

Summary
Dynamin is a large multidomain GTPase that assembles into helical arrays around the necks of deeply invaginated clathrin-coated pits and catalyzes membrane fission during the final stages of endocytosis. Although it is well established that the function of dynamin in vivo depends on its oligomerization and its capacity for efficient GTP hydrolysis, the molecular mechanisms governing these activities have remained poorly defined. In recent years, there has been an explosion of structural data that has provided new insights into the architecture, organization and nucleotide-dependent conformational changes of the dynamin fission machine. Here, we review the key findings of these efforts and discuss the implications of each with regard to GTP hydrolysis, dynamin assembly and membrane fission.
Key words: Membrane fission, Dynamin, Structure, Endocytosis, GTPase, Hydrolysis
Introduction
Dynamin is a multidomain GTPase that forms helical assemblies at the necks of deeply invaginated clathrin-coated pits (CCPs) and catalyzes membrane scission during the final stages of clathrin-mediated endocytosis (CME) (Mettlen et al., 2009). It is the prototypical member of a family of large atypical GTPases that share the common properties of low affinity for guanine nucleotides (apparent Km values of ∼10–100 µM), high basal turnover (∼0.4–1 min−1) and the propensity for oligomerization (Song and Schmid, 2003). Dynamin-related proteins participate in a wide variety of cellular processes including mitochondrial fission (DRP1 and Dnm1) and fusion (Opa1, Mgm1 and mitofusins), vacuolar fission (Vps1), interferon-induced anti-viral protection (Mx proteins), and plant cell cytokinesis and membrane fission (Arabidopsis thaliana DRP proteins) (Praefcke and McMahon, 2004). In vitro, basal GTP hydrolysis can be observed when using purified dynamin tetramers; they readily oligomerize into rings and spirals in low ionic strength buffer (Hinshaw and Schmid, 1995) or into helical arrays in the presence of anionic lipid scaffolds (Sweitzer and Hinshaw, 1998; Stowell et al., 1999), structures that are reminiscent of the dynamin collars formed at the necks of invaginated clathrin-coated pits in vivo (Takei et al., 1995; Hinshaw and Schmid, 1995). Similar assemblies have been observed with other dynamin family members, including Dnm1 (Ingerman et al., 2005; Mears et al., 2011), MxA (Kochs et al., 2002) and a bacterial dynamin like protein (BDLP) from Nostoc punctiforme (Low and Löwe, 2006; Low et al., 2009). Oligomerization stimulates the basal hydrolysis rate of dynamin more than 100-fold (Warnock et al., 1996; Stowell et al., 1999) and is essential for CME, evidenced by the fact that mutations that impair GTP binding, self-assembly or assembly-stimulated GTP hydrolysis also cause defects in endocytic uptake in vivo (Schmid and Frolov, 2011).
Dynamin has five functional domains (Fig. 1). The N-terminal G domain binds and hydrolyzes GTP and shows homology to the catalytic domains of other GTPases (van der Bliek, 1999). The ‘middle’ domain is largely a coiled-coil structure and mediates the higher-order dynamin oligomerization in conjunction with the GTPase effector domain (GED) (Ramachandran et al., 2007). Mutagenesis data initially suggested that the GED also functioned as an intramolecular GTPase-activating protein (GAP) (Sever et al., 1999); however, our subsequent biochemical and structural studies revealed that it exerts its effects through a non-classical mechanism in which its C-terminus (CGED) associates with the N- and C-terminal helices of the G domain (NGTPase and CGTPase, respectively), located far away from the active site (Chappie et al., 2009; Chappie et al., 2010). The resulting three-helix bundle is termed the ‘bundle-signaling element’ (BSE), as mutations within its hydrophobic core indirectly modulate the GTPase activity of dynamin in vitro and shift the kinetics of endocytosis in vivo such that fission, rather than coated-pit maturation, becomes rate limiting (Chappie et al., 2009). Dynamin also contains a membrane-binding pleckstrin homology (PH) domain that preferentially targets phosphatidylinositol (4,5)-bisphosphate [PtdIns(4,5)P2] lipids (Salim et al., 1996), and a C-terminal proline-arginine-rich domain (PRD) that binds the SH3 domains of accessory partner proteins that are important for CME (see supplementary Table S1 in Ferguson and De Camilli, 2012).
Fig. 1.
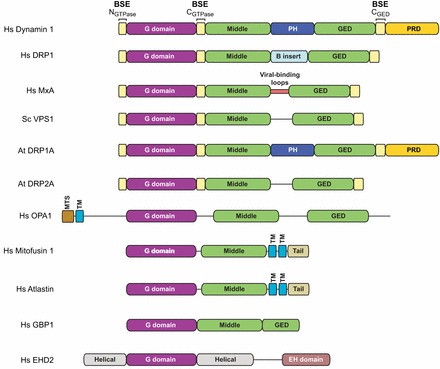
Domain architecture of dynamin family members. Representative members of different dynamin-related proteins are shown. Hs, Homo sapiens; Sc, Saccharomyces cerevisiae; At, Arabidopsis thaliana; BSE, bundle-signaling element; PH, pleckstrin homology domain; GED, GTPase effector domain; PRD, proline- and arginine-rich domain; MTS, mitochondrial-targeting sequence; TM, transmembrane domain; EH, epsin homology domain.
The G domain is the most highly conserved region among dynamin homologs. Even distant relatives, such as BDLP, the interferon-induced human guanylate binding protein 1 (GBP1), the endoplasmic reticulum fusion proteins atlastin, yeast Sey1 and plant RHD3, and the cellular trafficking EHD ATPases show significant sequence and structural similarity in this region (Low and Löwe, 2006; Ghosh et al., 2006; Daumke et al., 2007; Byrnes and Sondermann, 2011; Bian et al., 2011). The middle domain and GED are also present in dynamin-related proteins, underscoring their functional importance in oligomerization. Additions to this core framework, i.e. the PH domain and PRD in dynamin, the B-insert in DRP1 and Dnm1, the viral-binding loops in Mx proteins and membrane anchors found in Opa1, Mgm1 and mitofusins, represent function-specific modifications (Fig. 1).
Cell biologists have long struggled to understand the mechanisms underlying dynamin function. A structural-biology-led renaissance has occurred in recent years, which has provided numerous insights into the intricate architectural organization and hydrolysis activities of this complex fission machine. In this Commentary, we will review these results, emphasizing key findings from X-ray crystallography, cryoelectron microscopy and biochemistry. Our aim is not only to present the significant advances heralded by these studies, but also to inform those unfamiliar with structural biology about the limitations of the resulting models and important considerations for future experiments.
Dynamin structure at atomic resolution
X-ray crystallography can reveal detailed information about the architecture and mechanism of a macromolecule provided well-ordered diffraction-quality crystals can be obtained. This proved to be a challenge for dynamin, due to its size, complex folding and inherent propensity for oligomerization at high concentrations. For years, crystallographic studies were limited to a subset of isolated domains that retained a stable, compact structure, namely the G domains of human dynamin 1 and Dictyostelium discoideum DynA, which were made as fusions to myosin motors (Niemann et al., 2001; Reubold et al., 2005) and the PH domain (Ferguson et al., 1994; Timm et al., 1994). Recently, however, the structures of the intact dynamin molecule (Faelber et al., 2011; Ford et al., 2011) and of its relative MxA (Gao et al., 2011) were solved (Fig. 2A). The key to generating conformationally homogeneous monodisperse samples that were suitable for crystallization was the identification and incorporation of assembly-defective mutations that hinder the formation of helical arrays, while concomitantly preserving the underlying structural integrity. For human dynamin 1 and MxA, stretches of alanine mutations – IHGIR395–399 to AAAAA and YRGR440–443 to AAAA, respectively – were introduced based on previous success in crystallizing the isolated middle–GED MxA stalk (Gao et al., 2010); for rat dynamin, a single point mutation (G397D) was sufficient to enable suitable samples to be made. These substitutions, like the other previously described assembly-defective mutations in yeast mitochondrial Dnm1 (G385D, Ingerman et al., 2005) and human dynamin 1 (I690K, Sever et al., 2006; R361S and R399A, Ramachandran et al., 2007), shift the oligomeric state from tetramers to dimers. Additionally, the PRD was removed from human and rat dynamin and the viral substrate-binding loop (residues 533–561) of MxA was deleted. Together, these alterations produced crystals that diffracted to 3.7 Å (human dynamin 1; Faelber et al., 2011), 3.1 Å (rat dynamin; Ford et al., 2011), and 3.5 Å (MxA; Gao et al., 2011).
Fig. 2.
