

Abstract
The secreted molecule fibroblast growth factor 9 (FGF9) plays a critical role in testis determination in the mouse. In embryonic gonadal somatic cells it is required for maintenance of SOX9 expression, a key determinant of Sertoli cell fate. Conditional gene targeting studies have identified FGFR2 as the main gonadal receptor for FGF9 during sex determination. However, such studies can be complicated by inefficient and variable deletion of floxed alleles, depending on the choice of Cre deleter strain. Here, we report a novel, constitutive allele of Fgfr2, hobbyhorse (hob), which was identified in an ENU-based forward genetic screen for novel testis-determining loci. Fgr2hob is caused by a C to T mutation in the invariant exon 7, resulting in a polypeptide with a mis-sense mutation at position 263 (Pro263Ser) in the third extracellular immunoglobulin-like domain of FGFR2. Mutant homozygous embryos show severe limb and lung defects and, when on the sensitised C57BL/6J (B6) genetic background, undergo complete XY gonadal sex reversal associated with failure to maintain expression of Sox9. Genetic crosses employing a null mutant of Fgfr2 suggest that Fgr2hob is a hypomorphic allele, affecting both the FGFR2b and FGFR2c splice isoforms of the receptor. We exploited the consistent phenotype of this constitutive mutant by analysing MAPK signalling at the sex-determining stage of gonad development, but no significant abnormalities in mutant embryos were detected.
Introduction
Fibroblast growth factors (FGF) function in numerous processes throughout embryonic development, such as the induction and patterning of germ cell layers, body axis formation and organogenesis [1]. Out of 22 human and mouse FGFs, 18 bind to a distinct set of cell-surface FGF receptors (FGFRs) to initiate intra-cellular signalling that results in cellular responses, including cell proliferation and differentiation [2]. Our understanding of the physiological roles of FGF ligands and their receptors has been helped enormously by the study of mouse knockouts [1]. Ranging from early embryonic lethality to adult metabolic abnormalities, these mutant phenotypes reveal the breadth of impact of FGF signalling.
Testis determination in the embryo normally requires the Y-linked gene Sry to initiate the commitment of somatic cells in the developing bipotential gonad to the Sertoli cell fate [3]. SRY effects this commitment through its positive effects on the expression of Sox9, a gene that is itself necessary and sufficient for testis development [4]. The analysis of mice lacking FGF9 first revealed a role for this signalling pathway in testis determination. Fgf9-deficient animals die around birth due to severe lung hypoplasia and, on a mixed genetic background, XY embryos exhibit a range of gonadal abnormalities ranging from testicular hypoplasia to complete sex reversal [5]. On the C57BL/6J (B6) background, which is sensitised to disruptions to testis determination, XY Fgf9-deficient embryos consistently exhibit gonadal sex reversal, indicating that some of the earliest processes in testis determination are disrupted by the absence of FGF9 [6]. Subsequent studies revealed an important role for FGF9 in maintaining high levels of Sox9 expression in the developing XY gonad, mediated at least partly by its inhibitory effects on ovary-determining genes such as Wnt4 [7], [8]. It has also been proposed that the rapid diffusion of secreted FGF9 along the long, thin gonad at around 11.5 dpc prevents any appreciable delay in the gonadal poles receiving the masculinising signal begun by expression of SRY at the centre of the gonad [9]. Any such delay may result in ovotestis or ovary development in an XY embryo due to the restricted time window that is thought to define the competence of cells to respond to SRY and its downstream effectors [10].
FGF9 acts as a paracrine FGF, mediating its effects locally by binding to and activating one of four tyrosine kinase FGFRs, using heparin sulphate proteoglycan (HSPG) cofactor-association as a means of regulating ligand distribution and receptor binding [11]. Loss-of-function genetic studies have identified FGFR2 as the likely receptor for embryonic gonadal FGF9. Embryos lacking FGFR2 die mid-gestation, at around 10.5 dpc, precluding a study of the effects of this loss on testis determination. Conditional gene targeting revealed partial XY gonadal sex reversal when Fgfr2 deletion was restricted temporally, from around 10.5 dpc onwards, or spatially, to gonadal somatic cells [12]. Similar XY gonadal defects, including ovotestis and ovary development, were observed after mosaic deletion of Fgfr2 in the embryo using Ck19Cre [13]. Limb and lung defects were also common in these embryos. Expression analysis in isolated pre-Sertoli cells indicates that one of the two isoforms of Fgfr2, Fgfr2c, is found in this lineage [12]. In contrast, both the Fgfr2b and Fgfr2c isoforms are detectable in other gonadal cells. Thus, these data suggest that FGFR2 is required for Sertoli cell differentiation and testis development through its activation by FGF9 ligand.
Despite the wealth of data concerning FGF signalling and its role in testis determination, the mechanism by which the gonadal FGF signal is transduced intracellularly remains unclear. In other contexts, a number of pathways are implicated, including PI3K-AKT and RAS-MAPK signalling [2]. Here, we report the identification of a novel, constitutive mutation of Fgfr2, hobbyhorse (hob). Fgfr2hob homozygotes exhibit severe lung hypoplasia and absence of limbs, in addition to complete XY gonadal sex reversal on the sensitised B6 background. We exploit the phenotypic robustness of this model of FGF-dependent sex reversal to address the role of MAPK signalling in transducing the FGF signal during testis determination.
