networks.
Figure 2.
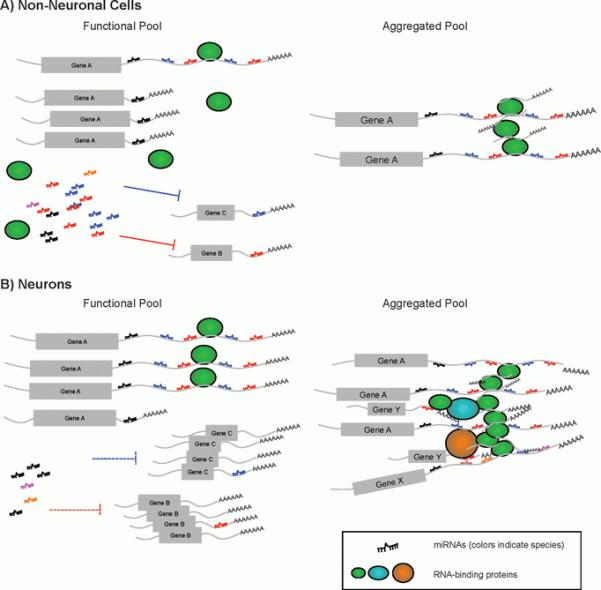
Illustration of potential impact of extended 3′UTRs on miRNA regulation networks and RNP aggregates. A: In non-neuronal cells where dominant 3′UTR isoform of gene A utilizes a proximal polyadenylation signal, the functional pool of miRNAs has an ample quantity of miRNAs (shown as ssRNA in multiple colors), which are sufficient to target gene B and gene C. The transcripts of gene A that are sequestered in the aggregation pool are not significant, and do not interfere with the correspondent miRNA targeting (miRNAs colored black, blue and red). B: In neurons, the long 3′UTR isoform of gene A is drastically upregulated. It may lead to one of two outcomes. First, it serves as a decoy to deplete miRNAs from the functional pool (miRNAs colored black, blue and red), which leads to upregulation of gene B and gene C. Secondly, more gene A transcripts are sequestered to form RNP aggregates. In turn, it depletes other RNA-binding proteins from their functional pool (oval shapes, colored green, cyan, and orange) as well as RNA transcripts of gene X and Y that are associated with these RNA-binding proteins. Collectively, these abnormal aggregates may cause toxicity in pathological conditions.
Long 3′UTRs and RNA aggregates in neural tissues play important roles in physiological and disease contexts
Both Miura et al. and other researchers have reported 3′UTR lengthening [1, 34-37] or preference for novel distal polyadenylation signals in neural cells [4, 38]. It is well characterized that 3′UTRs play important roles in RNA transportation and local protein translation in dendrite of neurons [22, 23]. Mechanistic studies of brain-derived neurotrophic factor (BDNF) further highlight the distinct roles of short and long 3′UTRs in neural cells. When at rest, the short, 0.35 kb 3′UTR mediates basal level production of BDNF whereas the long, 2.85 kb 3′UTR suppresses BDNF translation. Upon activation, the long 3′UTR isoform is activated by the stimuli and rapidly turns on BDNF production before the transcriptional activation [39]. Furthermore, the long isoform is required to regulate spine morphology and synaptic plasticity [40]. On the other hand, these locally translated mRNAs are often packed with RBPs to form ribonucleoprotein (RNP) aggregates, or RNA granules. These RNPs have been shown to be required for synaptic plasticity [41-43]. Interestingly, a recent study has reported that in both brain tissues and U2OS cells mRNAs enriched in RNA granules preferentially express significantly longer 3′UTRs than those not precipitated with the RNPs. In addition, functional classification of the mRNAs deposited in the RNPs revealed the enrichment in cell adhesion- and axon guidance-associated sequences, which are associated with polarized transportation and local translation [41]. Furthermore, both the 3′UTRs and the coding regions of these precipitated RNAs are enriched with the nanos response element that is recognized by Pum1 and Pum2. Strikingly, both Pum1 and Pum2 contain a low complexity (LC) domain, which is self-sufficient to mediate RNA aggregation [44]. Altogether, these results suggest that the 3′UTRs of neural mRNA may play particularly important roles in mediating neural functions. Thus, the finding of extremely long 3′UTRs in mammalian brain elicits a tantalizing possibility that these mRNAs are engineered for local translation but at the same time increasing the probability for RNA aggregation.
