

Abstract
We previously reported genome-wide significant evidence for linkage between chromosome 6q and bipolar I disorder (BPI) by performing a meta-analysis of original genotype data from 11 genome scan linkage studies. We now present follow-up linkage disequilibrium mapping of the linked region utilizing 3,047 single nucleotide polymorphism (SNP) markers in a case–control sample (N = 530 cases, 534 controls) and family-based sample (N = 256 nuclear families, 1,301 individuals). The strongest single SNP result (rs6938431, P=6.72× 10−5) was observed in the case–control sample, near the solute carrier family 22, member 16 gene (SLC22A16). In a replication study, we genotyped 151 SNPs in an independent sample (N = 622 cases, 1,181 controls) and observed further evidence of association between variants at SLC22A16 and BPI. Although consistent evidence of association with any single variant was not seen across samples, SNP-wise and gene-based test results in the three samples provided convergent evidence for association with SLC22A16, a carnitine transporter, implicating this gene as a novel candidate for BPI risk. Further studies in larger samples are warranted to clarify which, if any, genes in the 6q region confer risk for bipolar disorder.
Keywords: bipolar disorder, genetic, association, SLC22A16, 6q
Introduction
Bipolar disorder (BPD) is a chronic and often disabling disorder that, in broadest conceptualization, affects up to 4% of the US population and, in the most narrow (Type I) classification, consistently affects about 1% of adults worldwide [Kessler et al., 2005]. Despite the availability of effective treatments for mania and depression, the recurrence rate and disability associated with BPD remain substantial even among adequately treated patients [Fagiolini et al., 2005]. Thus, there is intense interest in determining the etiology of BPD, with the goal of developing more effective treatment and prevention strategies. Family and twin studies have consistently documented that BPD is familial and heritable [Smoller and Finn, 2003], but efforts to identify specific susceptibility genes have been complicated by the genetic and phenotypic complexity of the disorder.
For the most part, two parallel approaches have been exploited in the search for BPD susceptibility genes. The first has focused on biologically relevant candidate genes thought to underlie BPD based on prior knowledge of the biological basis or mechanism of action of drug treatments for BPD. For example, variants within the serotonin transporter gene (SLC6A4) have been implicated in BPD by meta-analyses of association studies [Cho et al, 2005; Lasky-Su et al., 2005]. However, the identification of such candidates is constrained by the state of knowledge about the biology of the disorder. The second strategy involves “unbiased” genome-wide approaches, including whole genome linkage and association methods that do not require pre-specified hypotheses about genes of interest. Genome-wide linkage scans can localize chromosomal regions harboring susceptibility loci which can then be examined using linkage disequilibrium (LD) methods to identify the relevant genes, a strategy that has proven successful in other complex diseases [e.g., Crohn's disease; Hugot et al., 2001; Rioux et al., 2001]. More recently, genome-wide association studies (GWAS) have become feasible, permitting LD analysis across the entire genome. Initial results have implicated several putative loci in BPD, though none has yet been established [Baum et al., 2007; Wellcome Trust Case Control Consortium, 2007; Sklar et al., 2008].
We have been pursuing both of these strategies, and here we report the primary results of a large-scale effort to combine linkage mapping and LD analyses to identify BPD susceptibility variants. We began by performing a meta-analysis of genome scan linkage studies in which we combined original genotype data from 11 linkage studies, comprising more than 5,000 individuals from 1,067 families [McQueen et al., 2005]. We identified two regions that achieved genome-wide significance: the first, at chromosome 6q, achieved a LOD score of 4.19 for BPI disorder (narrow phenotypic definition); and the second on chromosome 8q yielded a LOD score of 3.4 for a broad phenotypic definition (bipolar I + bipolar II) [McQueen et al., 2005]. Here we report LD analyses aimed at identifying susceptibility variants within the 6q locus.We employed a staged approach beginning with a screening set of 530 BPI cases and 534 unscreened controls and an independent family-based sample comprising 256 nuclear families in which offspring were BPI cases. The strongest results from stage 1 were followed up in an independent sample of 622 BPI cases and 1,181 screened controls. We also examined two sub-phenotypes of BPD (psychotic BP and early-onset BP) that have previously been shown to be familial subtypes of the disorder {Potash et al., 2003 #2102; Saunders et al., 2008 #8782}.
Methods
Samples
Case–control sample (STEP1)
A sample of 530 individuals with BPI were obtained from the genetic repository of the Systematic Treatment Enhancement Program for BPD (STEP-BD), a longitudinal cohort study designed to examine the effectiveness of treatments and their impact on the course of BPD [Sachs et al., 2003]. STEP-BD enrolled participants across the United States who met the Diagnostic and Statistical Manual of Mental Disorders-IV (DSM-IV) criteria for BPI, BPII, BP NOS, schizoaffective manic or BP type, or cyclothymic disorder on the basis of diagnostic interviews. Only self-defined Caucasian participants that met consensus diagnosis of BPI on both the Affective Disorders Evaluation and the Mini-International Neuropsychiatric Interview were included in the current genetic study. Caucasian control subjects (n = 534) were obtained from the NIMH Genetics Initiative through the NIMH Center for Collaborative Studies (http://zork.wustl.edu/nimh/). Control samples were derived from anonymous cord blood samples and thus were phenotypically unscreened [Mansour et al., 2005]. Such samples are normally discarded following normal delivery.
Family-based sample (NIMH)
Nuclear families (n = 256 families, 1,301 individuals) were selected from samples previously collected for linkage studies by the National Institute of Mental Health (NIMH) Genetics Collaborative Study of BPD waves 1 through 4 [Edenberg et al., 1997; Stine et al., 1997; Dick et al., 2003; McInnis et al., 2003]. In brief, diagnosis was determined using the Diagnostic Interview for Genetic Studies [Nurnberger et al., 1994] with best-estimate diagnosis assigned by two independent psychiatrists based on the Diagnostic Interview for Genetic Studies result, family informants, and review of medical records. For the present study, we initially identified all complete affected parent-proband trios for whom DNA was available from the Rutgers University repository (http://www.nimhgenetics.org) using abroad definition that included BPI, BPII, or SAB probands; from these, the families which offspring affected with BPI, based on phenotypic data (NIMH release 3.05; http://www.nimhgenetics.org), were selected for primary analyses. The same group of families were included in additional studies by our group [Perlis et al., 2008; Sklar et al., 2008].
