

Abstract
The Repeat Expansion Diseases (REDs) are human genetic disorders that arise from expansion of a tandem repeat tract. The Fragile X-related disorders are members of this disease group in which the repeat unit is CGG/CCG and is located in the 5′ untranslated region of the FMR1 gene. Affected individuals often show mosaicism with respect to repeat number resulting from both expansion and contraction of the repeat tract, however, the mechanism responsible for these changes in repeat number are unknown. Work from a variety of model systems suggests that Transcription Coupled Repair (TCR) may contribute to repeat instability in diseases resulting from CAG/CTG-repeat expansion. To test whether TCR could contribute to repeat instability in the Fragile X-related disorders, we tested the effect of mutations in Csb (Cockayne Syndrome group B), a gene essential for TCR, in a knock-in mouse model of these disorders. We found that the loss of CSB affects expansions in a gender and cell type-specific manner. Our data also show an unanticipated gender difference in instability even in Csb+/+ animals that may have implications for our understanding of the mechanism of repeat expansion in the FX mouse model and perhaps for humans as well.
Keywords: CSB, ERCC6, Repeat Expansion, Fragile X-related, ataxia, primary ovarian insufficiency, transcription coupled repair
Introduction
The Repeat Expansion Diseases (REDs) are a large group of inherited human disorders that arise from expansion of a specific tandem repeat tract (see (Orr and Zoghbi, 2007; Usdin, 2008) for reviews). The Fragile X-related disorders, which include Fragile X syndrome (FXS; MIM# 300624), Fragile X-associated tremor/ataxia syndrome (FXTAS; (MIM# 300623)), and Fragile X-associated primary ovarian insufficiency (FXPOI), are members of this group (see (Willemsen et al., 2011) for recent review). All three disorders are caused by the inheritance of large numbers of CGG/CCG-repeats in the 5′ untranslated region of the X-linked Fragile X mental retardation 1 gene (FMR1; MIM# 309550). Most normal alleles have ~30 repeats (Strom et al., 2007). Such alleles are relatively stable. However, as the repeat number increases, so the instability of this repeat tract increases as does the likelihood that an individual will exhibit symptoms characteristic of one or more of the Fragile X-related disorders.
FXTAS is an adult onset neurodegenerative condition whose symptoms include intention tremor, cerebellar gait ataxia, parkinsonism, autonomic dysfunction, cognitive deficits and ultimately dementia (Hagerman and Hagerman, 2004). FXPOI is an ovarian dysfunction disorder involving infertility, irregular menses and an early menopause (Sherman, 2000). These disorders are seen in carriers of FX premutation (PM) alleles, alleles that have 55–200 repeats. In both cases, pathology is thought to be related to some, as yet unknown, deleterious consequence of expression of FMR1 mRNA with large numbers of CGG-repeats, a problem that is exacerbated by the repeat mediated hyper-expression of the PM allele (Tassone et al., 2000). FXTAS disease severity is correlated with the number of repeats in the allele. Larger repeat numbers are also associated with higher risk of both germ-line and somatic expansion (Fu et al., 1991). Germ-line expansion can generate Full mutation (FM) alleles that have >200 repeats. Such alleles are associated with repeat-mediated epigenetic silencing of the allele and this results in FXS, the most common heritable cause of intellectual disability and the most common monogenic cause of autism (Oberle et al., 1991; Pieretti et al., 1991; Verkerk et al., 1991).
In addition to expansions, the regression of PM alleles to normal alleles has also been reported (Cabanes et al., 2004; Jin et al., 2010; Likhite et al., 2004). Furthermore, many PM and FM carriers are mosaic, often showing a complex mixture of different PM and FM alleles (Araneda et al., 2005; Berger et al., 2012; Dietrich et al., 2013; Murdoch and Van Kirk, 1997; Newman et al., 2006; Taylor et al., 1999; Tome et al., 2013). This mosaicism is thought to arise from a series of expansion and contraction events that occur during the individual’s lifetime.
Most models for repeat expansion and contraction are based on the fact that all the repeats associated with the REDs form intrastrand structures like hairpins (reviewed in (Mirkin, 2006; Pearson et al., 2005)). These secondary structures are thought to form when the DNA is unpaired, for example during replication, transcription or DNA repair that involves strand-displacement synthesis or gap-repair. Studies in yeast and bacteria suggest that problems with DNA replication, repair or recombination could all potentially result in expansion and contractions (reviewed in (Mirkin, 2006; Pearson, et al., 2005)). However, the mechanism or mechanisms responsible for instability in the Repeat Expansion Diseases remains unknown.
Data from Drosophila, mouse and human cells in tissue culture has led to the suggestion that Transcription Coupled Repair (TCR), the nucleotide excision repair (NER) pathway that operates in transcribed regions of the genome, is important for repeat instability in the case of Repeat Expansion Disorders resulting from expansion of CAG/CTG repeats (Jung and Bonini, 2007; Kovtun et al., 2011; Lin et al., 2009; Lin and Wilson, 2007; Salinas-Rios et al., 2011). A role for TCR in repeat expansion in the Fragile X-related disorders would be consistent with the observation that FM alleles, the majority of which are transcriptionally silent, are more stable than the smaller, transcriptionally active PM alleles from which they are derived or those FM alleles that escape silencing (Wohrle et al., 1998; Wohrle et al., 2001; Wohrle et al., 1996). We have previously generated an FX PM knock-in mouse model, in which the normal CGG/CCG-repeat tract present in the WT mouse Fmr1 gene was replaced with an expanded tract in the PM size range (Entezam et al., 2007; Entezam et al., 2010; Entezam and Usdin, 2008; Entezam and Usdin, 2009; Lokanga et al., 2013a). To better understand the relationship between TCR and repeat instability in the FX PM mouse, we crossed these mice to mice with a null mutation in Csb (Cockayne Syndrome group B, a.k.a. ERCC6 (MIM# 609413)). CSB is DNA-dependent ATPase that is essential for TCR but whose mode of action is still unclear. Our data demonstrate that CSB does play a role in repeat expansion but its effect varies with gender and cell type. We also demonstrate a previously unappreciated gender difference in the intergenerational transmission of expanded alleles that may have interesting ramifications for the expansion mechanisms and risk assessment in humans.