Materials and Methods
Mouse strains used and genotyping
All animal experimentation was approved by the Animal Welfare and Ethical Review Body (AWERB) at MRC Harwell and mice used in this study were bred with licensed approval from the UK Home Office (PPL 30/2877). Mice were housed in individually ventilated cages (IVCs) in a specific pathogen-free (SPF) environment. Further details of micro- and macro-environmental conditions are available on request. Adult mice were humanely sacrificed by dislocation of the neck, confirmed by palpation, and embryos were decapitated in ice-cold, phosphate-buffered saline solution. Mice harbouring Fgfr2tm1.1Dor were generated by breeding a floxed allele of Fgfr2 (Fgfr2tm1Dor/J, The Jackson Laboratory) with the ubiquitous Cre deleter FVB/N-Tg(ACTB-Cre)2Mrt/J (The Jackson Laboratory) after at least three generations of backcrossing of each strain to C57BL/6J. Cre-mediated deletion of the floxed locus removes exons 7–10, generating a non-functional Fgfr2 allele. Presence of the Fgfr2hob allele was detected with a LightScanner assay using the primers Fgfr2ex7F 5′-CCTTTCTCCATCAGAACGGTCA-3′ and Fgfr2ex7R 5′-CTGTAAACCTTGCAGACAAACTC-3′ with mutant probe: Fgfr2ex7 PrR 5′-GAGGCATTTGCAGACAGTCCAGCTT-3′.
Details of ENU mutagenesis, the three-generation breeding scheme and genome-wide mapping of developmental mutants identified have been described elsewhere [14]–[16]. Adult mice and embryos were sexed by a PCR assay that simultaneously amplifies the Ube1y1 and Ube1x genes, using the following primer pair: 5′-TGGATGGTGTGGCCAATG-3′ and 5′-CACCTGCACGTTGCCCTT-3′ [17].
Generation of embryos and expression analyses
Noon on the day of the copulatory plug was counted as 0.5 dpc. Embryos were staged accurately based on the number of tail somites (ts) or limb and gonad morphology. In the case of hob/hob embryos, somites were counted from the presumptive hindlimb bud, which appears as a thickening of the body wall in the area of the hindlimb field and is present up to approximately 12.5 dpc. Wholemount in situ hybridization (WMISH) analysis of embryonic tissues was performed as previously described [18]. Probes for Sox9 [19], Sry [20], Insl3 [17], Wnt4 and Stra8 [14], [21] have been previously described. WMISH was performed on at least three independent gonad samples from each embryonic class.
Immunohistochemistry
Antibodies to the following proteins were utilised in this study: SOX9 (Millipore, #AB5535); AMH (Santa Cruz, #sc28912); FOXL2 (a kind gift from Dagmar Wilhelm and Peter Koopman [22]); SRY (a kind gift from Makoto Tachibana, Kyoto University [23]); Cell Signaling antibodies - p38 MAPK (CST9212), phospho-p38 MAPK (CST9215), ERK (CST9102) and phospho-ERK (CST4377); 12G10 alpha-tubulin (Developmental Studies Hybridoma Bank, University of Iowa). Immunostaining was performed on sectioned, paraffin wax-embedded tissue from two independent gonad samples using the above primary antibodies (1∶100) and Alexa Fluor 594 (AMH, SOX9) or 488 (FOXL2, SRY) conjugated secondary antibodies (1∶200). Images were captured using a Zeiss 710 multiphoton microscope.
Protein detection by Simple Western analysis
Pooled sub-dissected gonads (n = 8) were lysed using 50 µl RIPA buffer (150 mM NaCl, 1% NP40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 7.5) supplemented with Aqueous and DMSO inhibitor cocktails (Protein Simple), and cell debris removed by centrifugation. Lysates (7.5 µl) were mixed with 2.5 µl of Simple Western sample dilution buffer (Protein Simple) containing reducing agent and fluorescent standards, and denatured at 95°C for 5 min. Lysates (10 µl), primary antibodies, horseradish peroxidase–conjugated secondary antibodies, separation matrix, stacking matrix and chemiluminescent substrate were dispensed into designated wells in a 384-well assay plate. Simple Western assay buffers, capillaries and the prepared assay plate were placed in Peggy instrument (Protein Simple), which carries out all assay steps automatically for up to 8 cycles. Briefly, proteins were separated through a size-resolving matrix in capillaries, immobilized to the inner capillary wall, incubated with primary and secondary antibodies before detection using chemiluminescence. Signal and quantitation of immunodetected proteins were generated automatically at the end of the run. Statistical analysis was performed on data from three biological replicates (24 gonad samples in total).
Results
Identification of the hobbyhorse (hob) mutation: an allele of Fgfr2 that disrupts limb, lung and gonad development
We have previously described a forward genetic screen employing N-ethyl-N-nitrosourea (ENU) mutagenesis aimed at identifying novel loci required for mouse development. This screen identified recessive mutations in genes functioning in neural tube closure and patterning [15], left-right patterning [16] and sex determination [14]. We identified a second pedigree in this screen (RECB/135) containing embryos with gonadal abnormalities at 14.5 dpc. Affected XY gonads exhibited a range of morphological abnormalities, ranging from disruption to cord formation through to complete absence of testis cords (Fig 1A). This variability is likely to be due to the mixed genetic background on which the screen was performed. Affected embryos also lacked limbs and had severely hypoplastic lungs (Fig 1B,C); due to their external appearance the mutants were called hobbyhorse (hob).
Figure 1. The hobbyhorse (hob) mutation disrupts XY sex determination and is caused by an ENU-induced point mutation of Fgfr2.