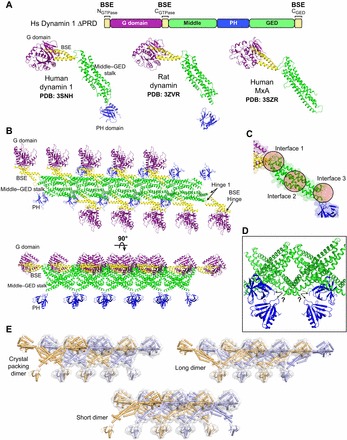
Structures and crystal packing of dynamin family members. (A) X-ray structures of intact human dynamin 1, rat dynamin and human MxA. Each protein contains different mutations that yield stable dimers in solution. Only the monomers are shown. The linear domain arrangement of human dynamin 1 is shown above for comparison. Coloring is as follows: G domain, purple; BSE, yellow; middle–GED stalk, green; PH domain, blue. (B) Dynamin-related proteins crystallize in linear arrays that are mediated primarily by stalk–stalk interactions. Rat dynamin crystal packing (PDB: 3ZVR) is depicted in two orientations (top view above, side view below). (C) Magnified view of the dynamin stalk illustrating the three interfaces (circles) that mediate intermolecular interactions within the crystallized linear dynmain array. Mutations that result in the monodisperse dimers used for crystallization map to interface 3. (D) Disordered regions at the base of the stalk yield an ambiguity (black arrows) in determining the stalk–PH connectivity. (E) Different putative domain connectivities within the mutant dynamin dimers can conform to the same crystal packing. Alternate dimers are colored orange and light blue. The light gray surface illustrates how different arrangements all yield the same overall shape and packing.
Dynamin and MxA each form extended monomers in which the polypeptide folds back on itself. As a result, the three-dimensional domain organization does not directly correlate with the linear sequence of functional domains (Fig. 1; Fig. 2A). The two termini of the polypeptides come together to form parts of the BSE, which in turn connects the G domain to a helical stalk comprised of the middle domain and GED. The PH domain sits at the bottom of the stalk in dynamin and is replaced by the viral-binding loops in MxA. In the crystals, these monomers pack into linear arrays and contact each other through three distinct interfaces in the stalk (Fig. 2B,C). Mutations that facilitated crystallization all map to a highly disordered region at the end of interface 3. None of the structures contain nucleotide and represent single, structural snapshots of the apo state (Faelber et al., 2011; Ford et al., 2011; Gao et al., 2011).
The connectivity between the structural domains is a significant aspect of these models. Two hinge regions (Fig. 2B) that link the BSE to the stalk (hinge 1) and the G domain (BSE hinge) are of crucial importance as they dictate the relative orientation of these domains with respect to one another. The hinge 1 conformation differs among the three structures: it is partially disordered in human dynamin (Faelber et al., 2011), whereas it forms a more rigid connection in MxA (Gao et al., 2011) and rat dynamin (Ford et al., 2011). Hinge 1 mutants impair GTPase activity, while they enhance the efficiency of endocytosis in dynamin 1 (Sever et al., 1999) and abolish antiviral activity and alter nucleotide dependent-oligomerization in Mx proteins (Johannes et al., 1997), suggesting an important functional role for this region. The BSE hinge contains a highly conserved stretch of amino acids that kinks the CGTPase helix around a key proline residue (294 in human dynamin 1, 294 in rat dynamin, and 340 in MxA) at the C-terminal end of the G domain. The flexibility of this segment is vital to a hydrolysis-dependent movement of the BSE (discussed below).
The electron density at the base of the stalk is weak in these structures, with extended stretches of middle domain and GED residues being completely disordered. This prevents the accurate modeling of the stalk–PH connectivity or the viral-binding loops, and creates an inherent ambiguity in determining the overall trajectory of the individual polypeptide chains. Multiple PH domains pack close to the stalk in the dynamin crystals and each could theoretically connect (Fig. 2D). The published representations of the monomer structures reflect this uncertainty, because different PH domains were selected in the human and rat models (Faelber et al., 2011; Ford et al., 2011). In this Commentary, we have taken the liberty to change the assignment of the PH domain in human dynamin 1 to that of rat dynamin for simplicity and consistency (Fig. 2A). Both arrangements, however, are equally probable. The interpretation of these connections becomes increasingly more complex when mutagenesis and biochemical data are also considered. For example, chemical crosslinking results impose additional structural constraints, which in turn generate two alternative subunit geometries (Fig. 2E; Fig. 3). Regardless of the underlying connectivity, all of these geometries are compatible with the subunit organization observed in both the crystal lattice (Fig. 2E) and the helical dynamin polymer (Fig. 3A,B). Further structural efforts are needed to decipher the connectivity unambiguously.
Fig. 3.
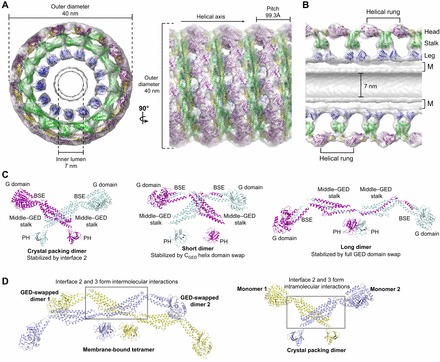
Dynamin assembly and subunit architectures. (A) Pseudo-atomic model of the assembled dynamin polymer (PDB: 3ZYS) that has been derived from computationally fitting GGGMPPCP (purple and yellow; PDB: 3ZYC), the MxA stalk (green, PDB: 3LJB) and the human dynamin 1 PH domain (blue, PDB: 1DYN) into the 12.2 Å GMPPCP-stabilized ΔPRD cryo-EM map (gray, EMD-1949). End-on and side-on views are shown in the left and right panels, respectively, with the dimensions and helical axis marked. (B) Cross-section view of the assembled dynamin polymer oriented as in A. The G domain, middle–GED stalk and PH domain occupy the head, stalk and leg density regions, respectively. The inner luminal diameter is indicated. M, membrane bilayer. (C) Different model representations of the minimal dynamin dimer building blocks. Monomers are colored purple and cyan. Left, dimer based on crystal packing (PDB: 3ZVR) that is stabilized by interface 2 interactions; center, X-shaped short dimer based on chemical crosslinking and computational docking that is stabilized by a domain swap of the CGED helix; right, M-shaped long dimer based on chemical crosslinking and computational docking that is stabilized by a full domain swap of the GED. (D) Putative structure of membrane-bound dynamin tetramer. Underlying dimers are colored yellow and light blue. This model assumes the entire GED is domain-swapped in each monomer (see text). In this context, portions of interface 2 and 3 mediate inter-dimer interactions (gray box). The structure of the crystal packing dimer is shown on the right for comparison with each monomer colored yellow and light blue. Note that in this case, interface 2 and 3 form intra-dimer interactions (gray box).
Structural organization of the dynamin polymer
Although the propensity of dynamin for oligomerization hindered crystallographic endeavors, it has aided in the structural characterization of the dynamin polymer. Mixing a functional, truncated form of dynamin (ΔPRD) (Muhlberg et al., 1997) with anionic liposomes produces helical tubes that are ideally suited for cryo-electron microscopy (EM), and numerous studies have produced three-dimensional reconstructions of these assemblies in different nucleotide states at moderate resolutions (12–23 Å) (Zhang and Hinshaw, 2001; Chen et al., 2004; Mears et al., 2007; Chappie et al., 2011). Without nucleotide, ΔPRD tubes have a diameter of 50 nm and 14.2 subunits per turn (Chen et al., 2004) and, upon the addition of the non-hydrolyzable GTP analog GMPPCP, they constrict to 40 nm and 13.2 subunits per turn (Zhang and Hinshaw, 2001) (Fig. 3A). In both cases, the asymmetric unit that is radially repeated within each helical rung is a T-shaped dimer consisting of three prominent densities: the leg, the stalk and the head (Zhang and Hinshaw, 2001) (Fig. 3B). Structural comparison shows that changes in the stalk density, presumably induced by nucleotide binding, drive the tube constriction (Chen et al., 2004). However, other data indicates that constriction alone is not sufficient to produce scission (Bashkirov et al., 2008; Shnyrova et al., 2013), suggesting that these EM models represent an intermediate along the endocytic scission pathway. Saccharomyces cerevisiae Dnm1 tubes also constrict, but exhibit a more dramatic narrowing from ∼129 nm to ∼67 nm that is associated with GTP hydrolysis (Mears et al., 2011). The magnitude of this change and its occurrence at a different step in the hydrolysis cycle might reflect the specialized role of Dmn1 in mitochondrial fission (Ingerman et al., 2005). While these observations are informative, the limited resolution of the EM density precludes a detailed description of the architecture and domain organization of assembled ΔPRD helices.