Materials and Methods
Mice breeding and maintenance
The generation of the FX PM mice was described previously (Entezam, et al., 2007). Mice were maintained in accordance with the guidelines of the NIDDK Animal Care and Use Committee and with the Guide for the Care and Use of Laboratory Animals (NIH publication no. 85-23, revised 1996). Four Csb+/+ and 5 Csb−/− breeding pairs were used to generate a total of 132 Csb+/+ and 148 Csb−/− paternal transmissions. Older females were replaced with younger ones when the litter frequency dropped. Five Csb+/+ and 5 Csb−/− breeding pairs were used to generate a total of 123 Csb+/+ and 66 Csb−/− maternal transmissions. To avoid the confounding effect of age on expansion, comparisons between Csb+/+ and Csb−/− animals were carried out using a subset of Csb+/+ animals that had an age distribution that was indistinguishable from that of Csb−/− parents.
Determination of genotype and repeat number
Genomic DNA from mouse tail was prepared using KAPA Mouse Genotyping Kits (KAPA biosystems). Genomic DNA from other mouse tissue was prepared using Maxwell®16 Mouse Tail DNA Purification Kits (Promega). FX PM genotyping was carried out using the mouse Repeat PCR assay as described previously (Lokanga, et al., 2013a). Csb genotyping was carried out as described elsewhere (Berg et al., 2000). The determination of the repeat number was done using a fluorescent polymerase chain reaction (PCR) assay with primers whose binding sites were located immediately adjacent to the repeat tract and their 3′ ends were unique to the PM allele (Lokanga, et al., 2013a). Thus only the PM allele is amplified. The reaction products were then run on a 3130XL Genetic Analyzer and analyzed using GeneMapper® 4.0 (Life Technologies).
Statistical Analysis
Statistical analysis was carried out using a web-based version of the GraphPad QuickCalcs Software (http://www.graphpad.com/quickcalcs) and VassarStats (http://vassarstats.net). Since evidence suggests that expansions and contractions occur via a different mechanism (Entezam, et al., 2010; Entezam and Usdin, 2008; Entezam and Usdin, 2009; Lokanga et al., 2014) they were considered separately. The expansion and contraction frequencies were scored as the number of transmitted alleles that were larger or smaller than the parental allele divided by the total number of alleles scored. The significance of differences in these frequencies was assessed using Fisher’s exact test. The distribution of repeat length changes was evaluated using both the Students t test and the Mann-Whitney test. Somatic instability was assessed using DNA isolated from the organs of 6-month-old and 12-month-old animals. The repeat profile was assessed using the same fluorescent PCR assay/GeneMapper analysis referred to above. The SII was determined as previously described (Lee et al., 2010).
Quantitation of mRNA
Mouse organs were homogenized using Precellys® lysing Kits (Bertin technologies). Total RNA was isolated using Maxwell®16 LEV simply RNA purification Kits (Promega). The RNA concentration and quality was determined using an Agilent Bioanalyzer. The RNA was reverse transcribed using a SuperScript®VILO™ cDNA synthesis kit (Life Technologies). Real time PCR was done in triplicate using TaqMan® Fast universal PCR master mix and the appropriate Taqman probe-primer pairs (Life Technologies). The Taqman probe primer pairs were as follows: Fmr1: Mm01339582_m1; Csb: Mm01221908_m1; Msh2: Mm00500563_m1; Msh3: Mm00487756_m1; Atm: Mm01177457_m1; Atr: Mm01223626_m1. Gapdh was used as endogenous control to compare the expression level in the livers of different animals (Mouse GAPDH Endogenous Control (VIC®/MGB Probe, primer limited); Applied Biosystems®). However, the expression levels of Gapdh differed in various organs. Thus for comparison of mRNA expression in different tissue equal amounts of RNA were used for each determination. Normalization was carried out by comparing the Ct value for the mRNA in different organs to the Ct value obtained from brain.
Results
To test the role of TCR in instability of FX PM alleles in our mouse model, we crossed FX PM mice containing ~150 CGG repeats in the 5′-UTR of the Fmr1 gene with mice carrying a Csb null mutation. Breeding pairs were set up in which one parent carried the FX PM allele and both parents were either WT or nullizygous for Csb. The repeat size in the offspring that inherited the PM allele was then determined as previously described (Lokanga, et al., 2013a). These data were then used to calculate the number of offspring inheriting alleles that were larger, smaller or the same size as the parental alleles. We also determined the number of repeats added or lost from each allele and examined the effect of Csb nullizygosity on somatic instability by comparing the repeat profiles seen in the organs of Csb+/+ and Csb−/− mice carrying one copy of the PM allele.
In the course of doing this work we found that the Csb+/+ males transmit larger alleles at a frequency of close to 100%, irrespective of paternal age (Fig. 1 and 2A). Furthermore, the size of each transmitted allele increases with paternal age (Fig. 2B). Thus alleles that have expanded in the germ-line of young fathers likely undergo additional rounds of expansion in the father’s germ-line that add ~5 repeats every 6 months. In contrast, the frequency of maternal transmission of larger alleles, while initially high, declines with age (Fig. 1 and 3A). Since it is unlikely that there is female-specific selection against alleles that are only a few repeats larger than the original allele, we attribute the declining frequency of larger alleles in the offspring of older females to alleles that had expanded in younger mothers and then subsequently contracted. Thus the category of alleles larger than the maternal allele likely represents a mixture of alleles some of which have merely expanded and some that have expanded and subsequently contracted. Similarly, the frequency of transmitted alleles that are the same size as the maternal allele increases with age. By the same logic, some of these alleles must reflect alleles that had initially expanded and then subsequently contracted. To reflect this we have tried to avoid the use of the potentially misleading labels “expansion” and “contraction” for transmitted alleles in favor of the terms “larger”, “smaller” and the “same” as the parental allele where appropriate.