A) A wild-type XY gonad (left) showing characteristic testicular morphology at 14.5 dpc, in contrast to two XY hobbyhorse mutants identified in a forward genetic screen, which have disrupted testis cords (centre) or lack cords entirely (right). All gonads shown are after wholemount in situ hybridisation (WMISH) with a Sox9 probe. B) A hobbyhorse mutant (right) lacks limbs. A wild-type embryo is also shown (left). C) Absence of lung development in a hobbyhorse embryo (right), in contrast to normal lungs at the same stage (left). D) Sequence trace showing homozygosity for a C to T mutation (asterisk) in exon 7 of Fgfr2 of a hobbyhorse embryo. Upper trace is wild-type, lower trace is hobbyhorse. E) The proline residue that is mutated in the hob allele is highly conserved in vertebrates. Mm, Mus musculus; Hs, Homo sapiens; Gg, Gallus gallus; Xl, Xenopus leavis; Dr, Danio rerio. F) Diagrammatic representation of FGFR2 and its domain structure in the FGFR2b and FGFR2c isoforms. The hob mutation (asterisk) resides in the third extracellular immunoglobulin-like domain, encoded by the invariant exon 7.
Using seven affected embryos and a genome-wide panel of polymorphic markers, we mapped the mutation responsible for this phenotype to distal mouse chromosome 7, between the markers rs6317573 and rs32097269 (data not shown). The phenotypic combination of lung, limb and gonadal abnormalities was strongly reminiscent of defects in FGF/FGFR signalling previously reported [5], [12], [13], and inspection of the critical linkage region identified the Fgfr2 gene. We amplified and sequenced Fgfr2 exons in genomic DNA from affected and unaffected embryos and identified a homozygous C to T mutation in exon 7 of all, and only, affected individuals (Fig 1D). This mutation results in a predicted proline to serine mis-sense amino acid substitution at position 263 (P263S) of the FGFR2 polypeptide chain. The affected proline residue is highly conserved in vertebrates (Fig1E) and resides in the third extracellular immunoglobulin-like domain (IgIII, D3) of FGFR2 (Fig 1F). Moreover, exon 7 is an invariant exon in Fgfr2 mRNA isoforms and so this mutation is predicted to affect both the Fgfr2b and Fgfr2c isoforms. The extracellular region of FGFRs encompassing the second and third Ig-like domains (D2, D3) and the intervening linker region is necessary and sufficient for FGF ligand binding [24]. Specificity of ligand binding by FGFR2 is determined by alternate splicing in the D3 domain.
Complete gonadal sex reversal in XY Fgfr2hob/hob embryos on C57BL/6J
We crossed the Fgfr2hob mutation to C57BL/6J (B6) for at least three generations to remove contaminating mutations and to examine gonad development in homozygotes on this sensitised genetic background. Adult heterozygotes were fertile and exhibited no overt abnormalities, suggesting that Fgfr2hob is a genuine recessive mutation. B6 XY embryos homozygous for the Fgfr2hob mutation exhibited complete gonadal sex reversal, as evidenced by the ovarian morphology and the absence of the Sertoli cell marker, Sox9, at 14.5 dpc and the high levels of the ovarian somatic marker Wnt4 (Fig 2A); in addition, expression of Stra8 was also observed in XY mutant gonads, indicating entry of germ cells into meiosis and thus germ cell sex-reversal (Fig 2A). Homozygous embryos still lacked limbs; a presumptive hindlimb bud, which appears as a thickening of the body wall in the area of the hindlimb field, was present up to approximately 12.5 dpc, as previously described in some embryos homozygous for a null allele of Fgfr2 (data not shown) [25]. Homozygous embryos also had hypoplastic lungs (data not shown) but were otherwise viable until late in gestation. In order to confirm that homozygosity for Fgfr2hob alone accounted for the mutant phenotype, we performed a genetic complementation test with an Fgfr2 null allele (Fgfr2tm1.1Dor). We first confirmed that embryos homozygous for Fgfr2tm1.1Dor were dying from around 10.5–11.0 dpc and lacked limb buds (Fig 2B). We then generated embryos carrying a single copy of both Fgfr2hob and Fgfr2tm1.1Dor. In contrast to Fgfr2tm1.1Dor homozygotes, doubly heterozygous embryos were alive at 14.5 dpc, though they were slightly smaller than wild-type littermates and had no limbs and exhibited lung hypoplasia (Fig 2C and data not shown). XY gonads from doubly heterozygous embryos had an ovarian morphology and exhibited negligible Sox9 expression at 13.5 dpc and Stra8-positive germ cells at 14.5 dpc (Fig 2D). From this complementation assay we conclude that homozygosity for the Fgfr2hob mutation accounts for limb, lung and gonadal abnormalities identified in the RECB/135 pedigree. Moreover, based on the fact that Fgfr2hob/m1.1Dor double heterozygotes survive for longer than Fgfr2tm1.1Dor homozygotes, we infer that Fgfr2hob is a hypomorphic allele. Genetic interaction tests involving Fgfr2hob and a null allele of Map3k4 [14] did not, in contrast, generate embryonic gonads with overt abnormalities (Fig S1).
Figure 2. Characterisation of XY Fgfr2hob/hob embryonic gonad development on the C57BL/6J (B6) background and complementation test with the Fgfr2tm1.1Dor null allele.