Protein aggregation in neurons is a common pathological characteristic of neurodegenerative diseases, two examples being beta-amyloid plaques in Alzheimer’s disease and Lewy bodies in Parkinson’s disease [45]. It has been reported that fragile X mental retardation protein (FMRP), which is required for localized translation in neurons, is clustered in cytoplasmic granules and stress granules [46]. Recent observations are beginning to implicate RBPs including TDP-43, FUS, Ataxin-2, and hnRNP1 in causative roles for a number of neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS) and frontotemporal lobar dementia with ubiquitin pathology (FTLD-U) [46-50]. Pathological mutations in TDP-43, FUS, Ataxin-2, etc. often are localized in prion-like domains or other LC domains. Interestingly, such mutant proteins are more prone to aggregate compared to their wildtype form, thus suggesting a causal relationship between aberrant RBP aggregation and pathogenesis of these diseases [46, 48-50]. Mutations altering RNA-binding ability of FUS and TDP-43 are shown to modify the toxicity of the pathogenic mutants. These observations indicate that the RNA-binding nature of these proteins plays an important role in pathogenesis [51-53]. Therefore, it is important to further investigate the interaction between RNA and RBPs during the pathogenesis of neurodegenerative diseases. One hypothesis is that in neurons, aggregation of RBPs sequesters the functional pool of RBP as well as their associated mRNAs (Fig. 2B). In turn, the sequestration may impair the translational output of the associated mRNAs (personal communication with Roy Parker). The extremely long 3′UTRs lead to a drastic increase in the binding sites of miRNAs and RBPs. Therefore, it would be interesting to experimentally determine the associated miRNAs and RBPs to these long 3′UTRs and examine their potential involvement in the formation of RNA granules in pathological conditions (Fig. 2B).
Concluding remarks
Rapid developments in RNA-seq technology and bioinformatics analysis have greatly improved our knowledge of the complexity of the transcriptome. The preferential 3′UTR extension in neural tissues as demonstrated by Miura et al. is likely to be an important mechanism to mediate cell type-specific functions. These exceptionally long 3′UTRs could serve as versatile platforms to recruit RNA binding complexes and regulate gene expression through cis and trans mechanisms. The challenge now is to experimentally characterize the functional significance of these long 3′UTRs in development, tissue homeostasis, and disease conditions. With an ever-growing arsenal of tools in molecular biology, biochemistry, genetics, and high-throughput sequencing at our disposal, we anticipate that many more exciting findings will extend the boundaries of our knowledge, further revealing the extent of 3′UTRs in controlling mRNA function in the brain.
Acknowledgments
We apologize for any omissions of relevant citations due to the page limitation. We would like to thank the members of the Yi and Dowell labs and Roy Parker for discussion. This publication was made possible by a grant from NIH/NIAMS (R01AR059697) and a grant from American Cancer Society (RSG-13-197-01-DDC) to R.Y. and by an NIH Predoctoral Training Grant T32 GM08759 to L.W.
Abbreviations
- ALS
amyotrophic lateral sclerosis
- APA
alternative polyadenylation
- BDNF
brain-derived neurotrophic factor
- ceRNA
competitive endogenous RNA
- FMRP
fragile X mental retardation protein
- FTLD-U
frontotemporalobar dementia with ubiquitin pathology
- FUS
fused in sarcoma
- LC domain
low-complexity domain
- lincRNA
long intergenic non-coding RNA
- Pax7
pair box 7
- Rbms1
RNA binding motif, single stranded interacting protein 1
- RBP
RNA-binding protein
- RNP
ribonucleoprotein
- TDP-43
TAR DNA-binding protein
- UTR
untranslated region
Footnotes
The authors have declared no conflict of interest.