Fig. 1. Intergenerational instability of the FX PM allele in WT males and females.

The frequency of the transmission of alleles that are larger, smaller or the same size as the parental allele were determined in the offspring of male and female PM mice that were 2–6 months old, 7–12 months old and 13–22 months old. There was no significant difference in the mean parental age in each age class by the Student t-test. No significant difference was seen in frequency of any allele class on paternally transmitted alleles by the Mann-Whitney U test. In females, there was a significant decline in the number of larger alleles that were transmitted with age (Mann-Whitney test: p=0.003 and p=0.0026). However, the number of offspring of mothers in 13–22 month category was very small (8 animals) and although the difference between these mice and the progeny of younger mothers was significant as indicated by the asterisks, our level of confidence in a sample size this small is low.
Fig. 2. Intergenerational instability of the FX PM allele in Csb+/+ and Csb−/− male mice.


A) The frequency of transmission of alleles that are larger, smaller or the same size as the parental allele in fathers of different ages. There was no significant difference in the mean paternal age in each age class for Csb+/+ and Csb−/− animals by the Student t-test. No significant differences were seen in the number of larger or smaller alleles or the number of alleles that were the same size as the paternal allele using the Mann-Whitney test of significance. The number above each bar corresponds to the number of animals observed in that category. B) Distribution of repeat length changes on paternal transmission in Csb+/+ and Csb−/− mice. There was no significant difference in the mean paternal age in each age class for Csb+/+ and Csb−/− animals by the Student t-test. The total number of alleles in each size class was plotted. Both Csb+/+ and Csb−/− males showed an age-related increase in the average number of repeats added with each transmission. However, no significant difference between Csb+/+ and Csb−/− animals was seen in the number of repeats added at any age.
Fig. 3. Intergenerational instability of the FX PM allele in Csb+/+ and Csb−/− mothers.
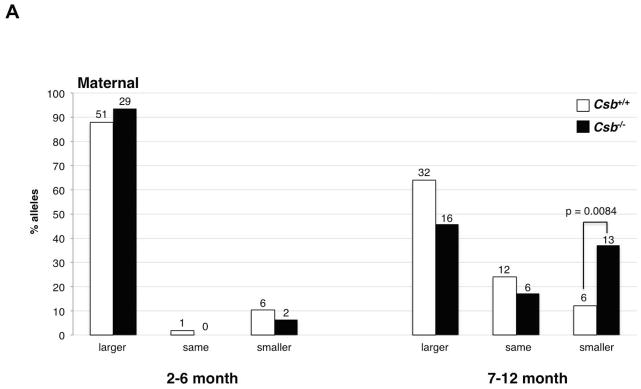

A) The frequency of transmission of alleles that were larger, smaller or the same size as the maternal allele in the offspring of female PM mice WT or nullizygous for Csb. There was no significant difference in the mean maternal age in each age class for Csb+/+ and Csb−/− animals by the Student t-test. The number above each bar corresponds to the number of animals observed in that category. B) Distribution of repeat length changes on maternal transmission in Csb+/+ and Csb−/− mice. The total number of alleles in each size class was plotted as a function of the change in repeat number from the maternal allele. The distribution of allele sizes was displaced towards smaller alleles in Csb−/− animals. The difference in the distribution of allele sizes for Csb+/+ and Csb−/− animals was significant for both young (2–6 months old) and old (7–12 months old) animals (Mann-Whitney: p=0.04 and p=0.02 respectively).
Loss of CSB does not affect instability in male carriers of PM alleles
There was no significant effect of Csb nullizygosity on the proportion of larger or smaller alleles that were paternally transmitted (Fig. 2A). This was true whether fathers of all ages were considered together or when they were stratified by age at breeding (2–6 months, 7–12 months and >12 months). The Csb genotype also did not affect the average number of repeats added with each intergenerational transmission (Fig. 2B). The failure of the Csb mutation to reduce the number of larger alleles or the size of these alleles indicates that CSB is not required to generate intergenerational expansions in males.
Loss of CSB affects the intergenerational expansion profile in old but not young females
The progeny of young Csb+/+ and Csb−/− females (2–6 months of age) also showed no significant difference in the frequency of transmission of alleles that were larger, smaller or the same size as the maternal allele (Fig. 3A). However, in the progeny of Csb−/− mothers that were 7–12 months old, there was a decline in the number of alleles that were larger or the same size as the maternal allele that was associated with a significant increase in the number of alleles that were smaller than the maternal allele (p=0.0084). There was also a significant change in the distribution of the repeat number changes in the progeny of both 2–6 month old and 7–12 month old mice (Fig. 3B; Mann-Whitney p= 0.04 for 2–6 month animals and p=0.02 for animals 7–12 months of age).
Csb nullizygosity reduces the extent of somatic instability in male mice
In addition to intergenerational expansions, PM alleles also expand somatically in male mice with organs like brain, liver and testes showing more expansion than organs such as kidney and heart (Lokanga, et al., 2013a). We quantified the somatic expansion frequencies in 6 and 12 month old Csb+/+ and Csb−/− male mice using the somatic instability index (SII), a combined measure of the frequency and size of repeat length changes (Lee, et al., 2010). No change in the organ-specificity of expansion was seen in Csb−/− animals (Fig. 4). However, the average number of repeats present on the derivative alleles was smaller in Csb−/− mice than in Csb+/+ mice (i.e., a lower SII, Fig. 4). At 6 months of age the SII differences were only significant for liver. However, at 12 months of age the differences were also significant for tail, kidney, testis and spleen. Thus despite the fact that the loss of CSB had no effect on intergenerational transmissions in males of any age, there is a significant effect on the extent of somatic instability in males that increases with age. The fact that an effect of a CSB deficiency is seen in testis but not in the progeny of male mice may reflect events occurring in somatic cells of the testis rather than the germ-line.
Fig. 4. Somatic expansion of the FX PM allele in male Csb+/+ and Csb−/− mice.