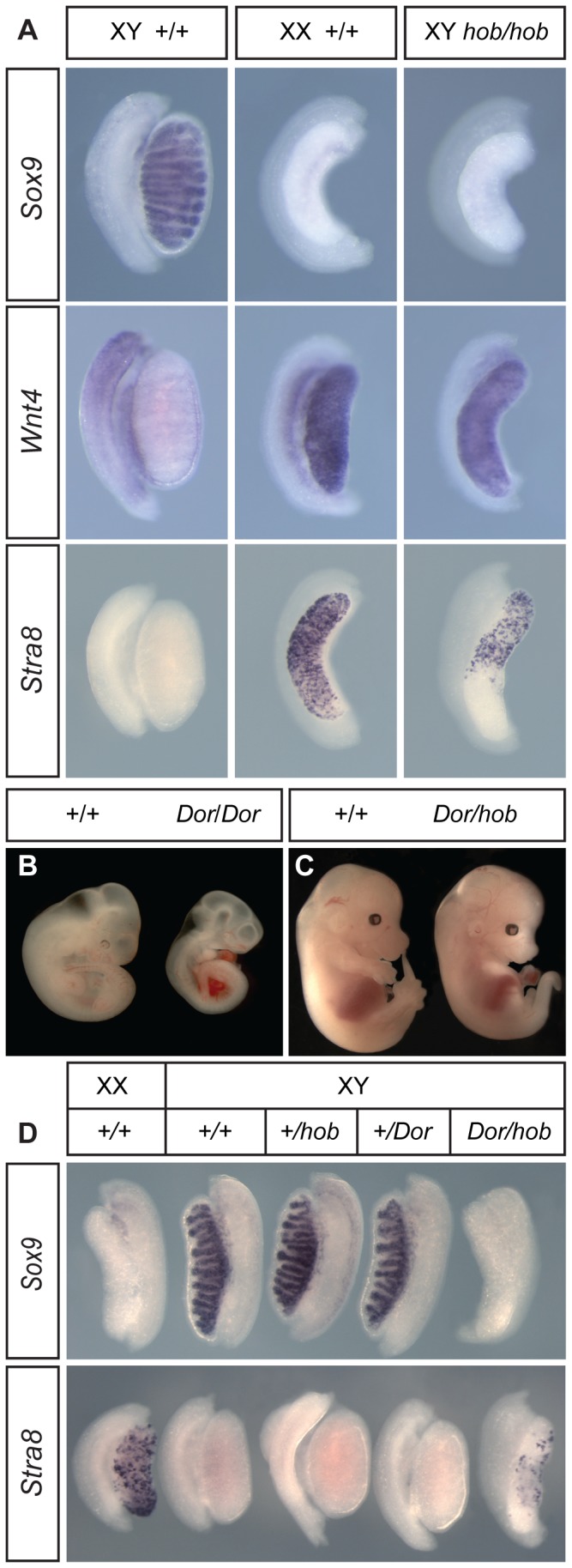
A) WMISH analysis of gonads at 14.5 dpc from XY wild-type, XX wild-type and XY Fgfr2hob/hob embryos using a marker of the Sertoli cell lineage (Sox9), ovarian somatic cells (Wnt4) and meiotic germ cells (Stra8). B) Embryos homozygous for the Fgfr2tm1.1Dor allele (Dor/Dor) are much smaller than wild-type controls (+/+) at 11.5 dpc and also lack limbs. C) Embryos at 14.5 dpc doubly heterozygous for the Fgfr2hob and Fgfr2tm1.1Dor alleles (Dor/hob) lack limbs and are noticeably smaller than wild-type controls (+/+). D) Upper panel: Sox9 WMISH of 13.5 dpc embryonic gonads from control and XY Fgfr2tm1.1Dor/hob doubly heterozygous embryos; lower panel: Stra8 WMISH of 14.5 gonads from embryos of same genotypes as upper panel. The developmental stage of the doubly heterozygous gonad in the lower panel appears significantly retarded when compared to the XX control.
Cellular and molecular phenotyping of XY Fgfr2hob/hob gonads
We then performed a more detailed analysis of gonad development in XY Fgfr2hob/hob homozygous embryos on B6 between 11.5 dpc and 14.5 dpc. Immunostaining revealed anti-Müllerian hormone (AMH), a marker of Sertoli cells, in the testis cords of wild-type gonads at 12.5 dpc, but none was detected in mutant gonadal tissue (Fig 3A–C). In contrast, the ovarian somatic marker FOXL2 was detected in mutant gonads at 12.5 dpc, but not in wild-type XY controls (Fig 3D–F). Sex reversal extended to the steroidogenic lineage, with no Insl3 transcript detected in XY mutants at 14.5 dpc (Fig 3G–I). However, Oct4 expression was detectable in both XY and XX Fgfr2hob/hob homozygotes at 11.5 dpc (Fig 3J–M) and 13.5 dpc (Fig 3N–Q), suggesting no overt disruption to germ cell development at this stage.
Figure 3. Complete XY gonadal sex reversal in Fgfr2hob/hob embryos on B6.