Follow-up case–control sample (STEP2)
For follow-up analyses, a second sample of BPI cases (n = 622) were obtained from the STEP-BD genetic repository, in the same way as STEP1. The control subjects (N = 1,181) were obtained from the NIMH Genetics Initiative through the NIMH Center for Collaborative Studies (http://zork.wustl.edu/nimh/). The control sample comprised a sub-sample of a panel of self-reported Caucasian individuals who completed an online self-administered psychiatric screen and were recruited via random-digit dialing by a marketing research company, Knowledge Networks (Menlo Park, CA) [Sanders et al., 2008] The panel provides a weighted probability sample, representative of the US population. The online screen included questions regarding demographics, ethnic ancestry, and DSM-IV criteria for depression, and anxiety disorders.
In addition, participants were queried about any history of schizophrenia, psychosis or BPD using a three-part question: “Have you ever received treatment for, or been diagnosed with, any of the following conditions: (a) Schizophrenia or schizoaffective disorder; (b) Hearing voices others could not hear or believing things that others said were not true (such as that people were trying to harm you); (c) BPD (manic-depression).” Controls were included only if they answered “no” to all three of these questions. In addition, controls who met lifetime criteria for recurrent major depressive disorder with impairment based on their responses to depression items were excluded. The STEP1 and STEP2 samples and the controls were also included in a recent GWAS of BPD [Sklar et al., 2008].
Gene and SNP Selection
Stage I: screening SNP selection and genotyping
Based on the results of our genome-scan meta-analysis [McQueen et al., 2005], single nucleotide polymorphism (SNP) genotyping focused on a ∼20 Mb core linkage peak region of chromosome 6q (spanning 105.34–125.73 Mb, NCBI build 34). The chromosome 6 linkage region boundaries were identified using a meta-analytic approach as described by McQueen et al. [2006]. In particular, the contiguous region on chromosome 6q for which the 95% confidence interval of the summary IBD estimate did not include null allele sharing (IBD = 0.5) defined the fine-mapping region. In addition, 22 genes (data shown in Supplement Table s1) located within 25 Mb flanking the core linkage peak region were selected for genotyping if they met at least one of two criteria: (1) known to be involved in neurotransmitter systems or central nervous system function; and/or (2) evidence of association with psychiatric disorders in prior genetic studies.
Overall, a ∼23 Mb region (core linkage peak region plus 22 candidate gene spanning 2.6 Mb) was selected for LD mapping. We initially used HapMap Phase I data (www.hapmap.org) to identify tagging SNPs that captured variation at common SNPs (minor allele frequency ≥5%) within the selected regions. The tagging SNPs were selected by H-clust software [Rinaldo et al., 2005] using an r2 threshold of 0.8 or greater.
A total of 3,047 6q SNPs were selected (1,503 in gene regions), with an average marker density in the core linkage peak region of 1 SNP per ∼7.6 kb (2,697 SNPs/20,393 kb). In total, 3,072 SNPs were genotyped in STEP1 and NIMH samples using two Oligo Pool Arrays (OPAs) by the Illumina BeadArray platform (San Diego, CA) at the Broad Institute. Between the two OPAs, eight SNPs (seven non-chr6 SNPs and one chr6 SNP) were duplicated for internal quality control purposes (data shown in Supplement Table s2). One control sample from the Centre d'E′ tude du Polymorphisme Humain set was also included on each 96-well plate as inter-plate controls.
Stage II: follow-up SNPs selection and genotyping
Based on the results observed in the STEP1 and NIMH samples, follow-up genotyping was performed for six gene regions that showed evidence of association with BPI in analyses of the STEP1 and/or NIMH trio samples (data shown in Supplement Table s3). First, SNPs in these six regions from the original OPAs were selected and genotyped in STEP2 sample (81 SNPs). Second, additional tagging SNPs for these loci were selected using Tagger [de Bakker et al, 2005] based on HapMap Phase II data (www.hapmap.org) and pair-wise tagging parameters, with a minimum r2 threshold of 0.8 and minimum minor allele frequency of 0.05. These SNPs were supplemented by known coding sequence SNPs in the gene regions derived from dbSNP (http://www.ncbi.nlm.nih.gov/SNP/). In total, 70 additional SNPs were genotyped in all three groups (STEP1, NIMH and STEP2). Third, an additional 56 SNPs were genotyped in NIMH sample only since they were genotyped in STEP1 and STEP2 samples in our GWAS [Sklar et al., 2008].
All SNPs in our follow-up tests were genotyped by an allele-specific primer extension of amplified products with detection by matrix-assisted laser desorption ionization time-of-flight mass spectroscopy using the Sequenom iPLEX genotyping platform (San Diego, CA) at the Broad Institute. Among them, 12 SNPs were genotyped in duplicate in both STEP1 and NIMH samples by Illumina OPAs and follow-up iPlex pools. The genotype concordance rate between Illumina OPAs and Sequenom iPLEX genotyping platforms was calculated using these duplicated SNPs.
Data Analyses
Quality control (QC) analyses
Genetic quality control analyses were performed using the PLINK software package [Purcell et al., 2007]. The original genetic dataset comprised 530 STEP1 cases, 534 anonymous controls, and 1,301 individuals from the NIMH nuclear families genotyped for 3,072 SNPs. We excluded 34 STEP1 individuals (17 cases and 17 controls), 57 NIMH individuals, and 269 SNPs that failed the OPAs quality control (QC) filters (QC data are shown in Supplement Tables s4 and s5). After QC, the STEP1 and NIMH datasets consisted of 513 STEP cases, 517 controls, and 1,244 individuals from the NIMH nuclear families genotyped for 2,803 SNPs for which genotype call rates exceeded 95%. For the analyzable duplicate individuals and SNPs, the within and between plate concordance exceeded 99.9%, and concordance with published HapMap CEPH genotypes was greater than 99.7%. The inter-OPAs concordance was greater than 99.9% on the basis of seven duplicated SNPs.