The average SII of different organs of Csb+/+ and Csb−/− mice with 145–160 repeats. The data represents the average of the individual SIIs from 6 Csb+/+ and 9 Csb−/− mice for the 6 month old mice and 6 Csb+/+ and 7 Csb−/− mice for the 12 months old animals. The asterisks indicate the organs in which the differences in SII for young and old animals were statistically significant (p<0.05).
Reduced transcription of the Fmr1 gene in Csb−/− mice does not account for the reduced extent of somatic expansion
Since CSB stimulates transcription (Selby and Sancar, 1997) and untranscribed FMR1 alleles are more stable than transcribed ones (Wohrle, et al., 1998; Wohrle, et al., 2001), reduced levels of CSB protein could potentially lead indirectly to reduced levels of instability. To evaluate this possibility we measured Fmr1 mRNA levels in FX PM mice WT or nullizygous for Csb. In liver, the organ showing the greatest effect of the loss of CSB on the extent of somatic expansion, the difference in the average levels of Fmr1 transcript between PM mice WT and nullizygous for Csb was not significant (Fig. 5A). For reasons that are not yet clear, WT Fmr1 transcript levels vary widely even in Csb+/+ mice (Brouwer et al., 2008). The SII we measured here also shows significant variability between animals. We therefore compared the levels of Fmr1 mRNA with the SII for each animal. Our data demonstrate that while there was considerable overlap in the Fmr1 mRNA levels in Csb+/+ and Csb−/− animals, the SII of Csb−/− animals and Csb+/+ animals showed much less overlap (Fig. 5B). Thus differences in transcription of Fmr1 do not account for differences in the SII seen between Csb+/+ and Csb−/− animals.
Fig. 5. The effect of CSB on the expression of genes potentially relevant for repeat expansion.

A) The Fmr1 mRNA levels in the livers of 12 month old Csb+/+ and Csb−/− male mice was determined by quantitative real time PCR as described in the Materials and Methods. The Fmr1 mRNA levels are expressed relative to the levels of a house keeping gene, Gapdh. The mRNA levels are an average of the levels from 6 Csb+/+ mice and 7 Csb−/− mice. The error bars indicate the standard deviations. The difference in Fmr1 mRNA expression between Csb+/+ and Csb−/− animals was not significant while the difference in SII was (p=0.331 and p=0.011 respectively). B) Comparison of Fmr1 mRNA levels and the SII of individual Csb+/+ and Csb−/− animals. The SII of each Csb+/+ and Csb−/− animal was plotted as a function of the Fmr1 mRNA level as described in the Materials and Methods. C) Csb expression in different mouse organs. Csb mRNA levels in different organs were determined as described in the Materials and Methods. The data shown represents the average of the Csb mRNA levels in 3 different animals. Since no housekeeping gene whose transcript levels are consistent in all tissue has been identified, we used the same amount of mRNA for all tissue and then normalized the Csb transcript levels in heart, liver, kidney, testis, spleen and lung to the levels seen in brain. The number above each bar represents the SII of that organ at 12 months of age. D) The effect of Csb on the expression of genes known to be involved in repeat expansion in the FX PM mouse. Transcript levels are expressed relative to the levels of a house keeping gene, Gapdh, multiplied by 10,000. The mRNA levels are an average of the levels from the livers 6 Csb+/+ and 7 Csb−/− mice all 12 months of age. The white bars indicate the mRNA levels in the Csb+/+ mice, the black bars the Csb−/− mice. The error bars indicate the standard deviations. No significant differences between the mRNA levels in Csb+/+ and Csb−/− mice were seen using the Student t test.
No simple relationship exists between the levels of Csb transcript and the effect of loss of CSB on somatic expansion in particular tissue
No antibodies known to reliably detect CSB in mouse tissue extracts are currently available. We thus assessed the level of CSB expression in different organs by real-time PCR. The levels of Csb mRNA in different tissue varied over a more than 10-fold range. However, organs with very similar levels of Csb RNA had very different SIIs (Fig. 5C). Thus there was no relationship between the level of Csb expression and the extent of somatic instability in the tissues examined. Thus the effect of the loss of CSB is not related to the level of CSB normally present in that organ. Rather it may reflect the levels of other proteins important for the expansion process that are rate-limiting in some tissue but not others.
Loss of CSB does not affect the transcription of the genes known to be important for repeat expansion or protecting the genome from repeat expansion
Since CSB can affect global transcription levels it may exert indirect effects on expansion by affecting the levels of transcripts produced from genes involved in the expansion process or in protecting the genome from these expansions. To date only 3 genes have been definitively implicated in this process in the FX PM mouse model; Atm and Atr are known to be protective (Entezam and Usdin, 2008; Entezam and Usdin, 2009) while Msh2 is required for expansion (Lokanga, et al., 2014). MSH2 also has a binding partner MSH3 that has been shown to be involved in expansions in mouse models of other Repeat Expansion Diseases (Foiry et al., 2006). We thus compared the levels of transcripts of these genes in the livers of Csb+/+ and Csb−/− animals. No significant difference was seen in the expression levels of any of these genes (Fig. 5D).