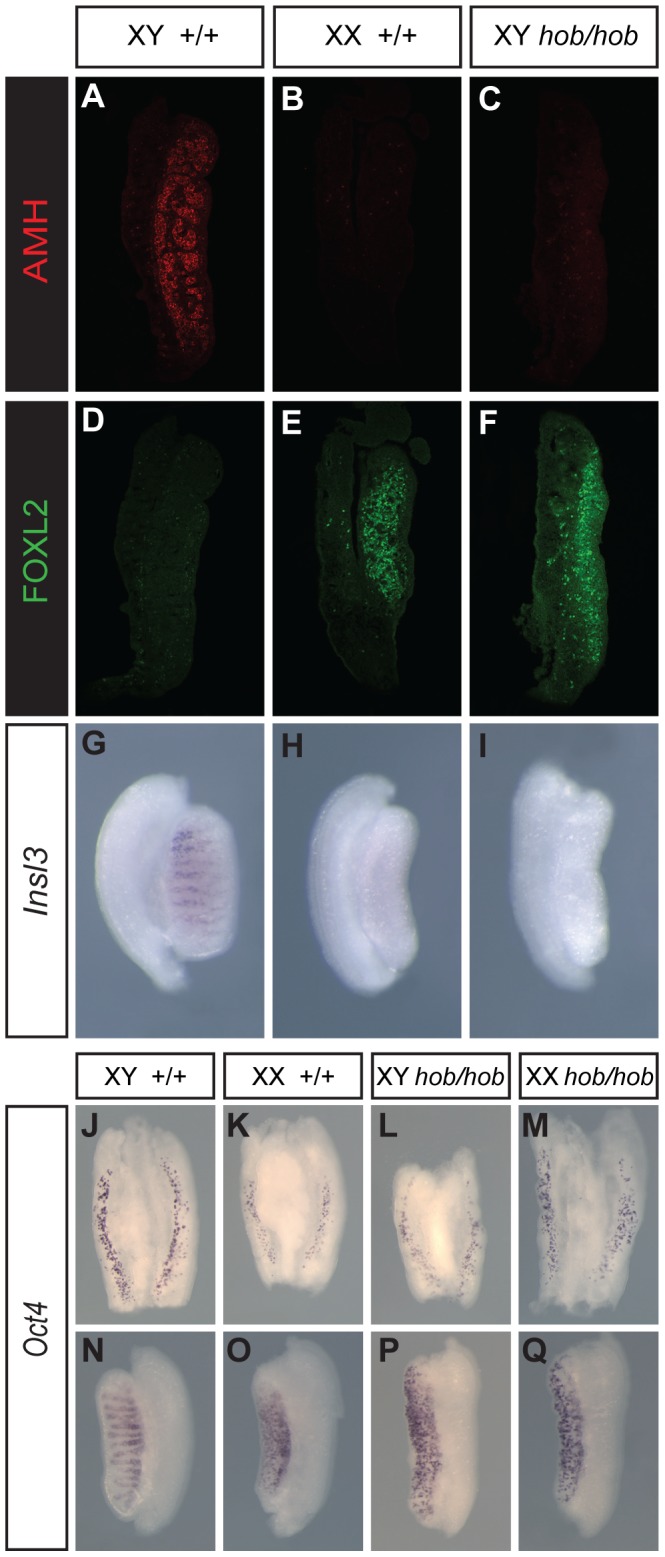
A–C) Immunostaining with anti-AMH antibody of gonadal sections from XY wild-type (A), XX wild-type (B) and XY Fgfr2hob/hob (C) embryos at 12.5 dpc. D–F) anti-FOXL2 immunostaining of samples equivalent to those in A–C. G–I) WMISH for Insl3 (a marker of Leydig cells) of gonads with same genotypes as A–C. J–M) Oct4 WMISH of 11.5 dpc (17 ts) gonads from control XY (J), XX (K), XY Fgfr2hob/hob and XX Fgfr2hob/hob gonads. N–Q) Oct4 WMISH of 13.5 dpc gonads from embryos of the same genotype as J-M.
Previously, loss of FGF9 has been associated with disruption to Sox9 expression, but Sry expression has been reported to be unaffected [7]. We first examined Sry in XY Fgfr2hob/hob embryos using WMISH and anti-SRY immunostaining of gonads at 11.5 dpc (from 16–18 tail somites (ts)). Neither of these methods revealed any disruption to Sry expression (Fig 4A–C). This is in agreement with a previous report on one Fgfr2 CKO [13]. In contrast, Sox9 expression, as assayed by WMISH and immunostaining, whilst initially up-regulated at 18 ts (Fig 4D–F), was observed at greatly reduced levels at 23 ts (around 11.75 dpc) in mutant gonads (Fig 4G–I) and at 12.5–13.0 dpc (Fig 4J–L), consistent with data reported concerning Sox9 expression in Fgf9-deficient gonads [7]. Thus, data concerning the molecular basis of sex reversal in XY Fgfr2hob homozygotes indicate normal up-regulation of Sox9, but a failure to maintain significant levels beyond 11.5 dpc.
Figure 4. Normal Sry expression, but disrupted Sox9 expression, in XY Fgfr2hob/hob embryonic gonads.
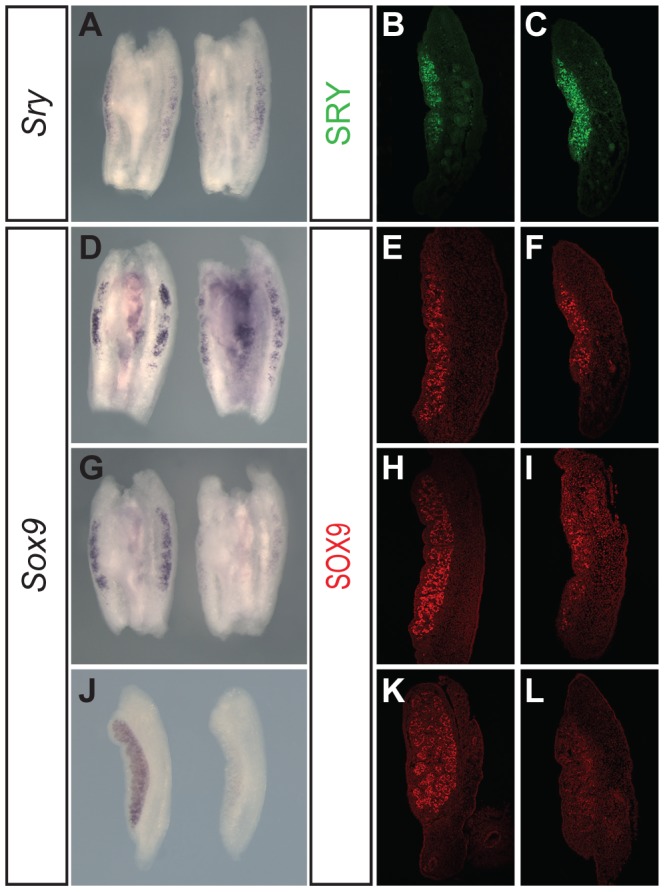
A) Sry WMISH at 11.5 dpc (16 ts) showing expression in XY wild-type (left) and XY Fgfr2hob/hob (right) gonads. B, C) anti-SRY immunostaining at 18 ts in wild-type (B) and XY Fgfr2hob/hob (C) gonads. D) Sox9 WMISH at 18 ts with tissue samples as described in (A). E, F) anti-SOX9 immunostaining at 18 ts in XY wild-type (E) and XY Fgfr2hob/hob (F) gonads. G) Sox9 WMISH at 23 ts in XY wild-type (left) and XY Fgfr2hob/hob (right) gonads. H, I) anti-SOX9 immunostaining at 23 ts in XY wild-type (H) and XY Fgfr2hob/hob (I) gonads. J) Sox9 WMISH at 12.5 dpc in XY wild-type (left) and XY Fgfr2hob/hob (right) gonads. K, L) anti-SOX9 immunostaining at 13.0 dpc in wild-type (K) and XY Fgfr2hob/hob (L) gonads.