Recent hybrid approaches in which X-ray structures were computationally fitted into the ΔPRD EM maps have yielded multiple pseudoatomic models for the dynamin polymer (Gao et al., 2010; Chappie et al., 2011; Ford et al., 2011; Faelber et al., 2011) (Fig. 3A). All these display the same overall topology – the PH domain occupying the leg density, the middle domain and GED in the stalk, and G domain in the head (Fig. 3B) – and, together with mutagenesis data (Sever et al., 2006; Ramachandran et al., 2007; Gao et al., 2010; Kenniston and Lemmon, 2010), suggest that a dimer is the basic building block of all dynamin assembly (Fig. 3C). A key difference among these models, however, is the interpretation of the connectivity within this dimer, owing to the fact that the residues linking the middle–GED stalk to the PH domain are disordered in all dynamin crystal structures. Two prevailing interpretations have emerged. In the first (Fig. 3C, left), dimers are defined based on crystal packing where an extensive interface between monomers (interface 2, see above) forms the primary lattice contact (Gao et al., 2010; Ford et al., 2011; Faelber et al., 2011). The second (Fig. 3C, center and right) defines dimers according to the constraints imposed by chemical crosslinking and limited proteolysis, which suggest that the CGED helix of the BSE is domain-swapped in the full-length protein and imply that the dynamin tetramer is a dimer of domain-swapped dimers (Chappie et al., 2011). Two putative arrangements are consistent with these data: a short X-shaped dimer with only the CGED helix swapped (Fig. 3C, center) or an extended M-shaped dimer with the entire GED swapped (Fig. 3C, right). We favor the extended M-shaped dimer as there is no evidence for CGED exchange at the top of the stalk in the full-length structures of human ΔPRD dynamin (Faelber et al., 2011), rat ΔPRD dynamin (Ford et al., 2011) or human MxA (Gao et al., 2011) (see Fig. 2A). The crystal packing of the ΔPRD X-ray structures is also compatible with the equivalent arrangement of the M-shaped domain-swapped dimers (Fig. 2E), and there is currently no definitive evidence to rule out this possibility given the disordered regions at the base of the stalk.
A similar M-shaped architecture has been proposed for a different mutant dimer (ΔPRD human dynamin 1 with R399A and I690K substitutions) based on small-angle X-ray scattering (SAXS) experiments (Kenniston and Lemmon, 2010). This model was introduced prior to the publication of the full-length dynamin crystal structures and independently suggested that obligatory mutant dimers can adopt an extended conformation in solution in the absence of intramolecular stalk interactions. It is feasible that these mutants are instead stabilized by a complete GED domain swap. This exchange would probably have to occur concomitantly with protein synthesis and/or folding, given the high energetic cost of disrupting the extensive hydrophobic interface between the middle domain and GED. In context of an extended GED-swapped dimer, interfaces 2 and 3 would facilitate intermolecular interactions between the two dimers within a tetramer (Fig. 3D, left) rather than forming intramolecular interactions within a dimer (Fig. 3D, right). Furthermore, these interfaces could facilitate higher-order interactions between tetramers once dynamin oligomerizes on the membrane (Chappie et al., 2011). All additional dynamin mutations that produce obligatory dimers map to interfaces 2 and 3, further supporting their role in high-order assembly (Sever et al., 2006; Ramachandran et al., 2007; Gao et al., 2010; Kenniston and Lemmon, 2010; Ford et al., 2011; Faelber et al., 2011).
Critics of the domain-swapped dimer models have argued that they are inconsistent with a subset of MxA interface 2 mutants that produce assembly-deficient monomers (Faelber et al., 2012). We feel it might be premature to assume that there is a one-to-one structural correlation between MxA and dynamin. For instance, the introduction of the corresponding MxA mutations into dynamin resulted in insoluble protein (Faelber et al., 2011). Thus, although the tetramers of each molecule might be held together by conserved intermolecular interactions, the presence of a domain-swap in dynamin, and its absence in MxA, could explain the biochemical incongruities. It would be interesting to see if crosslinking experiments in MxA would visualize any sort of GED exchange between monomers.
Although dimers constitute the minimal unit of dynamin assembly, we must emphasize that they are not free-floating autonomous entities under normal conditions; rather, they will exist as part of a stable tetramer. Subunit exchange within dynamin tetramers has thus far only been observed in the presence of harsh chemical denaturants (S. L. Schmid, personal communication), indicating that once a tetramer is formed it does not typically come apart. Furthermore, all current structural descriptions of the tetramer are derived from models of the assembled dynamin polymer, which is biased towards a membrane-bound conformation. Analysis of dynamin mutations linked to centronuclear myopathy suggests that the wild-type tetramer in solution is autoinhibited (Kenniston and Lemmon, 2010), adopting an assembly-incompetent configuration that is distinct from the assembly-competent membrane-bound form that is present in the polymer. The conversion between assembly-incompetent and assembly-competent states could serve as a key to the regulation of dynamin during endocytosis, with assembly being the switch that signifies the onset of fission (Mettlen et al., 2009). Further structural dissection of the dynamin tetramer will be necessary for understanding the range of its conformational flexibility.
Although dynamin can induce membrane curvature at high concentrations, nucleation of the dynamin polymer becomes strongly dependent on membrane curvature at what is thought to be a physiological protein concentration (Roux et al., 2010). Under these conditions, dynamin will preferentially bind highly curved regions of the plasma membrane (Ramachandran et al., 2009), which in vivo can be generated by BAR-domain-containing proteins (Mim and Unger, 2012). Once formed, the dynamin scaffold itself might contribute to lowering the energy barrier for fission by increasing the membrane elastic energy (Morlot et al., 2012).
Essential catalytic machinery and the mechanism of stimulated GTP hydrolysis
Dynamin assembles at the necks of CCPs during CME and harnesses the energy of GTP hydrolysis to catalyze membrane scission. Unraveling the mechanisms of the basal and assembly-stimulated GTPase activities of dynamin is therefore fundamental to understanding how dynamin functions as a fission machine. In all GTPases, efficient hydrolysis requires: (1) the correct positioning of a water molecule for an in-line SN2 attack on the γ-phosphate, (2) neutralization of a negative charge that develops between the β- and γ-phosphates in the transition state, and (3) stabilization of the conformationally flexible switch regions (switch I and switch II) within the catalytic core (Paduch et al., 2001; Li and Zhang, 2004). These conditions can be achieved either through interaction with a GTPase-activating protein (GAP) or through dimerization. In GAP-mediated hydrolysis, a stimulating partner binds the switch regions and facilitates the correct positioning of catalytic machinery in the active site (Vetter and Wittinghofer, 2001). This machinery typically includes a conserved glutamine residue in switch II that orients the nucleophilic water and a charge-compensating arginine finger (Scheffzek et al., 1997; Tesmer et al., 1997; Scheffzek et al., 1998). Dimerization accomplishes the same end-state by a symmetric association of the guanine-nucleotide-binding domains, resulting in the reciprocal activation of the individual monomers (Gasper et al., 2009).
The essential catalytic machinery and mechanism of assembly-stimulated GTP hydrolysis for dynamin long remained elusive despite extensive mutagenesis studies (see Table 1 in Schmid and Frolov, 2011) and the available crystal structures of the apo and GDP-bound G domain of dynamin A (Niemann et al., 2001) and the nucleotide-free rat dynamin G domain (Reubold et al., 2005). To address these issues, we engineered a minimal G-domain–GED fusion protein (GG) by connecting residues 6–320 and residues 726–750 from human dynamin 1 with a flexible linker (Chappie et al., 2009) (Fig. 4A). Encompassing the G domain core and the BSE, this construct reconstitutes the basal GTPase activity of dynamin but does not assemble, even at high concentrations, providing an ideal platform for structure-function studies (Chappie et al., 2009).
Fig. 4.