Discussion
In the course of this study we found that there were hitherto unappreciated gender and age related differences in the transmission of repeat expansions in FX PM allele mice even in a Csb+/+ background. Specifically, >90% of paternally transmitted alleles were larger than the parental allele irrespective of the father’s age (Fig. 1 and 2). Furthermore, the number of repeats in the transmitted allele continues to increase as the fathers increase in age with ~5 repeats being added every 6 months (Fig. 3). This suggests that in males PM alleles undergo multiple rounds of expansion and that expansion continues apparently unabated in the paternal germ-line throughout the father’s reproductive lifespan. This is quite different from what is seen in females. In females 2–6 months old, the frequency with which alleles that are larger than the maternal allele are transmitted also approaches 90%. Thus young males and females have a similar ability to generate germ-line expansions. However, in females that are 7–12 months old, the frequency with which larger alleles are transmitted drops by ~38% to 65% (Fig. 1; p=0.003 (Fisher’s exact test)). This drop cannot be explained simply by a reduced expansion frequency since otherwise the fraction of larger alleles would have remained constant. The loss of these alleles suggests that the frequency of contractions increases with age in females. Increased contractions during female germ-line transmission have been reported for mouse models of CAG/CTG-Repeat Expansion Diseases but this does not seem to follow a period of significant expansion as in the FX PM mouse (Kovtun et al., 2000; Mangiarini et al., 1996; Wheeler et al., 1999). A number of DNA repair genes, including those involved in DNA damage signaling, in mismatch repair and in the repair of oxidative DNA damage, are regulated by estrogen (Araneda, et al., 2005; Berger, et al., 2012; Cabanes, et al., 2004; Dietrich, et al., 2013; Jin, et al., 2010; Likhite, et al., 2004; Medunjanin et al., 2010; Miyamoto et al., 2006; Murdoch and Van Kirk, 1997; Murdoch and Van Kirk, 2001; Schultz-Norton et al., 2011). It may be that an age-related decline in estrogen contributes to the drop in maternally transmitted expansions thus allowing the contraction process to become more apparent. Whatever the basis of this gender difference, it may have implications for risk assessment in humans.
To assess the contribution of TCR to CGG/CCG-repeat instability, we have compared the frequency and extent of germ-line and somatic instability of the FX PM allele in Csb+/+ and Csb−/− mice. We have shown that loss of CSB had no significant effect on either the frequency of paternal germ-line transmission of any of the allele size classes irrespective of age (Fig. 1 and 2A) or on the magnitude of any of the repeat length changes (Fig. 2B). The failure to reduce either the incidence or size of larger alleles in fathers suggests that CSB is not acting to promote expansions in these animals in the same way that proteins like MSH2 do (Lokanga, et al., 2014). The loss of CSB also had no significant effect on the generation of larger or smaller alleles by younger mothers (Fig. 3A). However, it did lead to a significant increase in number of alleles smaller than the parental allele in the offspring of older mothers (Fig. 3A). Furthermore, loss of CSB significantly shifted the distribution of repeat length changes that were seen both for young and old mothers towards smaller alleles (Fig. 3B). Thus loss of CSB affected the mutation spectrum in both young and old females. This is unlikely to be due to a non-specific effect on cell viability or apoptosis since CSB loss does not affect the fertility or body weight of females (van der Horst et al., 1997). There is also no difference between the frequency with which the PM allele is transmitted in Csb+/+ animals and Csb−/− animals (data not shown) suggesting that there is no specific deleterious effect of the Csb mutation on the transmission/viability of cells with the PM allele. In any event, the difference between large and small alleles is relatively small and thus there is unlikely to be selection against these marginally larger alleles.
The fact that loss of CSB affects maternally but not paternally transmitted germ-line expansions would suggest either that a different mechanism is responsible for these expansions in males and females or that the role of CSB in expansions only becomes apparent when a factor normally required for expansion becomes rate limiting and this factor becomes rate-limiting in the female, but not male, germ-line. It is also possible that CSB is involved in protecting the genome against repeat contractions, an effect that may only become apparent when the expansion frequency drops as it does in older females.
The differential effect of loss of CSB in some organs may suggest that different expansion mechanisms operate in different tissues as was proposed to explain the fact that in a mouse model of SCA1, a CAG/CTG-repeat expansion disease, a mutation in Xpa, a protein involved in both TCR and GG-NER, reduced expansions in brain but did not affect expansions in liver (Hubert et al., 2011). It is also feasible, as with intergenerational instability, that the lower efficacy of the expansion pathway in some cells may allow the effect of the loss of CSB on the generation of contractions to be apparent or that CSB is required for expansion in some tissues because of a drop in the levels of a protein that is normally rate-limiting for expansion. However, even in organs like liver where large effects of the loss of CSB are seen (Fig. 4), there is no increase in the proportion of alleles that are smaller than the original inherited allele. Thus the effect of the loss of CSB in somatic tissue, and perhaps by extension in female germ-line as well, is more likely to be due to its effect on expansions rather than contractions.
How CSB mediates this effect is unclear. In principle, since CSB significantly affects the transcription of many genes (Newman, et al., 2006), the loss of CSB could affect the expression of genes that are directly involved in expansion. While such a role cannot be definitively ruled out until all the players in the expansion process have been identified, we have shown that the loss of CSB does not affect the transcription of Fmr1 itself (Fig. 5A and B) or any of those genes currently known to be involved in either expansion or protecting the genome against expansion in the FX PM mouse model (Fig. 5D). Furthermore, it has been shown that in human cells no genes in the candidate expansion pathways including MMR, TCR or BER, are significantly upregulated or downregulated by CSB (Newman, et al., 2006).
A simple model that explains both the pattern of female germ-line transmission and that of male somatic expansion is that loss of CSB affects expansion directly but it’s effect is only apparent in cells in which one of the factors normally involved in expansion has become rate-limiting. What factor CSB could substitute for in the expansion process is not clear. Since its participation is not absolutely required, the process itself is presumably not TCR. CSB is known to be recruited to sites of oxidative damage independently of other TCR proteins (Menoni et al., 2012) and has a number of interacting partners that are involved in base excision repair (BER) of oxidative DNA damage (reviewed in (Aamann et al., 2013)). This is of interest since we have previously shown that oxidative damage increases the expansion frequency in the PM mouse (Entezam, et al., 2010). Amongst its interactions with BER proteins, CSB stimulates the incision and AP lyase activity of Neil1, a DNA glycosylase involved in BER (Muftuoglu et al., 2009). Neil1 has been shown to be a modifier of expansions in a mouse model of HD (Mollersen et al., 2012). CSB may contribute to expansion by facilitating the activity of enzymes like Neil1 when other factors fail to do so.