To determine the tagging performance of SNPs that passed QC thresholds in the core linkage peak region of chromosome 6q, we employed Tagger [de Bakker et al., 2005] using HapMap phase II data (HapMap Release 21). Pair-wise tagging parameters included a minimum r2 threshold of 0.8 and minimum minor allele frequency of 0.05. Of a total of 16,805 HapMap phase II SNPs in the core linkage peak region, 74% were captured with r2 ≥0.8, with mean r2 = 0.88, suggesting that our final set of SNPs adequately captured common variation in this region.
The original iPLEX genotype datasets (Stage 2) comprised 1,064 STEP1 individuals (530 cases, 534 controls), 1,301 members of NIMH nuclear families and 1,803 STEP2 individuals (622 cases, 1,181 controls) genotyped for 70, 126, and 151 SNPs, respectively. We excluded 92 individuals and 8 SNPs from STEP1 dataset, 57 individuals and 12 SNPs from NIMH dataset, 221 individuals and 18 SNPs from STEP2 dataset that failed the iPLEX QC filters (data shown in Supplement Table s5). After QC, all remaining individuals and SNPs had genotype call rates greater than 95%. The concordance between OPA and iPLEX genotyping platforms was greater than 99.9% on the basis of seven duplicated SNPs that passed both QC filters.
Single-marker analyses
Single-marker association analyses were conducted using PLINK [Purcell et al., 2007] for the case–control samples (STEP1 and STEP2) and FBAT for the family-based sample (NIMH) [Laird et al., 2000]. We report P-values under an additive model using the Cochran–Armitage trend test implemented in PLINK, and from the family-based test in FBAT. We also report P-values for Z-scores based on combined results from the three independent samples. The lambda value [Devlin and Roeder, 1999] for the STEP1 sample was 1.06, suggesting that adjustment for population substructure was not necessary.
Gene-based analyses
Gene regions were defined by all geno-typed SNPs within 10 kb up- and down-stream of gene transcription sequences based on NCBI build 36.1 (March 2006). Overall, there are 117 RefSeq genes in our 6q region with at least one SNP genotyped by Illumina OPAs. The set-based test implemented in the PLINK software package was performed in case–control samples (STEP1 and STEP2). The min-p test implemented in the FBAT software (which computes the P-value for the minimum observed marker P-value in a gene using Monte Carlo simulations) was performed in family-based samples (NIMH). We also report the Fisher's combined P-value, as a combined measure of results from the case–control and family-based samples.
The sequence of analyses are summarized in Figure 1.
Fig. 1.
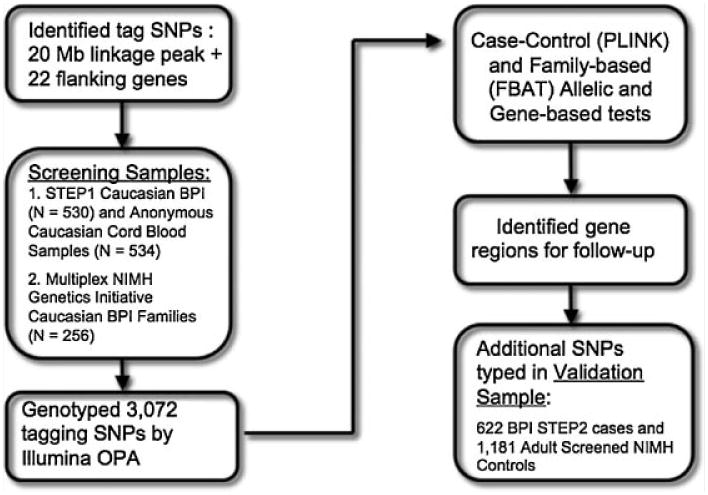
Flow chart of analytic approach.
Results
Primary Association Analyses
First, we performed single-SNP analyses for 2,803 SNPs genotyped in the Stage 1 samples (STEP1 and NIMH). Single-marker P-values from the Cochran–Armitage trend test in the STEP1 sample and from the FBAT test in the NIMH trio sample are shown in Figure 2, respectively. Supplementary Table s6 and s7 show the top 10 SNPs from these analyses, for STEP1 and NIMH, respectively, along with corresponding P-value, direction of effect (+ for OR > 1 and – for OR < 1), physical position and, where applicable, the gene region the SNP sits in.
Fig. 2.
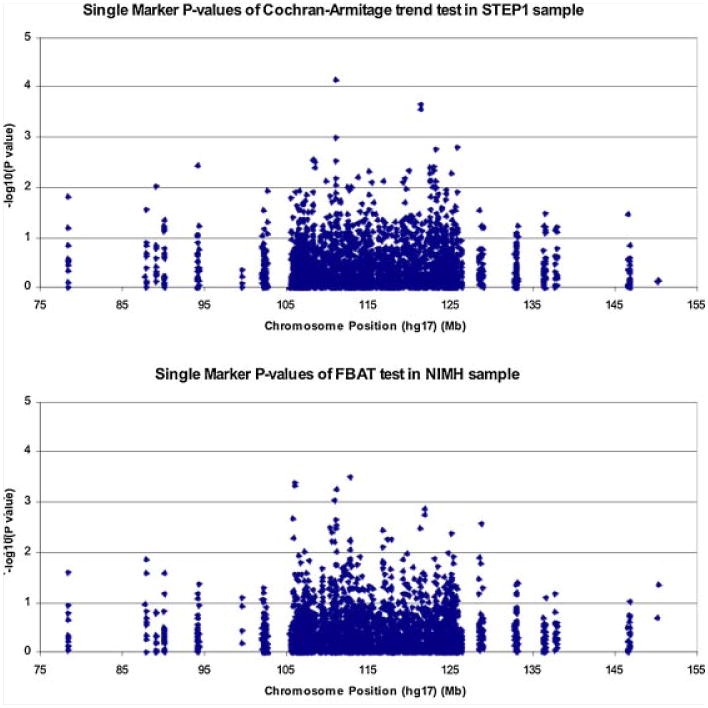
Single-marker P-values from FBAT test in NIMH sample.