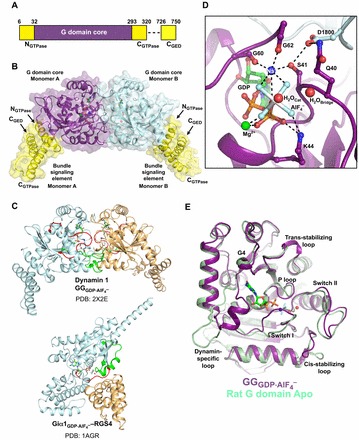
The catalytic machinery of dynamin. (A) Domain arrangement of the G-domain–GED fusion derived from human dynamin 1. Each monomer contains a G domain core (purple), three helical segments (NGTPase, CGTPase and CGED; yellow) that together form the bundle signaling element (BSE), and a flexible linker (dashed line). (B) Structure of the transition-state-stabilized  dimer (PDB: 2X2E). The monomer cores are colored purple and cyan, and the bundle signaling elements (BSEs) are in yellow. (C) Switch region I (red) and switch region II (green) are stabilized at the dimer interface in the GG transition-state complex. GG monomers are shown in cyan and orange. The structure of the Giα1–RGS4 transition-state complex is shown below for comparison (Giα1 in cyan, RGS4 GAP in orange). Note that in this case the switch regions are stabilized at the interface between the G-protein and the GAP. (D) Transition-state stabilization of the of the GG active site. Structural elements involved in this stabilization are labeled. Catalytic and bridging waters are depicted as red spheres; bound Mg2+ and charge-compensating Na+ ions are shown as green and blue spheres, respectively. Dashed lines indicate hydrogen-bonding interactions. Together, the charge compensating cation, the bound Mg2+ and the K44 side-chain act to neutralize the negative charge that develops when the bond is broken between the β- and γ-phosphates during hydrolysis. (E) Conformational changes in the active site that accompany the formation of a G domain dimer in dynamin.
dimer (PDB: 2X2E). The monomer cores are colored purple and cyan, and the bundle signaling elements (BSEs) are in yellow. (C) Switch region I (red) and switch region II (green) are stabilized at the dimer interface in the GG transition-state complex. GG monomers are shown in cyan and orange. The structure of the Giα1–RGS4 transition-state complex is shown below for comparison (Giα1 in cyan, RGS4 GAP in orange). Note that in this case the switch regions are stabilized at the interface between the G-protein and the GAP. (D) Transition-state stabilization of the of the GG active site. Structural elements involved in this stabilization are labeled. Catalytic and bridging waters are depicted as red spheres; bound Mg2+ and charge-compensating Na+ ions are shown as green and blue spheres, respectively. Dashed lines indicate hydrogen-bonding interactions. Together, the charge compensating cation, the bound Mg2+ and the K44 side-chain act to neutralize the negative charge that develops when the bond is broken between the β- and γ-phosphates during hydrolysis. (E) Conformational changes in the active site that accompany the formation of a G domain dimer in dynamin.  monomer colored purple; nucleotide free rat dynamin G domain structure (PDB: 2AKA) colored green. These structural changes serve to optimally position the catalytic machinery, which in turn promotes stimulated GTP hydrolysis.
monomer colored purple; nucleotide free rat dynamin G domain structure (PDB: 2AKA) colored green. These structural changes serve to optimally position the catalytic machinery, which in turn promotes stimulated GTP hydrolysis.
GG dimerizes in the presence of the transition-state mimic GDP·AlF4– but not with ground-state non-hydrolyzable GTP analogs such as GMPPCP (Chappie et al., 2010; Chappie et al., 2011). The 2.0 Å X-ray structure of the GG dimer ( ; Chappie et al., 2010) revealed that monomers associate exclusively through the G domains and bury a large surface of 2569 Å2 (Fig. 4B). The active sites face each other at the dimer interface, thereby sequestering the nucleotides from the solvent and reciprocally stabilizing the switch regions (Fig. 4C), whereas the BSEs extend in opposite directions from the dimer core. Together, the main chain atoms of G139 and T65 along with an additional bridging-water that interacts with Q40 in the P-loop position the catalytic water. The strict use of non-side chain atoms for this purpose is, to date, unique to dynamin and explains why all previous mutagenesis efforts failed to identify these crucial catalytic components (Fig. 4D). This also implies that basal GTP hydrolysis can only be disrupted by perturbing either the structural fold of dynamin or its nucleotide binding. Indeed, purported catalytic side chains such as T65 and S45 (Marks et al., 2001; Song et al., 2004) actually function as coordinating ligands for Mg2+, an essential cofactor required for GTP binding (Chappie et al., 2010; Chappie et al., 2011). Thus, the assumed catalytic role for these residues is likely to be a consequence of defects in nucleotide binding.
; Chappie et al., 2010) revealed that monomers associate exclusively through the G domains and bury a large surface of 2569 Å2 (Fig. 4B). The active sites face each other at the dimer interface, thereby sequestering the nucleotides from the solvent and reciprocally stabilizing the switch regions (Fig. 4C), whereas the BSEs extend in opposite directions from the dimer core. Together, the main chain atoms of G139 and T65 along with an additional bridging-water that interacts with Q40 in the P-loop position the catalytic water. The strict use of non-side chain atoms for this purpose is, to date, unique to dynamin and explains why all previous mutagenesis efforts failed to identify these crucial catalytic components (Fig. 4D). This also implies that basal GTP hydrolysis can only be disrupted by perturbing either the structural fold of dynamin or its nucleotide binding. Indeed, purported catalytic side chains such as T65 and S45 (Marks et al., 2001; Song et al., 2004) actually function as coordinating ligands for Mg2+, an essential cofactor required for GTP binding (Chappie et al., 2010; Chappie et al., 2011). Thus, the assumed catalytic role for these residues is likely to be a consequence of defects in nucleotide binding.
An unexpected feature of the GG active site is that a bound Na+ ion replaces the charge-compensating arginine finger and is coordinated by AlF4−, the β-phosphate, the carbonyl oxygens of G60 and G62 (in switch I) and the S41 side chain in the P-loop, in a manner similar to that of the in cis arginine finger of the related interferon-γ-induced human guanylate protein 1 (Fig. 4D). A number of other G-proteins including MnmE and FeoB have been identified that utilize K+ exclusively as a charge-compensating element (Ash et al., 2012). While both monovalent cations can support GG dimerization and the GTPase activity of dynamin in vitro (Chappie et al., 2010), K+ is likely to be more physiologically relevant in vivo, given its higher intracellular concentration. D180 of the trans-stabilizing loop reaches across the dimer interface and forms hydrogen bonds with Q40, S41 and the main chain nitrogen of G62, thereby stabilizing the P-loop and switch I conformations such that they are poised for catalysis (Fig. 4D). Q40, S41 and D180 are absolutely conserved among dynamin family members, and mutagenesis of these residues selectively impairs assembly-stimulated GTP hydrolysis without inhibiting basal turnover (Chappie et al., 2010).
By comparing different nucleotide-bound structures of the dynamin G domain, it has been possible to identify key conformational changes that are associated with GTP binding, G domain dimerization, and GTP hydrolysis. Superposition of  (Chappie et al., 2010) and the nucleotide-free rat dynamin G domain (Reubold et al., 2005) shows how numerous active site elements are reoriented upon the formation of the transition-state-stabilized G domain dimer (Fig. 4E): the P-loop tilts toward the α- and β-phosphates, switch I swings in to coordinate the charge-compensating cation, and the dynamin-specific loop converts from an extended unstructured conformation to a short helix, thereby capping the substrate and sequestering it from the solvent (Chappie et al., 2010). Additional changes are observed in the cis-stabilizing loop, which stabilizes the portion of switch II that does not make direct contact with the opposing monomer at the dimer interface (Chappie et al., 2010). G domain dimerization, thus, optimally positions the catalytic machinery of dynamin for rapid and efficient GTP hydrolysis.
(Chappie et al., 2010) and the nucleotide-free rat dynamin G domain (Reubold et al., 2005) shows how numerous active site elements are reoriented upon the formation of the transition-state-stabilized G domain dimer (Fig. 4E): the P-loop tilts toward the α- and β-phosphates, switch I swings in to coordinate the charge-compensating cation, and the dynamin-specific loop converts from an extended unstructured conformation to a short helix, thereby capping the substrate and sequestering it from the solvent (Chappie et al., 2010). Additional changes are observed in the cis-stabilizing loop, which stabilizes the portion of switch II that does not make direct contact with the opposing monomer at the dimer interface (Chappie et al., 2010). G domain dimerization, thus, optimally positions the catalytic machinery of dynamin for rapid and efficient GTP hydrolysis.
It is important to note that the stabilization of the GG dimer in the transition-state depends on both the interaction of D180 with P-loop and switch I residues across the dimer interface, as well as the presence of the γ-phosphate in each monomer. Together, these complete the coordination sphere for the charge-compensating cation and hold switch I in a locked position (Chappie et al., 2010). Loss of the γ-phosphate would presumably allow an increased mobility of switch I, thereby destabilizing the dimer interface and causing the interacting dynamin molecules to dissociate and recycle during the fission reaction. This notion is supported by the structure of the G domain from Dictyostelium dynamin A in complex with GDP (Niemann et al., 2001), where only the dynamin-specific loop and P-loop are visible and all the remaining catalytic machinery that is stabilized by dimerization is disordered.