Comparison of our data with data from models of other Repeat Expansion Diseases is complicated by the fact that conflicting results for these models have been reported. A mutation in the Drosophila homolog of Rad2/XPG, which is involved in both TCR and global genomic-NER (GG-NER), decreased both maternally and paternally transmitted germ-line expansions but had no effect on contractions in a fly model of a CAG/CTG-repeat expansion (Jung and Bonini, 2007). In contrast, siRNA knockdown of Csb in a cell-based model system for CAG/CTG repeat instability resulted in fewer contractions (Lin and Wilson, 2007). A small study in a mouse model of a CAG/CTG-repeat expansion disease showed that loss of CSB led to an increase in germ-line expansions and had no effect on somatic instability (Kovtun, Johnson, and McMurray, 2011). However, since this study was too small to produce statistically significant results, the role of CSB in CAG/CTG-repeat instability in mice still needs to be clarified. In any event, it is unclear whether CAG/CTG-repeats and CGG/CCG-repeats share the same mechanisms of instability and thus whether the effect of CSB in CAG/CTG models is relevant for our understanding of what is happening in the FX-related disorders is also unclear.
Targeting of the factors that promote expansion has been suggested as a therapeutic approach for the Repeat Expansion Disorders (Dragileva et al., 2009; Halabi et al., 2012; Mooney et al., 2001; Schultz-Norton, et al., 2011). Our data suggests that CSB could be added to the list of potential targets, at least for CGG/CCG-repeat expansion disorders.
Acknowledgments
Grant Sponsor/Funding source: This work was supported by the Intramural Program of the NIDDK, National Institutes of Health [DK057808-05 to Karen Usdin].
The Usdin lab would like to thank the members of the Lab Animals Sciences Section and the hard-working technicians that take care of our mouse colony, without whose efforts this work would not be possible. We would also like to thank Jia Chen, Orna Cohen-Fix, Bruce Hayward, Debbie Hinton and Daman Kumari for their careful reading of the manuscript and/or helpful suggestions.
Footnotes
Conflicts of Interest
The authors declare that no conflict of interest exists.
References
- Aamann MD, Muftuoglu M, Bohr VA, Stevnsner T. Multiple interaction partners for Cockayne syndrome proteins: implications for genome and transcriptome maintenance. Mechanisms of ageing and development. 2013;134:212–24. doi: 10.1016/j.mad.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araneda S, Pelloux S, Radicella JP, Angulo J, Kitahama K, Gysling K, Forray MI. 8-oxoguanine DNA glycosylase, but not Kin17 protein, is translocated and differentially regulated by estrogens in rat brain cells. Neuroscience. 2005;136:135–46. doi: 10.1016/j.neuroscience.2005.06.080. [DOI] [PubMed] [Google Scholar]
- Berg RJ, Rebel H, van der Horst GT, van Kranen HJ, Mullenders LH, van Vloten WA, de Gruijl FR. Impact of global genome repair versus transcription-coupled repair on ultraviolet carcinogenesis in hairless mice. Cancer Res. 2000;60:2858–63. [PubMed] [Google Scholar]
- Berger CE, Qian Y, Liu G, Chen H, Chen X. p53, a target of estrogen receptor (ER) alpha, modulates DNA damage-induced growth suppression in ER-positive breast cancer cells. The J Biol Chem. 2012;287:30117–27. doi: 10.1074/jbc.M112.367326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwer JR, Huizer K, Severijnen LA, Hukema RK, Berman RF, Oostra BA, Willemsen R. CGG-repeat length and neuropathological and molecular correlates in a mouse model for fragile X-associated tremor/ataxia syndrome. J Neurochem. 2008;107:1671–82. doi: 10.1111/j.1471-4159.2008.05747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabanes A, Wang M, Olivo S, DeAssis S, Gustafsson JA, Khan G, Hilakivi-Clarke L. Prepubertal estradiol and genistein exposures up-regulate BRCA1 mRNA and reduce mammary tumorigenesis. Carcinogenesis. 2004;25:741–8. doi: 10.1093/carcin/bgh065. [DOI] [PubMed] [Google Scholar]
- Dietrich AK, Humphreys GI, Nardulli AM. 17beta-Estradiol increases expression of the oxidative stress response and DNA repair protein apurinic endonuclease (Ape1) in the cerebral cortex of female mice following hypoxia. J Steroid Biochem Mol Biol. 2013;138:410–20. doi: 10.1016/j.jsbmb.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragileva E, Hendricks A, Teed A, Gillis T, Lopez ET, Friedberg EC, Kucherlapati R, Edelmann W, Lunetta KL, MacDonald ME, Wheeler VC. Intergenerational and striatal CAG repeat instability in Huntington’s disease knock-in mice involve different DNA repair genes. Neurobiol Dis. 2009;33:37–47. doi: 10.1016/j.nbd.2008.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Entezam A, Biacsi R, Orrison B, Saha T, Hoffman GE, Grabczyk E, Nussbaum RL, Usdin K. Regional FMRP deficits and large repeat expansions into the full mutation range in a new Fragile X premutation mouse model. Gene. 2007;395:125–34. doi: 10.1016/j.gene.2007.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Entezam A, Lokanga AR, Le W, Hoffman G, Usdin K. Potassium bromate, a potent DNA oxidizing agent, exacerbates germline repeat expansion in a fragile X premutation mouse model. Hum Mutat. 2010;31:611–6. doi: 10.1002/humu.21237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Entezam A, Usdin K. ATR protects the genome against CGG.CCG-repeat expansion in Fragile X premutation mice. Nucleic Acids Res. 2008;36:1050–6. doi: 10.1093/nar/gkm1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Entezam A, Usdin K. ATM and ATR protect the genome against two different types of tandem repeat instability in Fragile X premutation mice. Nucleic Acids Res. 2009;37:6371–7. doi: 10.1093/nar/gkp666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foiry L, Dong L, Savouret C, Hubert L, te Riele H, Junien C, Gourdon G. Msh3 is a limiting factor in the formation of intergenerational CTG expansions in DM1 transgenic mice. Hum Genet. 2006;119:520–6. doi: 10.1007/s00439-006-0164-7. [DOI] [PubMed] [Google Scholar]