MAPK signalling in Fgfr2hob/hob homozygous gonads at 11.5 dpc
The FGF signal is transduced intra-cellularly by means of a number of pathways, including PI3K-AKT and RAS-MAPK signalling [2]. Based on the profile of Sox9 expression in mutant gonads, which indicates defective expression from around 11.75 dpc, we performed a quantitative analysis of MAPK signalling in Fgfr2hob/hob homozygous gonads at around 11.5 dpc (16–18 ts) in order to identify potential abnormalities in signalling that might result in subsequent defects in Sox9 expression. Gonadal tissue from XY and XYhob/hob embryos was dissected away from the mesonephros and protein lysates were examined using a size-based, capillary electrophoresis method for immunodetection of the phosphorylated and non-phosphorylated forms of p38 MAPK and ERK 1/2. Both of these MAPKs are activated in developmental contexts by the FGF signal [26], [27]. At 16–18 ts no significant differences were observed between control and mutant gonadal tissue samples in the levels of phospho-p38 MAPK (Fig 5A, B). Unfortunately, we were unable to detect p-ERK at this stage in gonadal tissue, despite the fact that ERK itself was detectable and control samples from cultured cells indicated that the anti-p-ERK antibody was working reliably (data not shown).
Figure 5. Quantitation of phospho-p38 MAPK (p-p38) levels in gonadal samples at 11.5 dpc (16–18 ts) in XY wild-type and Fgfr2hob/hob gonads.
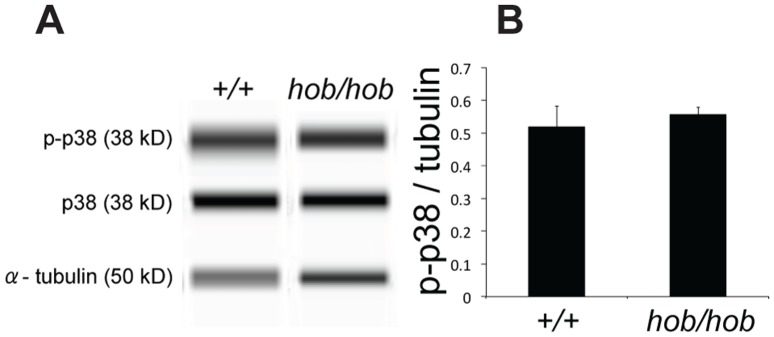
A) Lane view images showing Simple Western detection of p-p38, p38, and α-tubulin. B) Graph showing the ratio of p-p38 to tubulin in the two gonadal genotypes. The ratio of p-p38 to p38 was similarly unaltered. Errors were calculated using standard error mean.
Discussion
Here we report the identification, in a mouse forward genetic screen, of a novel sex-reversing mutant allele of Fgfr2. Previous studies of Fgfr2 function in testis determination have relied on conditional gene targeting, with the attendant variability in efficiency and site of gene ablation resulting in some phenotypic variation. Homozygous deletion of floxed Fgfr2 alleles using Sf1-Cre, which is expressed primarily in somatic cells within the gonad, resulted in partial XY sex reversal, with ovotestis formation commonly observed at 15.5 dpc [12]. Deletion of Fgfr2 with Ck19:Cre, a line that deletes in epiblast-derived cells in a mosaic fashion, also resulted in variable and partial XY gonadal sex reversal [13]. Complete sex reversal was observed after Fgfr2 deletion at 10.5 dpc with a heat-shock inducible Cre line, Hs-Cre, but there were variable numbers of SOX9-positive cells observed in mutant gonads [12]. Incomplete deletion of floxed alleles may contribute to phenotypic variability, as might residual genetic background variation. The constitutive Fgfr2hob allele affords a detailed study of the role of FGFR2 in mouse sex determination on a stable genetic and phenotypic background. The value of forward genetics in the identification of developmental loci, including the provision of new and useful alleles of genes of known function, is underlined by this study.