References
- 1.Ulitsky I, Shkumatava A, Jan CH, Subtelny AO, et al. Extensive alternative polyadenylation during zebrafish development. Genome Res. 2012;22:2054–66. doi: 10.1101/gr.139733.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Derti A, Garrett-Engele P, Macisaac KD, Stevens RC, et al. A quantitative atlas of polyadenylation in five mammals. Genome Res. 2012;22:1173–83. doi: 10.1101/gr.132563.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ozsolak F, Kapranov P, Foissac S, Kim SW, et al. Comprehensive polyadenylation site maps in yeast and human reveal pervasive alternative polyadenylation. Cell. 2010;143:1018–29. doi: 10.1016/j.cell.2010.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smibert P, Miura P, Westholm JO, Shenker S, et al. Global patterns of tissue-specific alternative polyadenylation in Drosophila. Cell Rep. 2012;1:277–89. doi: 10.1016/j.celrep.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jan CH, Friedman RC, Ruby JG, Bartel DP. Formation, regulation and evolution of Caenorhabditis elegans 3′UTRs. Nature. 2011;469:97–101. doi: 10.1038/nature09616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sherstnev A, Duc C, Cole C, Zacharaki V, et al. Direct sequencing of Arabidopsis thaliana RNA reveals patterns of cleavage and polyadenylation. Nat Struct Mol Biol. 2012;19:845–52. doi: 10.1038/nsmb.2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu X, Liu M, Downie B, Liang C, et al. Genome-wide landscape of polyadenylation in Arabidopsis provides evidence for extensive alternative polyadenylation. Proc Natl Acad Sci USA. 2011;108:12533–8. doi: 10.1073/pnas.1019732108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miura P, Shenker S, Andreu-Agullo C, Westholm JO, et al. Widespread and extensive lengthening of 3′UTRs in the mammalian brain. Genome Res. 2013;23:812–25. doi: 10.1101/gr.146886.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boutet SC, Cheung TH, Quach NL, Liu L, et al. Alternative polyadenylation mediates microRNA regulation of muscle stem cell function. Cell Stem Cell. 2012;10:327–36. doi: 10.1016/j.stem.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sandberg R, Neilson JR, Sarma A, Sharp PA, et al. Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer microRNA target sites. Science. 2008;320:1643–7. doi: 10.1126/science.1155390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mayr C, Bartel DP. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell. 2009;138:673–84. doi: 10.1016/j.cell.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ji Z, Tian B. Reprogramming of 3′ untranslated regions of mRNAs by alternative polyadenylation in generation of pluripotent stem cells from different cell types. PLoS One. 2009;4:e8419. doi: 10.1371/journal.pone.0008419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang L, Dowell RD, Yi R. Genome-wide maps of polyadenylation reveal dynamic mRNA 3′-end formation in mammalian cell lineages. RNA. 2013;19:413–25. doi: 10.1261/rna.035360.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mueller AA, Cheung TH, Rando TA. All’s well that ends well: alternative polyadenylation and its implications for stem cell biology. Curr Opin Cell Biol. 2013;25:222–32. doi: 10.1016/j.ceb.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, et al. Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods. 2008;5:877–9. doi: 10.1038/nmeth.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lionnet T, Czaplinski K, Darzacq X, Shav-Tal Y, et al. A transgenic mouse for in vivo detection of endogenous labeled mRNA. Nat Methods. 2011;8:165–70. doi: 10.1038/nmeth.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garber M, Grabherr MG, Guttman M, Trapnell C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat Methods. 2011;8:469–77. doi: 10.1038/nmeth.1613. [DOI] [PubMed] [Google Scholar]