- Fu YH, Kuhl DP, Pizzuti A, Pieretti M, Sutcliffe JS, Richards S, Verkerk AJ, Holden JJ, Fenwick RG, Jr, Warren ST, et al. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell. 1991;67:1047–58. doi: 10.1016/0092-8674(91)90283-5. [DOI] [PubMed] [Google Scholar]
- Hagerman PJ, Hagerman RJ. Fragile X-associated tremor/ataxia syndrome (FXTAS) Ment Retard Dev Disabil Res Rev. 2004;10:25–30. doi: 10.1002/mrdd.20005. [DOI] [PubMed] [Google Scholar]
- Halabi A, Ditch S, Wang J, Grabczyk E. DNA mismatch repair complex MutSbeta promotes GAA.TTC repeat expansion in human cells. The J Biol Chem. 2012;287:29958–67. doi: 10.1074/jbc.M112.356758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubert L, Jr, Lin Y, Dion V, Wilson JH. Xpa deficiency reduces CAG trinucleotide repeat instability in neuronal tissues in a mouse model of SCA1. Hum Mol Genet. 2011;20:4822–30. doi: 10.1093/hmg/ddr421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin P, Lu XJ, Sheng JQ, Fu L, Meng XM, Wang X, Shi TP, Li SR, Rao J. Estrogen stimulates the expression of mismatch repair gene hMLH1 in colonic epithelial cells. Cancer Prev Res (Phila) 2010;3:910–6. doi: 10.1158/1940-6207.CAPR-09-0228. [DOI] [PubMed] [Google Scholar]
- Jung J, Bonini N. CREB-binding protein modulates repeat instability in a Drosophila model for polyQ disease. Science. 2007;315:1857–9. doi: 10.1126/science.1139517. [DOI] [PubMed] [Google Scholar]
- Kovtun IV, Johnson KO, McMurray CT. Cockayne syndrome B protein antagonizes OGG1 in modulating CAG repeat length in vivo. Aging (Albany NY) 2011;3:509–14. doi: 10.18632/aging.100324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovtun IV, Therneau TM, McMurray CT. Gender of the embryo contributes to CAG instability in transgenic mice containing a Huntington’s disease gene. Hum Mol Genet. 2000;9:2767–75. doi: 10.1093/hmg/9.18.2767. [DOI] [PubMed] [Google Scholar]
- Lee JM, Zhang J, Su AI, Walker JR, Wiltshire T, Kang K, Dragileva E, Gillis T, Lopez ET, Boily MJ, Cyr M, Kohane I, et al. A novel approach to investigate tissue-specific trinucleotide repeat instability. BMC Syst Biol. 2010;4:29. doi: 10.1186/1752-0509-4-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Likhite VS, Cass EI, Anderson SD, Yates JR, Nardulli AM. Interaction of estrogen receptor alpha with 3-methyladenine DNA glycosylase modulates transcription and DNA repair. The J Biol Chem. 2004;279:16875–82. doi: 10.1074/jbc.M313155200. [DOI] [PubMed] [Google Scholar]
- Lin Y, Hubert L, Jr, Wilson JH. Transcription destabilizes triplet repeats. Mol Carcinog. 2009;48:350–61. doi: 10.1002/mc.20488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Wilson JH. Transcription-induced CAG repeat contraction in human cells is mediated in part by transcription-coupled nucleotide excision repair. Mol Cell Biol. 2007;27:6209–17. doi: 10.1128/MCB.00739-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lokanga RA, Entezam A, Kumari D, Yudkin D, Qin M, Smith CB, Usdin K. Somatic expansion in mouse and human carriers of Fragile X premutation alleles. Hum Mutat. 2013a;34:157–66. doi: 10.1002/humu.22177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lokanga RA, Zhao X-N, Usdin K. The mismatch repair protein, MSH2, is rate-limiting for repeat expansion in a Fragile X premutation mouse model. Hum Mutat. 2014;35:129–136. doi: 10.1002/humu.22464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- Medunjanin S, Weinert S, Poitz D, Schmeisser A, Strasser RH, Braun-Dullaeus RC. Transcriptional activation of DNA-dependent protein kinase catalytic subunit gene expression by oestrogen receptor-alpha. EMBO Rep. 2010;11:208–13. doi: 10.1038/embor.2009.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menoni H, Hoeijmakers JH, Vermeulen W. Nucleotide excision repair-initiating proteins bind to oxidative DNA lesions in vivo. The Journal of cell biology. 2012;199:1037–46. doi: 10.1083/jcb.201205149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirkin SM. DNA structures, repeat expansions and human hereditary disorders. Curr Opin Struct Biol. 2006;16:351–8. doi: 10.1016/j.sbi.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Miyamoto T, Shiozawa T, Kashima H, Feng YZ, Suzuki A, Kurai M, Nikaido T, Konishi I. Estrogen up-regulates mismatch repair activity in normal and malignant endometrial glandular cells. Endocrinology. 2006;147:4863–70. doi: 10.1210/en.2006-0632. [DOI] [PubMed] [Google Scholar]
- Mollersen L, Rowe AD, Illuzzi JL, Hildrestrand GA, Gerhold KJ, Tveteras L, Bjolgerud A, Wilson DM, 3rd, Bjoras M, Klungland A. Neil1 is a genetic modifier of somatic and germline CAG trinucleotide repeat instability in R6/1 mice. Human molecular genetics. 2012;21:4939–47. doi: 10.1093/hmg/dds337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooney LA, Perera FP, Van Bennekum AM, Blaner WS, Karkoszka J, Covey L, Hsu Y, Cooper TB, Frenkel K. Gender differences in autoantibodies to oxidative DNA base damage in cigarette smokers. Cancer Epidemiol Biomarkers Prev. 2001;10:641–8. [PubMed] [Google Scholar]