The membrane-bound conformation of dynamin prevents G domain dimers from forming within a tetramer and between neighboring tetramers in the same helical rung (Fig. 5A,B). Instead, dimerization can only occur between tetramers positioned in adjacent rungs of the helical assembly (Fig. 5B–D). Constraining the productive head-to-head interaction in this manner ensures that stimulated GTP hydrolysis is coupled to dynamin assembly.
Fig. 5.
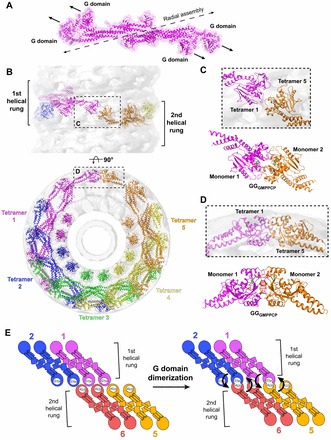
Structural constraints of G domain dimerzation. (A) Membrane-bound conformation of the dynamin tetramer (magenta). The dashed black line illustrates the axis of radial assembly within a helical rung. The arrows depict the relative orientation of the G domains within the tetramer. Note that the G domains do not form productive head-to-head interactions required for G domain dimerization. (B) Pseudoatomic model of the dynamin polymer generated by computationally docking crystallized dynamin domain structures into the GMPPCP-stabilized ΔPRD cryo-EM map (gray). The numbering and rainbow coloring (magenta to orange) denotes the sequential addition of tetramers to the assembly and terminates when the first G domain dimer is formed between magenta tetramer 1 and orange tetramer 5. The upper panel depicts a side view perpendicular to the helical axis; the lower panel is an end view looking down this axis. The sequential rungs of the dynamin helix are marked in the upper panel with black brackets. The areas indicated by the black boxes (see C and D) highlight that G domain dimerization only occurs between tetramers in adjacent helical rungs. (C,D) Magnified top (C) and side (D) views of G domain interactions within the assembled dynamin polymer. The crystallized GGGMPPCP dimer (monomers colored magenta and orange) is shown below each for comparison. (E) Illustration of G domain dimerization in the context of the helical polymer. Tetramers are colored and numbered as in B. Arrows denote the direction of the hydrolysis-dependent BSE conformational change that would accompany stimulated GTP hydrolysis in each G domain dimer pair.
The dynamin ‘powerstroke’ and conformational coupling
The structure of GG has also been solved in the presence of GMPPCP (GGGMPPCP). Although GGGMPPCP is entirely monomeric when analyzed by size exclusion chromatography (Chappie et al., 2010) and analytical ultracentrifugation (Chappie et al., 2011), it forms a head-to-head dimer in the crystal presumably owing to the high concentration of protein present during crystallization (Chappie et al., 2011). The non-hydrolyzable β-γ methylene linkage of GMPPCP does not provide the necessary H-bonding interactions to complete the coordination sphere of the charge-compensating monovalent cation, and instead a water molecule partially fills the vacant ion-binding site in its absence. The P-loop residue S41 rotates 90° to stabilize this water and in the process severs its hydrogen bond with D180 across the dimer interface. The remainder of the active site is essentially unchanged from that in  . The BSE helices, in contrast, assume a radically different position relative to the G domain core (Chappie et al., 2011): in GGGMPPCP the bundles are oriented upwards, extending out from the core; in
. The BSE helices, in contrast, assume a radically different position relative to the G domain core (Chappie et al., 2011): in GGGMPPCP the bundles are oriented upwards, extending out from the core; in  the bundles are rotated ∼69° downwards with a slight counter-clockwise twist, pivoting around the conserved, flexible BSE hinge (Fig. 6A,B). The central β-sheet shifts towards the nucleotide-binding pocket concomitantly with this change, which tightens the hydrophobic packing within the core and facilitates the formation of salt bridges and hydrophobic interactions that anchor the BSE. Together, these conformational changes suggest the possibility of a powerstroke in which the hydrolysis-dependent movement of the BSE converts the energy of G domain dimerization and/or stimulated GTP hydrolysis into large-scale rearrangements that could precipitate membrane fission (Chappie et al., 2011). These movements might also have a role in the direct conformational coupling that exists between the PH domain at the membrane surface and the G domain at the top of the molecule (Solomaha and Palfrey, 2005; Ramachandran and Schmid, 2008).
the bundles are rotated ∼69° downwards with a slight counter-clockwise twist, pivoting around the conserved, flexible BSE hinge (Fig. 6A,B). The central β-sheet shifts towards the nucleotide-binding pocket concomitantly with this change, which tightens the hydrophobic packing within the core and facilitates the formation of salt bridges and hydrophobic interactions that anchor the BSE. Together, these conformational changes suggest the possibility of a powerstroke in which the hydrolysis-dependent movement of the BSE converts the energy of G domain dimerization and/or stimulated GTP hydrolysis into large-scale rearrangements that could precipitate membrane fission (Chappie et al., 2011). These movements might also have a role in the direct conformational coupling that exists between the PH domain at the membrane surface and the G domain at the top of the molecule (Solomaha and Palfrey, 2005; Ramachandran and Schmid, 2008).
Fig. 6.
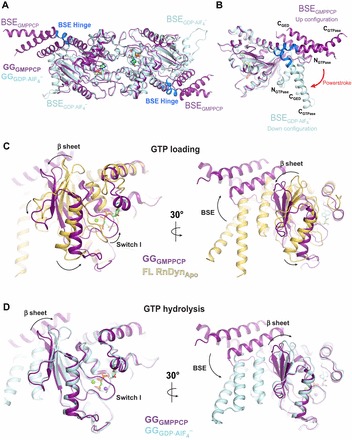
The dynamin powerstroke and conformational coupling. (A) Structural superposition of GGGMPPCP (purple, PDB: 3ZYC) and  (cyan, PDB: 2X2E) highlighting alternative BSE conformations. The BSE hinge is colored blue. (B) Side view of the hydrolysis-dependent BSE conformational change that constitutes the dynamin powerstroke. GGGMPPCP and
(cyan, PDB: 2X2E) highlighting alternative BSE conformations. The BSE hinge is colored blue. (B) Side view of the hydrolysis-dependent BSE conformational change that constitutes the dynamin powerstroke. GGGMPPCP and 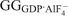 monomers are colored as in A and the BSE helices are labeled. The red arrow labeled ‘powerstroke’ depicts the 69° downwards rotation of the BSE in the transition-state complex. (C,D) Conformational coupling of the β-sheet, switch I and BSE movements during GTP loading (C) and GTP hydrolysis (D). GGGMPPCP (purple, PDB: 3ZYC),
monomers are colored as in A and the BSE helices are labeled. The red arrow labeled ‘powerstroke’ depicts the 69° downwards rotation of the BSE in the transition-state complex. (C,D) Conformational coupling of the β-sheet, switch I and BSE movements during GTP loading (C) and GTP hydrolysis (D). GGGMPPCP (purple, PDB: 3ZYC), 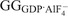 (cyan, PDB: 2X2E) and the G domain from the nucleotide-free, full-length rat dynamin crystal structure (yellow, PDB: 3ZVR) are superimposed. Arrows indicate the conformational changes between the different structures.
(cyan, PDB: 2X2E) and the G domain from the nucleotide-free, full-length rat dynamin crystal structure (yellow, PDB: 3ZVR) are superimposed. Arrows indicate the conformational changes between the different structures.
We find it significant that the BSE adopts a similar downwards conformation in all other dynamin structures, including those that are nucleotide free (Faelber et al., 2011; Ford et al., 2011; Reubold et al., 2005) and GDP-bound (Niemann et al., 2001). This implies that the up and down configurations of the BSE are coupled to the GTP loading and GTP hydrolysis steps, respectively, and that the G domain can somehow sense the nucleotide state in the active site and relay that information to the BSE. Small Arf-like GTPases use a distinct mechanism for this type of front-to-back communication wherein GTP engages switch I and induces an upwards shift of the central β-sheet. This movement displaces the N-terminal amphipathic helix, allowing it to interact with the membrane (Pasqualato et al., 2002). In dynamin, GTP loading engages switch I and causes the central β-sheet to tilt away from the active site, which pushes against the BSE shifting it upwards (Fig. 6C) The central β-sheet then tilts back towards the active site upon GTP hydrolysis and the formation of the transition state, thus allowing the bundle to swing down (Fig. 6D). Switch I remains engaged until the γ-phosphate is released, after which it becomes highly flexible. The coordination of distant movements through the central β-sheet is a common feature of both protein families and might thus represent a conserved mechanism for conformational coupling. We speculate that the nucleotide state in dynamin is sensed by S41 in the P-loop, because it adopts multiple conformations throughout the hydrolysis cycle and is the only side chain that makes direct contact with the charge-compensating cation in the transition state (Fig. 4D).