As shown in Supplementary Table s6, no single marker achieved statistical significance after Bonferroni correction for the full set of markers tested (Bonferroni-corrected threshold 0.05/2,803= 1.8 × 10−5). The strongest evidence for association was seen for rs6938431 (P=6.7 × 10−5), which is located 8.9 kb 5′ of d-aspartate oxidase gene (DDO) and 304 bp 3′ of Solute carrier family 22 (organic cation/carnitine transporter), member 16 (SLC22A16). Of note, another SNP in the top 10 lists (rs9400393) is also in the SLC22A16 region (the r2 is 0.094 between these two SNPs).
Supplementary Table s7 shows the top 10 SNP results for the NIMH family-based sample. While none of the SNPs that showed the strongest association in STEP1 were associated with BPI in the NIMH sample, another SNP in the SLC22A16 region, rs17071722, showed evidence of association in the NIMH sample. This SNP is not in LD with the SLC22A16 SNPs in Supplementary Table s6 (rs17071722 has an r2 of 0.019 with rs6938431, and 0.045 with rs9400393). The strongest association in the NIMH sample was observed for rs6931341 (P=0.0003), located ∼28 kb upstream of the transcription start point of alpha chain isoform 4 of laminin gene (LAMA4), In addition, two SNPs (r2 between them is 0.431) in the gene encoding prolyl endopeptidase (PREP), a gene implicated in the action of mood stabilizers [Williams et al., 2002], were among the top 10 results for the NIMH sample.
In Supplementary Table s8, we show the top 10 results from the pooled analysis of both the NIMH and STEP1 samples, combined according to Z-scores. There are two SNPs in this list that are in the DDO/SLC22A16 region, and two SNPs in this list that are in the 5′-nucleotidase domain containing one gene region (NT5DC1).
Gene-Based Tests
As an additional analysis, we performed gene-based tests for 117 genes. In Supplementary Tables s9 and s10 we show the top 10 genes in the STEP1 and NIMH samples, respectively.
As expected from the findings reported above, SLC22A16 was in the top 10 lists for both samples. In Supplementary Table s11, we show the combined results from both samples, combining P-values using Fisher's method. The leading gene-based results were observed for SLC22A16 (P=0.00048), DDO (P=0.0033), and PREP (P=0.0148), although none survive Bonferroni correction for all the marker tests we performed.
Follow-Up and Replication Analyses
Based on the pattern of findings from the NIMH and STEP1 samples, we performed follow-up analyses focusing on six loci, including five gene regions—SLC22A16, DDO, PREP, NT5DC1, GPR6, and the region around rs794854 (the top SNP in the combined analysis: Supplementary Table s8).
None of the additional SNPs in our follow-up study achieved lower P-values than the original set of SNPs within the NIMH and STEP1 samples. However, genotyping in the STEP2 sample added new, independent evidence for SLC22A16. Notably, 5 of the top 10 SNPs in STEP2 (Supplementary Table s12) are in the SLC22A16 region. In addition, one of these SNPs (rs17071722) was nominally associated with BPI in all three (STEP1, NIMH, and STEP2) samples (P=0.016, 0.002, and 0.02, respectively), although the associated allele is flipped between STEP1 (major allele) and the NIMH (minor allele)/STEP2 (minor allele) samples. Results for single marker tests in the SLC22A16 region are summarized in Figure 3.
Fig. 3.
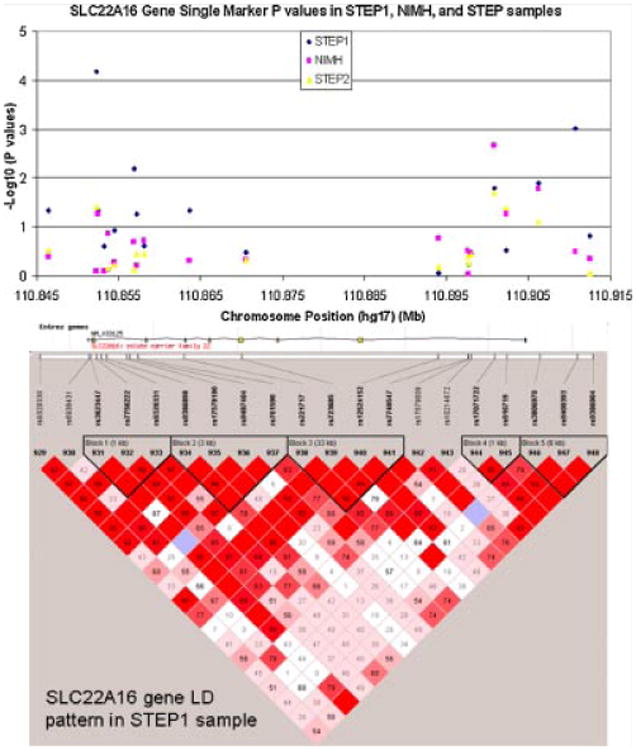
Single-marker P-values in the SLC22A16 gene region for the STEP1, NIMH, and STEP2 samples. Also shown: LD pattern in STEP1 sample.
The gene-based test for STEP2 resulted in nominally significant P-values for two genes: SLC22A16 (P=0.03) and C6orf204 (P=0.036). It is interesting that SLC22A16 is nominally significant in all three samples (with P-values 0.001, 0.03, and 0.03 in STEP1, NIMH, and STEP2), although the top ranking SNP from STEP1, rs6938431 (Supplementary Tables s6), has an opposite direction of effect in STEP2 (Supplementary Tables s12).