Our data suggest that Fgfr2hob is a hypomorphic mutant allele, perhaps disrupting interaction of FGFR2 with its ligand, given the position of the altered amino acid in the extracellular immunoglobulin-like domain. This prediction requires testing in a future study. The extended viability of Fgfr2hob/hob embryos, in comparison to Fgfr2tm1.1Dor/tm1.1Dor homozygotes that completely lack FGFR2 function, is also suggestive of an overcoming of an early placental defect in Fgfr2hob/hob embryos. It is this placental defect that is thought to account for the death of Fgfr2tm1.1Dor/tm1.1Dor homozygotes at around 11.5 dpc. The amelioration of this placental defect is in contrast to the persistent absence of limbs and severe lung hypoplasia that is a common phenotypic feature of homozygotes for both alleles. This discrepancy might be explained by a lower threshold of FGFR2 function in the developing placenta, greater functional redundancy with respect to FGF receptor signalling in this tissue or distinct ligand sensitivities to the hob mutation. With respect to the relative importance of the Fgfr2b and Fgfr2c isoforms in testis determination, our data do not allow us to discriminate because we predict that the hob allele disrupts both isoforms of the receptor. It will be important to carefully analyse gonad development in mice specifically lacking either the Fgfr2b or Fgfr2c isoform on the B6 genetic background in order to determine their individual contributions to testis determination.
Gonadal sex reversal in XY Fgfr2hob/hob embryos is associated with a failure to maintain high levels of the key testis determinant, SOX9, after 11.5 dpc. In contrast, the pro-ovarian gene Wnt4 is elevated, indicating sex reversal of the supporting cell lineage. These data are consistent with observations of failure to maintain Sox9 expression in Fgf9-deficient XY gonads [7]. In contrast, Sry expression appears to remain unaffected by disruption of FGF9/FGFR2 signalling. Recent studies involving phenotypic analysis of XY embryos lacking Fgf9 and Wnt4 suggest that the primary role of FGF9 may be to inhibit the activity of WNT/β-catenin signalling [8]. The mechanistic basis of this at the molecular level remains unclear, but Fgfr2hob/hob embryos may offer a useful tool for investigating such antagonistic interactions of the testis- and ovary-determining gene regulatory networks (Carre & Greenfield). However, an inhibitory role for FGF9/FGFR2 signalling may suggest that the search for targets of FGF-dependent signalling in the gonad should be extended to proteins associated with ovary development and canonical WNT signalling more generally.
We have exploited the relative stability of the XY Fgfr2hob/hob embryonic phenotype to perform careful quantitation of MAPK signalling during the testis-determining stages of gonadogenesis and report no significant deficits at early stages (16–18 ts). We have previously reported a role for p38 MAPK signalling in regulation of Sry expression and testis determination [28]. The unaltered levels of phospho-p38 MAPK at 16-18 ts in Fgfr2hob/hob embryonic gonads are therefore consistent with the observation of normal Sry expression in this sex-reversing mutant. Technical improvements may be required to detect any p-ERK present and thereby determine whether a role exists for this MAPK in regulating expression of Sox9 during testis determination, as reported in other contexts [29]. It will also be important to analyse the phosphorylation status of any transcription factor implicated in regulation of Sox9 or Wnt4 expression in Fgfr2hob/hob embryos, or cellular models derived from these, in future studies.
Supporting Information
Absence of genetic interaction between Fgfr2hob (hob) and Map3k4tm1Flv ( M4 ). Stra8 WMISH of control gonads and gonads from doubly heterozygous embryos (+/hob, +/M4). Expression is only detected in control XX gonads.
(TIF)
Acknowledgments
We thank staff of the Mary Lyon Centre (MLC) at Harwell for animal husbandry support, in particular Jackie Harrison and Lee Kent in Ward 5. We thank Peter Koopman and Dagmar Wilhelm for kindly providing anti-FOXL2 antibody and Makoto Tachibana for anti-SRY antibody. We thank staff of the MLC histology facility for sectioning, staff of the GEMS facility for genotyping, and Martin Fray and his staff in the FESA Core for line rederivations.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are included within the paper.
Funding Statement
This work was funded by the UK Medical Research Council through Core funding to AG at its Mammalian Genetics Unit, Harwell (MC_A390_5RX50). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Itoh N, Ornitz DM (2011) Fibroblast growth factors: from molecular evolution to roles in development, metabolism and disease. J Biochem 149: 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Goetz R, Mohammadi M (2013) Exploring mechanisms of FGF signalling through the lens of structural biology. Nat Rev Mol Cell Biol 14: 166–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Koopman P, Gubbay J, Vivian N, Goodfellow P, Lovell-Badge R (1991) Male development of chromosomally female mice transgenic for Sry . Nature 351: 117–121. [DOI] [PubMed] [Google Scholar]
- 4. Sekido R, Lovell-Badge R (2008) Sex determination involves synergistic action of SRY and SF1 on a specific Sox9 enhancer. Nature 453: 930–934. [DOI] [PubMed] [Google Scholar]
- 5. Colvin JS, Green RP, Schmahl J, Capel B, Ornitz DM (2001) Male-to-female sex reversal in mice lacking fibroblast growth factor 9. Cell 104: 875–889. [DOI] [PubMed] [Google Scholar]
- 6. Schmahl J, Kim Y, Colvin JS, Ornitz DM, Capel B (2004) Fgf9 induces proliferation and nuclear localization of FGFR2 in Sertoli precursors during male sex determination. Development 131: 3627–3636. [DOI] [PubMed] [Google Scholar]