Structural comparison of a GG fusion from Arabidopsis thaliana DRP1A in complex with GDP and GDP·AlF4− shows a similar conformational change of the BSE (Yan et al., 2011); the movement, however, appears to be coupled to a different nucleotide state such that the bundles are up in the transition state and down in the presence of bound GDP. This conformational difference could reflect different mechanistic requirements of CME in plants. Recent evidence suggests that DRP1A functions with DRP2B to catalyze coated-vesicle release in Arabidopsis (Fujimoto et al., 2010). These two proteins display distinct domain features: the G domain of Arabidopsis DRP1 is more similar to human dynamin 1, yet it lacks the PH domain and PRD regions that are present in Arabidopsis DRP2B (Hong et al., 2003). This raises the possibility that they act together in a heteromeric complex, with each fulfilling a distinct functional role that is already pre-encoded within each of the individual metazoan dynamins (Fujimoto et al., 2010). It remains to be seen whether Arabidopsis GG DRP2B–DRP1A heterodimers or GG DRP2B homodimers can support the formation of a transition-state complex and exhibit the same BSE conformational change.
Although mutagenesis studies have established an essential role for the transition-state-stabilized G domain dimer in both dynamin-catalyzed fission (Liu et al., 2013) and endocytosis (Chappie et al., 2010), the functional relevance of the accompanying BSE movement has yet to be confirmed. Whether this movement produces a direct physical change, as we proposed (Chappie et al., 2011), or manifests its effects indirectly in these processes remains to be seen. Nucleotide-dependent BSE changes have thus far only been visualized in the context of G-domain–GED fusions (Chappie et al., 2011; Yan et al., 2011). Our knowledge concerning dynamin conformational changes is therefore skewed towards only a select region of the molecule in a specific context. Elucidating the structural changes that occur in other domains upon GTP hydrolysis will dramatically improve our understanding of the powerstroke and its role in the fission reaction.
Conclusions
Structural biology has provided the following crucial insights into dynamin GTP hydrolysis and assembly: (1) the dynamin polypeptide folds back on itself and assumes an extended conformation in the folded state; (2) the underlying building block of dynamin assembly and oligomerization is a domain-swapped dimer, two of which stably associate to form a tetramer in solution; (3) dynamin tetramers are likely to adopt different conformations when they are free in solution and when bound to the membrane surface; (4) dynamin uses a bound monovalent cation as the charge-compensating element in the transition state of the hydrolysis reaction and orients the catalytic water with main chain atoms; (5) transition-state-dependent G domain dimerization optimally positions the catalytic machinery in dynamin, leading to stimulated GTP hydrolysis; (6) the BSE undergoes a hydrolysis-dependent conformational change that might act as a powerstroke during fission events; and (7) in the context of the assembled polymer, G domain dimerization occurs between tetramers in adjacent rungs of the dynamin helix. The timing and functional consequences of these activities during the fission reaction still need to be clarified.
Early models for dynamin-catalyzed membrane fission suggested that dynamin functioned purely as a mechanochemical enzyme, expanding (Stowell et al., 1999), constricting (Chen et al., 2004) or twisting (Roux et al., 2006) with GTP hydrolysis to sever the membrane. Recent studies argue that fission might instead be driven by partial disassembly of the dynamin polymer (Pucadyil and Schmid, 2008; Bashkirov et al., 2008), curvature-dependent changes in elastic energy at the edge of the dynamin polymer (Morlot et al., 2012) or PH domain tilting within short metastable dynamin collars (Shnyrova et al., 2013). These models disagree on the role of stimulated GTP hydrolysis, despite the fact that this activity is absolutely required for fission in vitro (Liu et al., 2013) and efficient endocytosis in vivo (Chappie et al., 2010). Testing the behavior of Q40, S41 and D180 mutants in different minimal biophysical systems (Bashkirov et al., 2008; Morlot et al., 2012; Shnyrova et al., 2013) could help in this regard.
An emerging consensus seems to be that, in addition to facilitating G domain dimerization, dynamin assembly and constriction generate high membrane curvature and localized stress (Bashkirov et al., 2008; Ramachandran et al., 2009; Roux et al., 2010), which asymmetrically impose a greater strain on the inner monolayer of the membrane neck (Bashkirov et al., 2008; Schmid and Frolov, 2011). This can lead to the formation of a hemifission intermediate if the inner luminal diameter of the neck approaches the bilayer thickness (∼4 nm) (Bashkirov et al., 2008; Shnyrova et al., 2013). The current models for the dynamin polymer all have an inner luminal diameter of ∼7 nm and therefore probably represent an intermediate along the fission pathway rather than the final structural state (Chappie et al., 2010). This discrepancy limits the direct comparisons and interpretations that can be made when integrating structural and biophysical data. Furthermore, the extended nature of this helical structure is an artifact of the cryo-EM reconstruction methodology and represents a non-physiological extreme. Indeed, estimations of the minimal fission apparatus suggest that only two helical turns are necessary for maximal fission efficiency (Liu et al., 2013; Shnyrova et al., 2013), which correlates with the short dynamin collars observed in vivo (Kosaka and Ikeda, 1983) and the observation that long dynamin scaffolds inhibit fission in vitro (Bashkirov et al., 2008). The structural models we describe here thus provide an atomic-resolution template that can assist in interpreting the low-resolution, and often indirect, read out from various cellular and biochemical assays.
We anticipate that future structures of other dynamin-related proteins will represent variations on a common structural theme, especially given the shared features and motifs present in dynamin, MxA and distant cousins, such as human GBP1 (Prakash et al., 2000; Ghosh et al., 2006), atlastin (Byrnes and Sondermann, 2011; Bian et al., 2011), BDLP (Low and Löwe, 2006) and EHD2 (Daumke et al., 2007), as well as the observed dimerization-dependent activation of these GTPases (Wittinghofer and Vetter, 2011). The function and cellular localization of many of these proteins differ from that of dynamin, and it will be interesting to see what aspects of the mechanisms and/or associated conformational changes are also conserved. New findings might necessitate sub-dividing the ‘dynamin family’ further into specified groupings that more accurately reflect the distinct structural and functional properties of its members.
Acknowledgments
We thank Dr Jenny Hinshaw for insightful discussions and Dr Sandra Schmid for the communication of unpublished data.
Footnotes
Funding
The work of our laboratory was supported by the Intramural Program of the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). J.S.C. is supported by a Nancy Nossal Fellowship award from NIDDK. Deposited in PMC for release after 12 months.