Phenotypic Subtype Analyses (Psychosis and Early-Onset BPI)
In light of the suggestive evidence of association observed for SLC22A16, we explored whether a stronger association might be observed within genetically relevant subtypes of BPD. In particular, psychotic BPD and early-onset BPD have been shown to be familial and heritable subtypes that may reflect more genetically homogeneous influences [Smoller and Gardner-Schuster, 2007]. Early-onset BPD was defined as having a first manic or depressive episode before age 18, and psychosis was defined as having had psychotic symptoms or a psychotic diagnosis on any of the STEP-BD instruments that assessed psychosis. For the NIMH sample, psychosis was identified in individuals reporting lifetime history of either delusions or hallucinations, as assessed by the DIGS [Nurnberger et al., 1994]. The resulting sample sizes for the three datasets and the two sub-phenotypes are shown in Supplementary Table s13.
For the subtype of psychotic BPI, nominally significant association for the gene-based test of SLC22A16 was observed only for the NIMH sample (P=0.0073) sample. For the early-onset BPD sub-phenotype, the STEP1 and STEP2 both showed nominally significant P-values (STEP1 P=0.0008, STEP2 P=0.027) (data shown in Supplementary Table s13).
Discussion
We report results from a large-scale LD mapping study of BPI disorder using independent case–control and family-based samples. Although we did not detect single marker or gene signals that meet stringent criteria for study-wide significance, we identified several interesting loci that warrant further examination in much larger samples.
Our strongest association signal was observed in the STEP1 sample at rs6938431 (P=6.72 × 10−5), which is ∼300 bp 3′ of SLC22A16. Another SNP in intron 1 of SLC22A16 showed a nominally significant association with BPI in all three samples (STEP1, NIMH, and STEP2; P=0.016, 0.002, and 0.02, respectively). Although no single SNP showed consistent (in terms of directionality of effect) evidence of association across samples, SLC22A16 appears in the top 10 results from single-marker and gene-based tests in all three samples. Further studies of the role of SLC22A16 polymorphisms using large well-characterized clinical samples are warranted. The estimated odds ratios for the seven nominally significant SLC22A16 SNPs in either STEP1 or STEP2 range from 1.35 to 1.63.
The association found between variants in SLC22A16 and BPI is intriguing since it is located directly under the linkage peak identified by our previous meta-analysis of genome scan linkage studies [McQueen et al., 2005]. The SLC22A16 gene, which encodes a high affinity carnitine transporter that also transports various organic cations, has not previously been highlighted as a candidate gene for BPD. It is one of a family of organic ion transporters (referred to as amphiphilic solute facilitators, ASFs), that transport various medically and physiologically important compounds, including pharmaceuticals, toxins, hormones, neurotransmitters, and cellular metabolites [Koepsell et al., 2007]. It is expressed in a variety of tissues including brain [Eraly and Nigam, 2002], although its role in the central nervous system has not been characterized. Carnitine itself is essential for the transport of fatty acids into mitochondria for β oxidation; mitochondrial dysfunction, and abnormal bioenergetics have been implicated in the pathophysiology of BPD [Stork and Renshaw, 2005], and preliminary evidence suggests that carnitine may have neuroprotective effects [Soczynska et al., 2008].
The strategy pursued in this study—dense LD mapping of genomic regions implicated by linkage mapping—has been successful in other areas of complex diseases including Crohn's disease, alcoholism, and Alzheimer's disease [Corder et al., 1993; Hugot et al., 2001; Rioux et al., 2001; Dick et al., 2006]. However, the recent advent of genome-wide association studies (GWAS) and their success in identifying susceptibility genes for a variety of complex disorders have highlighted the advantages of the GWAS approach. Nevertheless, there may still be a role for positional mapping given that current GWAS platforms do not comprehensively assay variation throughout the genome. At the same time, one lesson of recent GWAS studies has been that common susceptibility variants confer modest effects (odds ratios in the range of 1.1–1.5) and thus require very large sample sizes to be successful. Thus, one reason that we may not have detected consistent evidence of association with variants in the 6q region may have been insufficient power. The samples included in the present study also contributed to a GWAS of BPI that we recently reported [Sklar et al., 2008]. In that study (which was undertaken after the analyses reported here), no evidence of association was observed in the 6q region examined in the current study. Data from the Wellcome Trust GWAS of BPD also did not support association with the region encompassing SLC22A16 [Wellcome Trust Case Control Consortium, 2007]. Of note, only 407 of the 3,047 SNPs genotyped in the current study were included on the Affymetrix GeneChip 500K array used in the GWAS. The most strongly associated SNP at SLC22A16 in the present study (rs6938431) was not on the 500K Array. Using the same tagging settings described in the Methods Section, the LD coverage of the 500K array for SLC22A16 is only 64%. The LD coverage of the Illumina OPAs (Stage I of the current study) for SLC22A16 is 76%, and the LD coverage of Chr6 OPAs plus the SLC22A16 SNPs genotyped in Stage II is 90%.
In addition to the power constraints of our sample, several additional limitations that may have increased the risk of Type II error in this study should be noted. First, SNP selection focused on common variants (minor allele frequency ≥5%). It is possible that that the genome-wide significant linkage that we observed in our meta-analysis for the 6q region was driven by rare variants that were not well captured by our tagging SNPs. This scenario would be consistent with the strong linkage signal we detected previously, and the weaker association signals we report in the current article. In this scenario, further examination of the region, including re-sequencing for detection of rare variants, may lead to the discovery of additional susceptibility variants. In any case, there is no reason to believe that the common variants we report in the current study account for all of the genetic variance at this locus. Second, differences in ascertainment criteria and diagnostic assessments for the STEP-BD and NIMH Genetics Initiative sample may have resulted in heterogeneity that obscured positive findings. Third, the inclusion of unscreened controls in the STEP1 case–control analysis may have reduced power if occult cases of BPI were present, though the impact of this should be negligible given the low-population prevalence of BPI [Moskvina et al., 2005]. The risk of confounding and spurious association due to population stratification is another concern in case–control samples; however this is unlikely to play a role in our analyses due the small genomic inflation factor (based on mean Chi-squared values) of 1.06 in the STEP1 sample, and the use of family-based samples.