- Muftuoglu M, de Souza-Pinto NC, Dogan A, Aamann M, Stevnsner T, Rybanska I, Kirkali G, Dizdaroglu M, Bohr VA. Cockayne syndrome group B protein stimulates repair of formamidopyrimidines by NEIL1 DNA glycosylase. The J Biol Chem. 2009;284:9270–9. doi: 10.1074/jbc.M807006200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murdoch WJ, Van Kirk EA. Oestradiol inhibits spontaneous and cisplatin-induced apoptosis in epithelial ovarian cancer cells: relationship to DNA repair capacity. Apoptosis. 1997;2:478–84. doi: 10.1023/a:1026426212366. [DOI] [PubMed] [Google Scholar]
- Murdoch WJ, Van Kirk EA. Estrogenic upregulation of DNA polymerase beta in oocytes of preovulatory ovine follicles. Mol Reprod Dev. 2001;58:417–23. doi: 10.1002/1098-2795(20010401)58:4<417::AID-MRD9>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Newman JC, Bailey AD, Weiner AM. Cockayne syndrome group B protein (CSB) plays a general role in chromatin maintenance and remodeling. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:9613–8. doi: 10.1073/pnas.0510909103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberle I, Rousseau F, Heitz D, Kretz C, Devys D, Hanauer A, Boue J, Bertheas M, Mandel J. Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science. 1991;252:1097–102. doi: 10.1126/science.252.5009.1097. [DOI] [PubMed] [Google Scholar]
- Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu Rev Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- Pearson CE, Nichol Edamura K, Cleary JD. Repeat instability: mechanisms of dynamic mutations. Nat Rev Genet. 2005;6:729–42. doi: 10.1038/nrg1689. [DOI] [PubMed] [Google Scholar]
- Pieretti M, Zhang FP, Fu YH, Warren ST, Oostra BA, Caskey CT, Nelson DL. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell. 1991;66:817–22. doi: 10.1016/0092-8674(91)90125-i. [DOI] [PubMed] [Google Scholar]
- Salinas-Rios V, Belotserkovskii BP, Hanawalt PC. DNA slip-outs cause RNA polymerase II arrest in vitro: potential implications for genetic instability. Nucleic Acids Res. 2011;39:7444–54. doi: 10.1093/nar/gkr429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz-Norton JR, Ziegler YS, Nardulli AM. ERalpha-associated protein networks. Trends Endocrinol Metab. 2011;22:124–9. doi: 10.1016/j.tem.2010.11.005. [DOI] [PubMed] [Google Scholar]
- Selby CP, Sancar A. Cockayne syndrome group B protein enhances elongation by RNA polymerase II. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:11205–9. doi: 10.1073/pnas.94.21.11205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman SL. Premature ovarian failure in the fragile X syndrome. Am J Med Genet. 2000;97:189–94. doi: 10.1002/1096-8628(200023)97:3<189::AID-AJMG1036>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Strom CM, Crossley B, Redman JB, Buller A, Quan F, Peng M, McGinnis M, Fenwick RG, Jr, Sun W. Molecular testing for Fragile X Syndrome: lessons learned from 119,232 tests performed in a clinical laboratory. Genet Med. 2007;9:46–51. doi: 10.1097/gim.0b013e31802d833c. [DOI] [PubMed] [Google Scholar]
- Tassone F, Hagerman RJ, Taylor AK, Gane LW, Godfrey TE, Hagerman PJ. Elevated levels of FMR1 mRNA in carrier males: a new mechanism of involvement in the fragile-X syndrome. Am J Hum Genet. 2000;66:6–15. doi: 10.1086/302720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AK, Tassone F, Dyer PN, Hersch SM, Harris JB, Greenough WT, Hagerman RJ. Tissue heterogeneity of the FMR1 mutation in a high-functioning male with fragile X syndrome. Am J Med Genet. 1999;84:233–9. [PubMed] [Google Scholar]
- Tome S, Manley K, Simard JP, Clark GW, Slean MM, Swami M, Shelbourne PF, Tillier ER, Monckton DG, Messer A, Pearson CE. MSH3 polymorphisms and protein levels affect CAG repeat instability in Huntington’s disease mice. PLoS Genetics. 2013;9:e1003280. doi: 10.1371/journal.pgen.1003280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usdin K. The biological effects of simple tandem repeats: lessons from the repeat expansion diseases. Genome Res. 2008;18:1011–9. doi: 10.1101/gr.070409.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Horst GT, van Steeg H, Berg RJ, van Gool AJ, de Wit J, Weeda G, Morreau H, Beems RB, van Kreijl CF, de Gruijl FR, Bootsma D, Hoeijmakers JH. Defective transcription-coupled repair in Cockayne syndrome B mice is associated with skin cancer predisposition. Cell. 1997;89:425–35. doi: 10.1016/s0092-8674(00)80223-8. [DOI] [PubMed] [Google Scholar]
- Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–14. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- Wheeler VC, Auerbach W, White JK, Srinidhi J, Auerbach A, Ryan A, Duyao MP, Vrbanac V, Weaver M, Gusella JF, Joyner AL, MacDonald ME. Length-dependent gametic CAG repeat instability in the Huntington’s disease knock-in mouse. Hum Mol Genet. 1999;8:115–22. doi: 10.1093/hmg/8.1.115. [DOI] [PubMed] [Google Scholar]
- Willemsen R, Levenga J, Oostra BA. CGG repeat in the FMR1 gene: size matters. Clin Genet. 2011;80:214–25. doi: 10.1111/j.1399-0004.2011.01723.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohrle D, Salat U, Glaser D, Mucke J, Meisel-Stosiek M, Schindler D, Vogel W, Steinbach P. Unusual mutations in high functioning fragile X males: apparent instability of expanded unmethylated CGG repeats. J Med Genet. 1998;35:103–11. doi: 10.1136/jmg.35.2.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohrle D, Salat U, Hameister H, Vogel W, Steinbach P. Demethylation, reactivation, and destabilization of human fragile X full-mutation alleles in mouse embryocarcinoma cells. Am J Hum Genet. 2001;69:504–15. doi: 10.1086/322739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohrle D, Schwemmle S, Steinbach P. DNA methylation and triplet repeat stability: new proposals addressing actual questions on the CGG repeat of fragile X syndrome. Am J Med Genet. 1996;64:266–7. doi: 10.1002/ajmg.1320640202. [DOI] [PubMed] [Google Scholar]