References
- Ash M. R., Maher M. J., Mitchell Guss J., Jormakka M. (2012). The cation-dependent G-proteins: in a class of their own. FEBS Lett. 586, 2218–2224 10.1016/j.febslet.2012.06.030 [DOI] [PubMed] [Google Scholar]
- Bashkirov P. V., Akimov S. A., Evseev A. I., Schmid S. L., Zimmerberg J., Frolov V. A. (2008). GTPase cycle of dynamin is coupled to membrane squeeze and release, leading to spontaneous fission. Cell 135, 1276–1286 10.1016/j.cell.2008.11.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian X., Klemm R. W., Liu T. Y., Zhang M., Sun S., Sui X., Liu X., Rapoport T. A., Hu J. (2011). Structures of the atlastin GTPase provide insight into homotypic fusion of endoplasmic reticulum membranes. Proc. Natl. Acad. Sci. USA 108, 3976–3981 10.1073/pnas.1101643108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrnes L. J., Sondermann H. (2011). Structural basis for the nucleotide-dependent dimerization of the large G protein atlastin-1/SPG3A. Proc. Natl. Acad. Sci. USA 108, 2216–2221 10.1073/pnas.1012792108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappie J. S., Acharya S., Liu Y. W., Leonard M., Pucadyil T. J., Schmid S. L. (2009). An intramolecular signaling element that modulates dynamin function in vitro and in vivo. Mol. Biol. Cell 20, 3561–3571 10.1091/mbc.E09-04-0318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappie J. S., Acharya S., Leonard M., Schmid S. L., Dyda F. (2010). G domain dimerization controls dynamin's assembly-stimulated GTPase activity. Nature 465, 435–440 10.1038/nature09032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappie J. S., Mears J. A., Fang S., Leonard M., Schmid S. L., Milligan R. A., Hinshaw J. E., Dyda F. (2011). A pseudoatomic model of the dynamin polymer identifies a hydrolysis-dependent powerstroke. Cell 147, 209–222 10.1016/j.cell.2011.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. J., Zhang P., Egelman E. H., Hinshaw J. E. (2004). The stalk region of dynamin drives the constriction of dynamin tubes. Nat. Struct. Mol. Biol. 11, 574–575 10.1038/nsmb762 [DOI] [PubMed] [Google Scholar]
- Daumke O., Lundmark R., Vallis Y., Martens S., Butler P. J., McMahon H. T. (2007). Architectural and mechanistic insights into an EHD ATPase involved in membrane remodelling. Nature 449, 923–927 10.1038/nature06173 [DOI] [PubMed] [Google Scholar]
- Faelber K., Posor Y., Gao S., Held M., Roske Y., Schulze D., Haucke V., Noé F., Daumke O. (2011). Crystal structure of nucleotide-free dynamin. Nature 477, 556–560 10.1038/nature10369 [DOI] [PubMed] [Google Scholar]
- Faelber K., Held M., Gao S., Posor Y., Haucke V., Noé F., Daumke O. (2012). Structural insights into dynamin-mediated membrane fission. Structure 20, 1621–1628 10.1016/j.str.2012.08.028 [DOI] [PubMed] [Google Scholar]
- Ferguson S. M., De Camilli P. (2012). Dynamin, a membrane-remodelling GTPase. Nat. Rev. Mol. Cell Biol. 13, 75–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson K. M., Lemmon M. A., Schlessinger J., Sigler P. B. (1994). Crystal structure at 2.2 A resolution of the pleckstrin homology domain from human dynamin. Cell 79, 199–209 10.1016/0092-8674(94)90190-2 [DOI] [PubMed] [Google Scholar]
- Ford M. G., Jenni S., Nunnari J. (2011). The crystal structure of dynamin. Nature 477, 561–566 10.1038/nature10441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto M., Arimura S., Ueda T., Takanashi H., Hayashi Y., Nakano A., Tsutsumi N. (2010). Arabidopsis dynamin-related proteins DRP2B and DRP1A participate together in clathrin-coated vesicle formation during endocytosis. Proc. Natl. Acad. Sci. U S A. 107, 6094–6099 10.1073/pnas.0913562107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S., von der Malsburg A., Paeschke S., Behlke J., Haller O., Kochs G., Daumke O. (2010). Structural basis of oligomerization in the stalk region of dynamin-like MxA. Nature 465, 502–506 10.1038/nature08972 [DOI] [PubMed] [Google Scholar]
- Gao S., von der Malsburg A., Dick A., Faelber K., Schröder G. F., Haller O., Kochs G., Daumke O. (2011). Structure of myxovirus resistance protein a reveals intra- and intermolecular domain interactions required for the antiviral function. Immunity 35, 514–525 10.1016/j.immuni.2011.07.012 [DOI] [PubMed] [Google Scholar]
- Gasper R., Meyer S., Gotthardt K., Sirajuddin M., Wittinghofer A. (2009). It takes two to tango: regulation of G proteins by dimerization. Nat. Rev. Mol. Cell Biol. 10, 423–429 10.1038/nrm2689 [DOI] [PubMed] [Google Scholar]
- Ghosh A., Praefcke G. J., Renault L., Wittinghofer A., Herrmann C. (2006). How guanylate-binding proteins achieve assembly-stimulated processive cleavage of GTP to GMP. Nature 440, 101–104 10.1038/nature04510 [DOI] [PubMed] [Google Scholar]
- Hinshaw J. E., Schmid S. L. (1995). Dynamin self-assembles into rings suggesting a mechanism for coated vesicle budding. Nature 374, 190–192 10.1038/374190a0 [DOI] [PubMed] [Google Scholar]
- Hong Z., Bednarek S. Y., Blumwald E., Hwang I., Jurgens G., Menzel D., Osteryoung K. W., Raikhel N. V., Shinozaki K., Tsutsumi N. et al. (2003). A unified nomenclature for Arabidopsis dynamin-related large GTPases based on homology and possible functions. Plant Mol. Biol. 53, 261–265 10.1023/B:PLAN.0000007000.29697.81 [DOI] [PubMed] [Google Scholar]
- Ingerman E., Perkins E. M., Marino M., Mears J. A., McCaffery J. M., Hinshaw J. E., Nunnari J. (2005). Dnm1 forms spirals that are structurally tailored to fit mitochondria. J. Cell Biol. 170, 1021–1027 10.1083/jcb.200506078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannes L., Kambadur R., Lee-Hellmich H., Hodgkinson C. A., Arnheiter H., Meier E. (1997). Antiviral determinants of rat Mx GTPases map to the carboxy-terminal half. J. Virol. 71, 9792–9795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenniston J. A., Lemmon M. A. (2010). Dynamin GTPase regulation is altered by PH domain mutations found in centronuclear myopathy patients. EMBO J. 29, 3054–3067 10.1038/emboj.2010.187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochs G., Haener M., Aebi U., Haller O. (2002). Self-assembly of human MxA GTPase into highly ordered dynamin-like oligomers. J. Biol. Chem. 277, 14172–14176 10.1074/jbc.M200244200 [DOI] [PubMed] [Google Scholar]
- Kosaka T., Ikeda K. (1983). Possible temperature-dependent blockage of synaptic vesicle recycling induced by a single gene mutation in Drosophila. J. Neurobiol. 14, 207–225 10.1002/neu.480140305 [DOI] [PubMed] [Google Scholar]
- Li G., Zhang X. C. (2004). GTP hydrolysis mechanism of Ras-like GTPases. J. Mol. Biol. 340, 921–932 10.1016/j.jmb.2004.06.007 [DOI] [PubMed] [Google Scholar]
- Liu Y. W., Mattila J. P., Schmid S. L. (2013). Dynamin-catalyzed membrane fission requires coordinated GTP hydrolysis. PLoS ONE 8, e55691 10.1371/journal.pone.0055691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low H. H., Löwe J. (2006). A bacterial dynamin-like protein. Nature 444, 766–769 10.1038/nature05312 [DOI] [PubMed] [Google Scholar]
- Low H. H., Sachse C., Amos L. A., Löwe J. (2009). Structure of a bacterial dynamin-like protein lipid tube provides a mechanism for assembly and membrane curving. Cell 139, 1342–1352 10.1016/j.cell.2009.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks B., Stowell M. H. B., Vallis Y., Mills I. G., Gibson A., Hopkins C. R., McMahon H. T. (2001). GTPase activity of dynamin and resulting conformation change are essential for endocytosis. Nature 410, 231–235 10.1038/35065645 [DOI] [PubMed] [Google Scholar]