In summary, we performed LD mapping in three independent samples of a region of chromosome 6q that has been strongly linked to BPI disorder. Although consistent evidence of association with any single variant was not seen across samples, SNP-wise and gene-based tests provided convergent evidence for association with SLC22A16, implicating this gene as a novel candidate for BPI risk. Further studies in larger samples are warranted to replicate or refute an association of this gene with BPD.
Supplementary Material
Acknowledgments
This study was funded by National Institute of Mental Health grants MH063445 (PI: JWS; MH067288 (PI: PS), and MH63420 (PI: VLN) as well as support from a Charles A. King Trust Fellowship and NARSAD Young Investigator Award to JB Fan. “The STEP-BD project was funded in whole or in part with Federal funds from the National Institute of Mental Health (NIMH), National Institutes of Health, under Contract N01MH80001 to Gary S. Sachs, M.D. (PI), Michael E. Thase M.D. (Co-PI), Mark S. Bauer, M.D. (Co-PI). Active STEP-BD Sites and Principal Investigators included: Baylor College of Medicine (Lauren B. Marangell, M.D.); Case University (Joseph R. Calabrese, M.D.); Massachusetts General Hospital and Harvard Medical School (Andrew A. Nierenberg, M.D.); Portland VA Medical Center (Peter Hauser, M.D.); Stanford University School of Medicine (Terence A. Ketter, M.D.); University of Colorado Health Sciences Center (Marshall Thomas, M.D.); University of Massachusetts Medical Center (Jayendra Patel, M.D.); University of Oklahoma College of Medicine (Mark D. Fossey, M.D.); University of Pennsylvania Medical Center (Laszlo Gyulai, M.D.); University of Pittsburgh Western Psychiatric Institute and Clinic (Michael E. Thase, M.D.); University of Texas Health Science Center at San Antonio (Charles L. Bowden, M.D.).Collection of DNA from consenting participants in STEP-BD was supported by N01-MH-80001 (G Sachs, PI). We thank Beth Rosen-Sheidley and Laurie Silfies for their assistance with this effort. Control subjects from the National Institute of Mental Health Schizophrenia Genetics Initiative (NIMH-GI), data and biomaterials were collected by the “Molecular Genetics of Schizophrenia II” (MGS-2) collaboration. The investigators and co-investigators are: ENH/Northwestern University, Evanston, IL, MH059571, Pablo V. Gejman, M.D. (Collaboration Coordinator; PI), Alan R. Sanders, M.D.; Emory University School of Medicine, Atlanta, GA, MH59587, Farooq Amin, M.D. (PI); Louisiana State University Health Sciences Center; New Orleans, Louisiana, MH067257, Nancy Buccola APRN, BC, MSN (PI); University of California-Irvine, Irvine, CA,MH60870, William Byerley, M.D. (PI); Washington University, St. Louis, MO, U01, MH060879, C. Robert Cloninger, M.D. (PI); University of Iowa, Iowa, IA,MH59566, Raymond Crowe, M.D. (PI), Donald Black, M.D.; University of Colorado, Denver, CO, MH059565, Robert Freed-man, M.D. (PI); University of Pennsylvania, Philadelphia, PA, MH061675, Douglas Levinson M.D. (PI); University of Queensland, Queensland, Australia, MH059588, Bryan Mowry, M.D. (PI); Mt. Sinai School of Medicine, New York, NY,MH59586, Jeremy Silverman, Ph.D. 34 (PI). NIMH Family samples were collected as part of ten projects that participated in the National Institute of Mental Health (NIMH) Bipolar Disorder Genetics Initiative. From 1999 to 2003, the Principal Investigators and Co-Investigators were: Indiana University, Indianapolis, IN, R01 MH59545, John Nurnberger, M.D., Ph.D., Marvin J. Miller, M.D., Elizabeth S. Bowman, M.D., N. Leela Rau, M.D., P. Ryan Moe, M.D., Nalini Samavedy, M.D., Rif El-Mallakh, M.D. (at University of Louisville), Husseini Manji, M.D. (at Wayne State University), Debra A. Glitz, M.D. (at Wayne State University), Eric T. Meyer, M.S., Carrie Smiley, R.N., Tatiana Foroud, Ph.D., Leah Flury, M.S., Danielle M. Dick, Ph.D., Howard Edenberg, Ph.D.; Washington University, St. Louis, MO, R01 MH059534, John Rice, Ph.D,Theodore Reich, M.D., Allison Goate, Ph.D., Laura Bierut, M.D.; Johns Hopkins University, Baltimore, MD, R01 MH59533, Melvin McInnis M.D., J.Raymond DePaulo, Jr., M.D., Dean F. MacKinnon, M.D., Francis M. Mondimore, M.D., James B. Potash, M.D., Peter P. Zandi, Ph.D, Dimitrios Avramopoulos, and Jennifer Payne; University of Pennsylvania, PA, R01 MH59553, Wade Berrettini M.D.,Ph.D.; University of California at Irvine, CA, R01 MH60068, William Byerley M.D., and Mark Vawter M.D.; University of Iowa, IA, R01 MH059548, William Coryell M.D., and Raymond Crowe M.D.; University of Chicago, IL, R01 MH59535, Elliot Gershon, M.D., Judith Badner Ph.D., Francis McMahon M.D., Chunyu Liu Ph.D., Alan Sanders M.D., Maria Caserta, Steven Dinwiddie M.D., Tu Nguyen,Donna Harakal; University of California at San Diego, CA, R01 MH59567, John Kelsoe, M.D., Rebecca McKinney, B.A.; Rush University, IL, R01 MH059556, William Scheftner M.D., Howard M. Kravitz, D.O., M.P.H., Diana Marta, B.S., Annette Vaughn-Brown, MSN, RN, and Laurie Bederow, MA; NIMH Intramural Research Program, Bethesda, MD, 1Z01MH002810-01, Francis 35 J. McMahon, M.D., Layla Kassem, PsyD, Sevilla Detera-Wa-dleigh, Ph.D, Lisa Austin, Ph.D, Dennis L. Murphy, M.D.