- 7. Kim Y, Kobayashi A, Sekido R, DiNapoli L, Brennan J, et al. (2006) Fgf9 and Wnt4 act as antagonistic signals to regulate mammalian sex determination. PLoS Biol 4: e187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jameson SA, Lin YT, Capel B (2012) Testis development requires the repression of Wnt4 by Fgf signaling. Developmental Biology 370: 24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hiramatsu R, Harikae K, Tsunekawa N, Kurohmaru M, Matsuo I, et al. (2010) FGF signaling directs a center-to-pole expansion of tubulogenesis in mouse testis differentiation. Development 137: 303–312. [DOI] [PubMed] [Google Scholar]
- 10. Hiramatsu R, Matoba S, Kanai-Azuma M, Tsunekawa N, Katoh-Fukui Y, et al. (2008) A critical time window of Sry action in gonadal sex determination in mice. Development 136: 129–138. [DOI] [PubMed] [Google Scholar]
- 11. Harada M, Murakami H, Okawa A, Okimoto N, Hiraoka S, et al. (2009) FGF9 monomer-dimer equilibrium regulates extracellular matrix affinity and tissue diffusion. Nat Genet 41: 289–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim Y, Bingham N, Sekido R, Parker KL, Lovell-Badge R, et al. (2007) Fibroblast growth factor receptor 2 regulates proliferation and Sertoli differentiation during male sex determination. Proc Natl Acad Sci U S A 104: 16558–16563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bagheri-Fam S, Sim H, Bernard P, Jayakody I, Taketo MM, et al. (2008) Loss of Fgfr2 leads to partial XY sex reversal. Dev Biol 314: 71–83. [DOI] [PubMed] [Google Scholar]
- 14. Bogani D, Siggers P, Brixey R, Warr N, Beddow S, et al. (2009) Loss of mitogen-activated protein kinase kinase kinase 4 (MAP3K4) reveals a requirement for MAPK signalling in mouse sex determination. PLoS Biol 7: e1000196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Patterson VL, Damrau C, Grimes DT, Paudyal A, Reeve B, et al. (2009) Mouse hitchhiker mutants have spina bifida, dorso-ventral patterning defects and polydactyly: Identification of Tulp3 as a novel negative regulator of the Sonic hedghog pathway. Hum Mol Genet 18: 1719–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Field S, Riley KL, Grimes DT, Hilton H, Simon M, et al. (2011) Pkd1l1 establishes left-right asymmetry and physically interacts with Pkd2. Development 138: 1131–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Warr N, Siggers P, Bogani D, Brixey R, Pastorelli L, et al. (2009) Sfrp1 and Sfrp2 are required for normal male sexual development in mice. Dev Biol 326: 273–284. [DOI] [PubMed] [Google Scholar]
- 18. Grimmond S, Van Hateren N, Siggers P, Arkell R, Larder R, et al. (2000) Sexually dimorphic expression of protease nexin-1 and vanin-1 in the developing mouse gonad prior to overt differentiation suggests a role in mammalian sexual development. Hum Mol Genet 9: 1553–1560. [DOI] [PubMed] [Google Scholar]
- 19. Wright E, Hargrave MR, Christiansen J, Cooper L, Kun J, et al. (1995) The Sry-related gene Sox-9 is expressed during chondrogenesis in mouse embryos. Nature Genetics 9: 15–20. [DOI] [PubMed] [Google Scholar]
- 20. Bullejos M, Koopman P (2001) Spatially dynamic expression of Sry in mouse genital ridges. Dev Dyn 221: 201–205. [DOI] [PubMed] [Google Scholar]
- 21. Warr N, Bogani D, Siggers P, Brixey R, Tateossian H, et al. (2011) Minor abnormalities of testis development in mice lacking the gene encoding the MAPK signalling component, MAP3K1. PLos One 6: e19572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wilhelm D, Washburn LL, Truong V, Fellous M, Eicher EM, et al. (2009) Antagonism of the testis- and ovary-determining pathways during ovotestis development in mice. Mech Dev 126: 324–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Warr N, Siggers P, Carre GA, Bogani D, Brixey R, et al. (2014) Transgenic Expression of Map3k4 Rescues T-associated Sex Reversal (Tas) in Mice. Hum Mol Genet. 23: 3035–3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mohammadi M, Olsen SK, Ibrahimi OA (2005) Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev 16: 107–137. [DOI] [PubMed] [Google Scholar]
- 25. Xu X, Weinstein M, Li C, Naski M, Cohen RI, et al. (1998) Fibroblast growth factor receptor 2 (FGFR2)-mediated reciprocal regulation loop between FGF8 and FGF10 is essential for limb induction. Development 125: 753–765. [DOI] [PubMed] [Google Scholar]
- 26. Keren-Politansky A, Keren A, Bengal E (2009) Neural ectoderm-secreted FGF initiates the expression of Nkx2.5 in cardiac progenitors via a p38 MAPK/CREB pathway. Dev Biol 335: 374–384. [DOI] [PubMed] [Google Scholar]
- 27. Suzuki-Hirano A, Harada H, Sato T, Nakamura H (2010) Activation of Ras-ERK pathway by Fgf8 and its downregulation by Sprouty2 for the isthmus organizing activity. Dev Biol 337: 284–293. [DOI] [PubMed] [Google Scholar]
- 28. Warr N, Carre GA, Siggers P, Faleato JV, Brixey R, et al. (2012) Gadd45gamma and Map3k4 interactions regulate mouse testis determination via p38 MAPK-mediated control of Sry expression. Developmental Cell 23: 1020–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Murakami S, Kan M, McKeehan WL, de Crombrugghe B (2000) Up-regulation of the chondrogenic Sox9 gene by fibroblast growth factors is mediated by the mitogen-activated protein kinase pathway. Proc Natl Acad Sci U S A 97: 1113–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Absence of genetic interaction between Fgfr2hob (hob) and Map3k4tm1Flv ( M4 ). Stra8 WMISH of control gonads and gonads from doubly heterozygous embryos (+/hob, +/M4). Expression is only detected in control XX gonads.
(TIF)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are included within the paper.