- 18.Guttman M, Amit I, Garber M, French C, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;457:223–7. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ponjavic J, Oliver PL, Lunter G, Ponting CP. Genomic and transcriptional co-localization of protein-coding and long non-coding RNA pairs in the developing brain. PLoS Genet. 2009;5:e1000617. doi: 10.1371/journal.pgen.1000617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pelechano V, Wei W, Steinmetz LM. Extensive transcriptional heterogeneity revealed by isoform profiling. Nature. 2013;497:127–31. doi: 10.1038/nature12121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garneau NL, Wilusz J, Wilusz CJ. The highways and byways of mRNA decay. Nat Rev Mol Cell Biol. 2007;8:113–26. doi: 10.1038/nrm2104. [DOI] [PubMed] [Google Scholar]
- 22.Burgin KE, Waxham MN, Rickling S, Westgate SA, et al. In situ hybridization histochemistry of Ca2+/calmodulin-dependent protein kinase in developing rat brain. J Neurosci. 1990;10:1788–98. doi: 10.1523/JNEUROSCI.10-06-01788.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller S, Yasuda M, Coats JK, Jones Y, et al. Disruption of dendritic translation of CaMKIIalpha impairs stabilization of synaptic plasticity and memory consolidation. Neuron. 2002;36:507–19. doi: 10.1016/s0896-6273(02)00978-9. [DOI] [PubMed] [Google Scholar]
- 24.Xie X, Lu J, Kulbokas EJ, Golub TR, et al. Systematic discovery of regulatory motifs in human promoters and 3′UTRs by comparison of several mammals. Nature. 2005;434:338–45. doi: 10.1038/nature03441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–33. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Salmena L, Poliseno L, Tay Y, Kats L, et al. A ceRNA hypothesis: the Rosetta stone of a hidden RNA language? Cell. 2011;146:353–8. doi: 10.1016/j.cell.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chi SW, Zang JB, Mele A, Darnell RB. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature. 2009;460:479–86. doi: 10.1038/nature08170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seitz H. Redefining microRNA targets. Curr Biol. 2009;19:870–3. doi: 10.1016/j.cub.2009.03.059. [DOI] [PubMed] [Google Scholar]
- 29.Tay Y, Kats L, Salmena L, Weiss D, et al. Coding-independent regulationof the tumor suppressor PTEN by competing endogenous mRNAs. Cell. 2011;147:344–57. doi: 10.1016/j.cell.2011.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sumazin P, Yang X, Chiu H-S, Chung W-J, et al. An extensive microRNA-mediated network of RNA-RNA interactions regulates established oncogenic pathways in glioblastoma. Cell. 2011;147:370–81. doi: 10.1016/j.cell.2011.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karreth FA, Tay Y, Perna D, Ala U, et al. In vivo identification of tumor-suppressive PTEN ceRNAs in an oncogenic BRAF-induced mouse model of melanoma. Cell. 2011;147:382–95. doi: 10.1016/j.cell.2011.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cesana M, Cacchiarelli D, Legnini I, Santini T, et al. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell. 2011;147:358–69. doi: 10.1016/j.cell.2011.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Poliseno L, Salmena L, Zhang J, Carver B, et al. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature. 2010;465:1033–8. doi: 10.1038/nature09144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stark A, Brennecke J, Bushati N, Russell RB, et al. Animal microRNAs confer robustness to gene expression and have a significant impact on 3′UTR evolution. Cell. 2005;123:1133–46. doi: 10.1016/j.cell.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 35.Wang ET, Sandberg R, Luo S, Khrebtukova I, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–6. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramsköld D, Wang ET, Burge CB, Sandberg R. An abundance of ubiquitously expressed genes revealed by tissue transcriptome sequence data. PLoS Comput Biol. 2009;5:e1000598. doi: 10.1371/journal.pcbi.1000598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shepard PJ, Choi E-A, Lu J, Flanagan LA, et al. Complex and dynamic landscape of RNA polyadenylation revealed by PAS-Seq. RNA. 2011;17:761–2. doi: 10.1261/rna.2581711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hilgers V, Perry MW, Hendrix D, Stark A, et al. Neural-specific elongation of 3′UTRs during Drosophila development. Proc Natl Acad Sci USA. 2011;108:15864–9. doi: 10.1073/pnas.1112672108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lau AG, Irier HA, Gu J, Tian D, et al. Distinct 3′UTRs differentially regulate activity-dependent translation of brain-derived neurotrophic factor (BDNF) Proc Natl Acad Sci USA. 2010;107:15945–50. doi: 10.1073/pnas.1002929107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.An JJ, Gharami K, Liao G-Y, Woo NH, et al. Distinct role of long 3′UTR BDNF mRNA in spine morphology and synaptic plasticity in hippocampal neurons. Cell. 2008;134:175–87. doi: 10.1016/j.cell.2008.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Han TW, Kato M, Xie S, Wu LC, et al. Cell-free formation of RNA granules: bound RNAs identify features and components of cellular assemblies. Cell. 2012;149:768–79. doi: 10.1016/j.cell.2012.04.016. [DOI] [PubMed] [Google Scholar]
- 42.Kwak JE, Drier E, Barbee SA, Ramaswami M, et al. GLD2 poly(A) polymerase is required for long-term memory. Proc Natl Acad Sci USA. 2008;105:14644–9. doi: 10.1073/pnas.0803185105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McCann C, Holohan EE, Das S, Dervan A, et al. The Ataxin-2 protein is required for microRNA function and synapse-specific long-term olfactory habituation. Proc Natl Acad Sci USA. 2011;108:E655–62. doi: 10.1073/pnas.1107198108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kato M, Han TW, Xie S, Shi K, et al. Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell. 2012;149:753–67. doi: 10.1016/j.cell.2012.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taylor JP, Hardy J, Fischbeck KH. Toxic proteins in neurodegenerative disease. Science. 2002;296:1991–5. doi: 10.1126/science.1067122. [DOI] [PubMed] [Google Scholar]
- 46.Didiot M-C, Subramanian M,, Flatter E,, Mandel J-L, et al. Cells lacking the fragile X mental retardation protein (FMRP) have normal RISC activity but exhibit altered stress granule assembly. Mol Biol Cell. 2009;20:428–37. doi: 10.1091/mbc.E08-07-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen-Plotkin AS, Lee VM-Y, Trojanowski JQ. TAR DNA-binding protein 43 in neurodegenerative disease. Nat Rev Neurol. 2010;6:211–20. doi: 10.1038/nrneurol.2010.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ito D, Suzuki N. Conjoint pathologic cascades mediated by ALS/FTLD-U linked RNA-binding proteins TDP-43 and FUS. Neurology. 2011;77:1636–43. doi: 10.1212/WNL.0b013e3182343365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim HJ, Kim NC, Wang Y-D, Scarborough EA, et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013;495:467–73. doi: 10.1038/nature11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nonhoff U, Ralser M, Welzel F, Piccini I, et al. Ataxin-2 interacts with the DEAD/H-box RNA helicase DDX6 and interferes with P-bodies and stress granules. Mol Biol Cell. 2007;18:1385–96. doi: 10.1091/mbc.E06-12-1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Daigle JG, Lanson NA, Smith RB, Casci I, et al. RNA-binding ability of FUS regulates neurodegeneration, cytoplasmic mislocalization and incorporation into stress granules associated with FUS carrying ALS-linked mutations. Hum Mol Genet. 2013;22:1193–205. doi: 10.1093/hmg/dds526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sun Z, Diaz Z, Fang X, Hart MP, et al. Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS Biol. 2011;9 doi: 10.1371/journal.pbio.1000614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Voigt A, Herholz D, Fiesel FC, Kaur K, et al. TDP-43-mediated neuron loss in vivo requires RNA-binding activity. PLoS One. 2010;5:e12247. doi: 10.1371/journal.pone.0012247. [DOI] [PMC free article] [PubMed] [Google Scholar]