Grant sponsor: National Institute of Mental Health; Grant number: MH063445; Grant number: MH067288; Grant number: MH63420; Grant sponsor: National Institute of Mental Health (NIMH); Grant sponsor: National Institutes of Health; Grant number: N01MH80001
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- Baum AE, Akula N, Cabanero M, Cardona I, Corona W, Klemens B, Schulze TG, Cichon S, Rietschel M, Nothen MM, et al. A genome-wide association study implicates diacylglycerol kinase eta (DGKH) and several other genes in the etiology of bipolar disorder. Mol Psychiatry. 2007;13(2):197–207. doi: 10.1038/sj.mp.4002012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HJ, Meira-Lima I, Cordeiro Q, Michelon L, Sham P, Vallada H, Collier DA. Population-based and family-based studies on the serotonin transporter gene polymorphisms and bipolar disorder: A systematic review and meta-analysis. Mol Psychiatry. 2005;10(8):771–781. doi: 10.1038/sj.mp.4001663. [DOI] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261(5123):921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- de Bakker PI, Yelensky R, Pe'er I, Gabriel SB, Daly MJ, Altshuler D. Efficiency and power in genetic association studies. Nat Genet. 2005;37(11):1217–1223. doi: 10.1038/ng1669. [DOI] [PubMed] [Google Scholar]
- Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55(4):997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- Dick DM, Foroud T, Flury L, Bowman ES, Miller MJ, Rau NL, Moe PR, Samavedy N, El-Mallakh R, Manji H, et al. Genome-wide linkage analyses of bipolar disorder: A new sample of 250 pedigrees from the national institute of mental health genetics initiative. Am J Hum Genet. 2003;73(1):107–114. doi: 10.1086/376562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick DM, Jones K, Saccone N, Hinrichs A, Wang JC, Goate A, Bierut L, Almasy L, Schuckit M, Hesselbrock V, et al. Endophenotypes successfully lead to gene identification: Results from the collaborative study on the genetics of alcoholism. Behav Genet. 2006;36(1):112–126. doi: 10.1007/s10519-005-9001-3. [DOI] [PubMed] [Google Scholar]
- Edenberg HJ, Foroud T, Conneally PM, Sorbel JJ, Carr K, Crose C, Willig C, Zhao J, Miller M, Bowman E, et al. Initial genomic scan of the NIMH genetics initiative bipolar pedigrees: Chromosomes 3, 5, 15, 16, 17, and 22. Am J Med Genet. 1997;74(3):238–246. [PubMed] [Google Scholar]
- Eraly SA, Nigam SK. Novel human cDNAs homologous to Drosophila Orct and mammalian carnitine transporters. Biochem Biophys Res Commun. 2002;297(5):1159–1166. doi: 10.1016/s0006-291x(02)02343-4. [DOI] [PubMed] [Google Scholar]
- Fagiolini A, Kupfer DJ, Masalehdan A, Scott JA, Houck PR, Frank E. Functional impairment in the remission phase of bipolar disorder. Bipolar Disord. 2005;7(3):281–285. doi: 10.1111/j.1399-5618.2005.00207.x. [DOI] [PubMed] [Google Scholar]
- Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, Almer S, Tysk C, O'Morain CA, Gassull M, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature. 2001;411(6837):599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR, Walters EE. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):593–602. doi: 10.1001/archpsyc.62.6.593. [DOI] [PubMed] [Google Scholar]
- Koepsell H, Lips K, Volk C. Polyspecific organic cation transporters: Structure, function, physiological roles, and biopharmaceutical implications. Pharm Res. 2007;24(7):1227–1251. doi: 10.1007/s11095-007-9254-z. [DOI] [PubMed] [Google Scholar]
- Laird NM, Horvath S, Xu X. Implementing a unified approach to family-based tests of association. Genet Epidemiol. 2000;19(Suppl 1):S36–S42. doi: 10.1002/1098-2272(2000)19:1+<::AID-GEPI6>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Lasky-Su JA, Faraone SV, Glatt SJ, Tsuang MT. Meta-analysis of the association between two polymorphisms in the serotonin transporter gene and affective disorders. Am J Med Genet Part B. 2005;133B(1):110–115. doi: 10.1002/ajmg.b.30104. [DOI] [PubMed] [Google Scholar]
- Mansour HA, Talkowski ME, Wood J, Pless L, Bamne M, Chowdari KV, Allen M, Bowden CL, Calabrese J, El-Mallakh RS, et al. Serotonin gene polymorphisms and bipolar I disorder: Focus on the serotonin transporter. Ann Med. 2005;37(8):590–602. doi: 10.1080/07853890500357428. [DOI] [PubMed] [Google Scholar]
- McInnis MG, Dick DM, Willour VL, Avramopoulos D, MacKinnon DF, Simpson SG, Potash JB, Edenberg HJ, Bowman ES, McMahon FJ, et al. Genome-wide scan and conditional analysis in bipolar disorder: Evidence for genomic interaction in the National Institute of Mental Health genetics initiative bipolar pedigrees. Biol Psychiatry. 2003;54(11):1265–1273. doi: 10.1016/j.biopsych.2003.08.001. [DOI] [PubMed] [Google Scholar]