- Mears J. A., Ray P., Hinshaw J. E. (2007). A corkscrew model for dynamin constriction. Structure 15, 1190–1202 10.1016/j.str.2007.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mears J. A., Lackner L. L., Fang S., Ingerman E., Nunnari J., Hinshaw J. E. (2011). Conformational changes in Dnm1 support a contractile mechanism for mitochondrial fission. Nat. Struct. Mol. Biol. 18, 20–26 10.1038/nsmb.1949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mettlen M., Pucadyil T. J., Ramachandran R., Schmid S. L. (2009). Dissecting dynamin's role in clathrin-mediated endocytosis. Biochem. Soc. Trans. 37, 1022–1026 10.1042/BST0371022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mim C., Unger V. M. (2012). Membrane curvature and its generation by BAR proteins. Trends Biochem. Sci. 37, 526–533 10.1016/j.tibs.2012.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morlot S., Galli V., Klein M., Chiaruttini N., Manzi J., Humbert F., Dinis L., Lenz M., Cappello G., Roux A. (2012). Membrane shape at the edge of the dynamin helix sets location and duration of the fission reaction. Cell 151, 619–629 10.1016/j.cell.2012.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhlberg A. B., Warnock D. E., Schmid S. L. (1997). Domain structure and intramolecular regulation of dynamin GTPase. EMBO J. 16, 6676–6683 10.1093/emboj/16.22.6676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemann H. H., Knetsch M. L. W., Scherer A., Manstein D. J., Kull F. J. (2001). Crystal structure of a dynamin GTPase domain in both nucleotide-free and GDP-bound forms. EMBO J. 20, 5813–5821 10.1093/emboj/20.21.5813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paduch M., Jeleń F., Otlewski J. (2001). Structure of small G proteins and their regulators. Acta Biochim. Pol. 48, 829–850 [PubMed] [Google Scholar]
- Pasqualato S., Renault L., Cherfils J. (2002). Arf, Arl, Arp and Sar proteins: a family of GTP-binding proteins with a structural device for ‘front-back’ communication. EMBO Rep. 3, 1035–1041 10.1093/embo-reports/kvf221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praefcke G. J., McMahon H. T. (2004). The dynamin superfamily: universal membrane tubulation and fission molecules? Nat. Rev. Mol. Cell Biol. 5, 133–147 10.1038/nrm1313 [DOI] [PubMed] [Google Scholar]
- Prakash B., Praefcke G. J. K., Renault L., Wittinghofer A., Herrmann C. (2000). Structure of human guanylate-binding protein 1 representing a unique class of GTP-binding proteins. Nature 403, 567–571 10.1038/35000617 [DOI] [PubMed] [Google Scholar]
- Pucadyil T. J., Schmid S L. (2008). Real-time visualization of dynamin-catalyzed membrane fission and vesicle release. Cell 135, 1263–1275 10.1016/j.cell.2008.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R., Schmid S. L. (2008). Real-time detection reveals that effectors couple dynamin's GTP-dependent conformational changes to the membrane. EMBO J. 27, 27–37 10.1038/sj.emboj.7601961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R., Surka M., Chappie J. S., Fowler D. M., Foss T. R., Song B. D., Schmid S. L. (2007). The dynamin middle domain is critical for tetramerization and higher-order self-assembly. EMBO J. 26, 559–566 10.1038/sj.emboj.7601491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R., Pucadyil T. J., Liu Y. W., Acharya S., Leonard M., Lukiyanchuk V., Schmid S. L. (2009). Membrane insertion of the pleckstrin homology domain variable loop 1 is critical for dynamin-catalyzed vesicle scission. Mol. Biol. Cell 20, 4630–4639 10.1091/mbc.E09-08-0683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reubold T. F., Eschenburg S., Becker A., Leonard M., Schmid S. L., Vallee R. B., Kull F. J., Manstein D. J. (2005). Crystal structure of the GTPase domain of rat dynamin 1. Proc. Natl. Acad. Sci. USA 102, 13093–13098 10.1073/pnas.0506491102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux A., Uyhazi K., Frost A., De Camilli P. (2006). GTP-dependent twisting of dynamin implicates constriction and tension in membrane fission. Nature 441, 528–531 10.1038/nature04718 [DOI] [PubMed] [Google Scholar]
- Roux A., Koster G., Lenz M., Sorre B., Manneville J. B., Nassoy P., Bassereau P. (2010). Membrane curvature controls dynamin polymerization. Proc. Natl. Acad. Sci. USA 107, 4141–4146 10.1073/pnas.0913734107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salim K., Bottomley M. J., Querfurth E., Zvelebil M. J., Gout I., Scaife R., Margolis R. L., Gigg R., Smith C. I., Driscoll P. C., Waterfield M. D., Panayotou G. (1996). Distinct specificity in the recognition of phosphoinositides by the pleckstrin homology domains of dynamin and Bruton’s tyrosine kinase. EMBO J. 15, 6241–6250 [PMC free article] [PubMed] [Google Scholar]
- Scheffzek K., Ahmadian M. R., Kabsch W., Wiesmüller L., Lautwein A., Schmitz F., Wittinghofer A. (1997). The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science 277, 333–338 10.1126/science.277.5324.333 [DOI] [PubMed] [Google Scholar]
- Scheffzek K., Ahmadian M. R., Wittinghofer A. (1998). GTPase-activating proteins: helping hands to complement an active site. Trends Biochem. Sci. 23, 257–262 10.1016/S0968-0004(98)01224-9 [DOI] [PubMed] [Google Scholar]
- Schmid S. L., Frolov V. A. (2011). Dynamin: functional design of a membrane fission catalyst. Annu. Rev. Cell Dev. Biol. 27, 79–105 10.1146/annurev-cellbio-100109-104016 [DOI] [PubMed] [Google Scholar]
- Sever S., Muhlberg A. B., Schmid S. L. (1999). Impairment of dynamin's GAP domain stimulates receptor-mediated endocytosis. Nature 398, 481–486 10.1038/19024 [DOI] [PubMed] [Google Scholar]
- Sever S., Skoch J., Newmyer S., Ramachandran R., Ko D., McKee M., Bouley R., Ausiello D., Hyman B. T., Bacskai B. J. (2006). Physical and functional connection between auxilin and dynamin during endocytosis. EMBO J. 25, 4163–4174 10.1038/sj.emboj.7601298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shnyrova A. V., Bashkirov P. V., Akimov S. A., Pucadyil T. J., Zimmerberg J., Schmid S. L., Frolov V. A. (2013). Geometric catalysis of membrane fission driven by flexible dynamin rings. Science 339, 1433–1436 10.1126/science.1233920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomaha E., Palfrey H. C. (2005). Conformational changes in dynamin on GTP binding and oligomerization reported by intrinsic and extrinsic fluorescence. Biochem. J. 391, 601–611 10.1042/BJ20050707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song B. D., Schmid S. L. (2003). A molecular motor or a regulator? Dynamin's in a class of its own. Biochemistry 42, 1369–1376 10.1021/bi027062h [DOI] [PubMed] [Google Scholar]
- Song B. D., Leonard M., Schmid S. L. (2004). Dynamin GTPase domain mutants that differentially affect GTP binding, GTP hydrolysis, and clathrin-mediated endocytosis. J. Biol. Chem. 279, 40431–40436 10.1074/jbc.M407007200 [DOI] [PubMed] [Google Scholar]
- Stowell M. H., Marks B., Wigge P., McMahon H. T. (1999). Nucleotide-dependent conformational changes in dynamin: evidence for a mechanochemical molecular spring. Nat. Cell Biol. 1, 27–32 10.1038/8997 [DOI] [PubMed] [Google Scholar]
- Sweitzer S. M., Hinshaw J. E. (1998). Dynamin undergoes a GTP-dependent conformational change causing vesiculation. Cell 93, 1021–1029 10.1016/S0092-8674(00)81207-6 [DOI] [PubMed] [Google Scholar]
- Takei K., McPherson P. S., Schmid S. L., De Camilli P. (1995). Tubular membrane invaginations coated by dynamin rings are induced by GTP-gamma S in nerve terminals. Nature 374, 186–190 10.1038/374186a0 [DOI] [PubMed] [Google Scholar]
- Tesmer J. J. G., Berman D. M., Gilman A. G., Sprang S. R. (1997). Structure of RGS4 bound to AlF4—activated G(i α1): stabilization of the transition state for GTP hydrolysis. Cell 89, 251–261 10.1016/S0092-8674(00)80204-4 [DOI] [PubMed] [Google Scholar]
- Timm D., Salim K., Gout I., Guruprasad L., Waterfield M., Blundell T. (1994). Crystal structure of the pleckstrin homology domain from dynamin. Nat. Struct. Biol. 1, 782–788 10.1038/nsb1194-782 [DOI] [PubMed] [Google Scholar]
- van der Bliek A. M. (1999). Functional diversity in the dynamin family. Trends Cell Biol. 9, 96–102 10.1016/S0962-8924(98)01490-1 [DOI] [PubMed] [Google Scholar]
- Vetter I. R., Wittinghofer A. (2001). The guanine nucleotide-binding switch in three dimensions. Science 294, 1299–1304 10.1126/science.1062023 [DOI] [PubMed] [Google Scholar]
- Warnock D. E., Hinshaw J. E., Schmid S. L. (1996). Dynamin self-assembly stimulates its GTPase activity. J. Biol. Chem. 271, 22310–22314 10.1074/jbc.271.37.22310 [DOI] [PubMed] [Google Scholar]
- Wittinghofer A., Vetter I. R. (2011). Structure-function relationships of the G domain, a canonical switch motif. Annu. Rev. Biochem. 80, 943–971 10.1146/annurev-biochem-062708-134043 [DOI] [PubMed] [Google Scholar]
- Yan L., Ma Y., Sun Y., Gao J., Chen X., Liu J., Wang C., Rao Z., Lou Z. (2011). Structural basis for mechanochemical role of Arabidopsis thaliana dynamin-related protein in membrane fission. J. Mol. Cell Biol. 3, 378–381 10.1093/jmcb/mjr032 [DOI] [PubMed] [Google Scholar]
- Zhang P., Hinshaw J. E. (2001). Three-dimensional reconstruction of dynamin in the constricted state. Nat. Cell Biol. 3, 922–926 10.1038/ncb1001-922 [DOI] [PubMed] [Google Scholar]