- McQueen MB, Devlin B, Faraone SV, Nimgaonkar VL, Sklar P, Smoller JW, Abou Jamra R, Albus M, Bacanu SA, Baron M, et al. Combined analysis from 11 linkage studies of bipolar disorder provides strong evidence of susceptibility loci on chromosomes 6q and 8q. Am J Hum Genet. 2005;77(4):582–595. doi: 10.1086/491603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQueen MB, Blacker D, Laird NM. Variance calculations for identity-by-descent estimation. Am J Hum Genet. 2006;78(6):914–921. doi: 10.1086/503920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskvina V, Holmans P, Schmidt KM, Craddock N. Design of case–controls studies with unscreened controls. Ann Hum Genet. 2005;69(Pt 5):566–576. doi: 10.1111/j.1529-8817.2005.00175.x. [DOI] [PubMed] [Google Scholar]
- Nurnberger J, Jr, Blehar M, Kaufmann C, York-Coller C, Simpson S, Harkavy-Friedman J, Severe J, Malaspina D, Reich T Initiative acftNG. Diagnostic Interview for Genetic Studies: Rational, unique features, and training. Arch Gen Psychiatry. 1994;51:849–859. doi: 10.1001/archpsyc.1994.03950110009002. [DOI] [PubMed] [Google Scholar]
- Perlis RH, Purcell S, Fagerness J, Kirby A, Petryshen TL, Fan J, Sklar P. Family-based association study of lithium-related and other candidate genes in bipolar disorder. Arch Gen Psychiatry. 2008;65(1):53–61. doi: 10.1001/archgenpsychiatry.2007.15. [DOI] [PubMed] [Google Scholar]
- Potash JB, Chiu YF, MacKinnon DF, Miller EB, Simpson SG, McMahon FJ, McInnis MG, DePaulo JR., Jr Familial aggregation of psychotic symptoms in a replication set of 69 bipolar disorder pedigrees. Am J Med Genet. 2003;116B(1):90–97. doi: 10.1002/ajmg.b.10761. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldo A, Bacanu SA, Devlin B, Sonpar V, Wasserman L, Roeder K. Characterization of multilocus linkage disequilibrium. Genet Epidemiol. 2005;28(3):193–206. doi: 10.1002/gepi.20056. [DOI] [PubMed] [Google Scholar]
- Rioux JD, Daly MJ, Silverberg MS, Lindblad K, Steinhart H, Cohen Z, Delmonte T, Kocher K, Miller K, Guschwan S, et al. Genetic variation in the 5q31 cytokine gene cluster confers susceptibility to Crohn disease. Nat Genet. 2001;29(2):223–228. doi: 10.1038/ng1001-223. [DOI] [PubMed] [Google Scholar]
- Sachs GS, Thase ME, Otto MW, Bauer M, Miklowitz D, Wisniewski SR, Lavori P, Lebowitz B, Rudorfer M, Frank E, et al. Rationale, design, and methods of the systematic treatment enhancement program for bipolar disorder (STEP-BD) Biol Psychiatry. 2003;53(11):1028–1042. doi: 10.1016/s0006-3223(03)00165-3. [DOI] [PubMed] [Google Scholar]
- Sanders AR, Duan J, Levinson DF, Shi J, He D, Hou C, Burrell GJ, Rice JP, Nertney DA, Olincy A, et al. No significant association of 14 candidate genes with schizophrenia in a large European ancestry sample: Implications for psychiatric genetics. Am J Psychiatry. 2008;165(4):497–506. doi: 10.1176/appi.ajp.2007.07101573. [DOI] [PubMed] [Google Scholar]
- Saunders EH, Scott LJ, McInnis MG, Burmeister M. Familiality and diagnostic patterns of subphenotypes in the National Institutes of Mental Health bipolar sample. Am J Med Genet B Neuropsychiatr Genet. 2008;147B(1):18–26. doi: 10.1002/ajmg.b.30558. [DOI] [PubMed] [Google Scholar]
- Sklar P, Smoller JW, Fan J, Ferreira MA, Perlis RH, Chambert K, Nimgaonkar VL, McQueen MB, Faraone SV, Kirby A, et al. Whole-genome association study of bipolar disorder. Mol Psychiatry. 2008;13(6):558–569. doi: 10.1038/sj.mp.4002151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoller JW, Finn CT. Family, twin, and adoption studies of bipolar disorder. Am J Med Genet Part C. 2003;123C(1):48–58. doi: 10.1002/ajmg.c.20013. [DOI] [PubMed] [Google Scholar]
- Smoller JW, Gardner-Schuster E. Genetics of bipolar disorder. Curr Psychiatry Rep. 2007;9(6):504–511. doi: 10.1007/s11920-007-0069-8. [DOI] [PubMed] [Google Scholar]
- Soczynska JK, Kennedy SH, Chow CS, Woldeyohannes HO, Konarski JZ, McIntyre RS. Acetyl-L-carnitine and alpha-lipoic acid: Possible neurotherapeutic agents for mood disorders? Expert Opin Investig Drugs. 2008;17(6):827–843. doi: 10.1517/13543784.17.6.827. [DOI] [PubMed] [Google Scholar]
- Stine OC, McMahon FJ, Chen L, Xu J, Meyers DA, MacKinnon DF, Simpson S, McInnis MG, Rice JP, Goate A, et al. Initial genome screen for bipolar disorder in the NIMH genetics initiative pedigrees: Chromosomes 2, 11, 13, 14, and X. Am J Med Genet. 1997;74(3):263–269. [PubMed] [Google Scholar]
- Stork C, Renshaw PF. Mitochondrial dysfunction in bipolar disorder: Evidence from magnetic resonance spectroscopy research. Mol Psychiatry. 2005;10(10):900–919. doi: 10.1038/sj.mp.4001711. [DOI] [PubMed] [Google Scholar]
- Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447(7145):661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RS, Cheng L, Mudge AW, Harwood AJ. A common mechanism of action for three mood-stabilizing drugs. Nature. 2002;417(6886):292–295. doi: 10.1038/417292a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.