

Abstract
Rates of metabolic and cardiovascular diseases have increased at an astounding rate in recent decades. While poor diet and physical inactivity are central drivers, these lifestyle changes alone fail to fully account for the magnitude and rapidity of the epidemic. Thus, attention has turned to identifying novel risk factors, including the contribution of environmental endocrine disrupting chemicals. Epidemiological and preclinical data support a role for various contaminants in the pathogenesis of diabetes. In addition to the vascular risk associated with dysglycemia, emerging evidence implicates multiple pollutants in the pathogenesis of atherosclerosis and cardiovascular disease. Reviewed herein are studies linking endocrine disruptors to these key diseases that drive significant individual and societal morbidity and mortality. Identifying chemicals associated with metabolic and cardiovascular disease as well as their mechanisms of action is critical for developing novel treatment strategies and public policy to mitigate the impact of these diseases on human health.
Keywords: atherosclerosis, cardiovascular disease, endocrine disruptors, diabetes, pollution, environment, energy metabolism, cardiovascular risk
Introduction
Type 2 diabetes (T2DM) exerts a tremendous individual and societal toll. Patients with diabetes have a risk of death approximately double that of their healthy peers, and T2DM remains the leading cause of kidney failure, blindness, and non-traumatic amputations [1]. Consequently, recent estimates suggest that total costs associated with diagnosed T2DM amount to a staggering $245 billion annually in the United States alone [2]. This massive economic and societal burden has increased at an alarming rate, with both disease incidence and associated costs exceeding that of even the most recent projections. As many as a third of U.S. adults are expected to have T2DM by the year 2050 [3], and even optimal intervention strategies are projected to achieve only moderate success at reversing these trends at their current rates of growth [4]. Finally, these estimates do not account for the global burden of the disease, which is expected to rise from 382 million to 592 million individuals worldwide by the year 2035 [5].
In addition to the clear association with microvascular complications such as retinopathy, neuropathy, and nephropathy; T2DM, type 1 diabetes, and other pre-diabetic conditions (e.g. impaired fasting glucose and glucose intolerance) are major risk factors for the development of macrovascular complications, including atherosclerosis, stroke, coronary artery disease, and peripheral vascular disease [6,7,8]. In fact, diabetes is considered an independent risk factor for cardiovascular disease (CVD) and mortality [9,10]. T2DM is associated with several common CVD risk factors, including obesity, diabetic dyslipidemia, hyperglycemia, and insulin resistance. The clustering of these metabolic risk factors in T2DM works in an additive fashion to promote vascular disease.
While the genetic contributions to T2DM and cardiovascular disease are substantial, environmental factors are often cited as the major drivers of risk for these chronic disease states. Increased energy intake, particularly consumption of calorically-dense foods common to the Western diet, combined with an increasingly sedentary lifestyle clearly contribute to the current metabolic disease epidemic [11]. However, these factors alone fail to fully account for the magnitude of the metabolic disease epidemic. As such, attention has turned to the increasing body of evidence linking the risk for diabetes and CVD to additional exogenous factors, including environmental contaminants [12]. Voluntary and involuntary exposures to myriad synthetic chemicals are a common feature of modern society, and these exposures form the basis for many recent studies examining the links between environmental chemicals and the etiology of multiple chronic diseases [13,14,15].
Endocrine disrupting chemicals (EDCs) are a broad class of structurally diverse compounds that have the capacity to modulate endogenous hormonal signaling pathways. These chemicals include industrial pollutants, waste products, pharmaceuticals, phytochemicals, pesticides, consumer products, and plastics; and they vary widely in both structure and mode of action [16]. A wide variety of EDCs have been previously associated with an increased risk of T2DM and other metabolic disorders in both epidemiological studies and experimental animal models [17,18,19]. Furthermore, these findings are supported by an increasing body of cell-based and biochemical studies demonstrating the capacity of these compounds to modulate insulin production in pancreatic β cells as well as insulin action in target tissues, which would be predicted to drive systemic metabolic dysfunction [20,21](Sargis, Diabetes & Metabolism Journal, in press).
The growing body of evidence suggesting that EDCs have the capacity to augment the development of metabolic diseases such as obesity and diabetes is compelling; however, less well studied is the role of these toxins in the pathogenesis of atherosclerosis and CVD. Only more recently have studies begun to examine links between environmental pollutants and macrovascular disease [22]. This data implicates several compounds that may accelerate the development of atherosclerosis through their effects on established risk factors common to both diabetes and CVD (e.g. obesity, dyslipidemia). Given the terrible burden of T2DM and macrovascular disease in modern society, understanding the links between environmental pollutants and these disease states is critical for formulating effective prevention strategies and identifying novel therapeutic interventions.
Epidemiological Studies Linking EDCs with Diabetes and Cardiovascular Disease
Epidemiological studies have provided intriguing links between environmental contaminants and the development of diabetes and other metabolic diseases (reviewed in [13,17,19,22]). To date, the majority of studies connecting environmental exposures to diabetes and metabolic dysfunction have focused on a narrow group of compounds for which exposure data is most complete. A recent meta-analysis by the National Toxicology Program (NTP) determined that there is sufficient evidence to support a positive association between T2DM and persistent organic pollutants (POPs) [23]. These diabetogenic POPs include the pesticide dichlorodiphenyltrichloroethane (DDT) and its metabolite dichlorodiphenyldichloroethylene (DDE), as well as pollutants from the dioxin and polychlorinated biphenyl (PCBs) families [23]. A similar NTP analysis concluded that there was a potential connection between arsenic, a common groundwater contaminant, and diabetes [24]. Although the data was somewhat inconclusive, especially at lower levels of exposure, the data showed a relatively robust association at high levels of exposure. Urinary levels of bisphenol A (BPA), a monomer used in polycarbonate plastics that has widespread exposure [25], has also been shown to be significantly associated with diabetes in the 2003–4 NHANES data set [26].
In addition to links between EDCs and diabetes per se, other studies have identified associations between various toxins and risk factors for diabetes such as obesity, the metabolic syndrome and insulin resistance. Several POPs, including organochlorine pesticides and their metabolites (e.g. DDE) as well as various PCB congeners, positively associate with obesity, abdominal adiposity, and components of the metabolic syndrome [15,27,28,29]. Phthalates are used in the plastics industry as well as in various consumer goods and medical devices [30], and phthalate metabolites have been associated with insulin resistance and abdominal obesity [31,32]. Insulin resistance has also been shown to correlate with urinary concentrations of BPA [33] and serum dioxin levels [34]. Finally, air pollution has been implicated in metabolic derangements with exposure to particulate matter (PM) of either the 2.5 μm (PM2.5) [35,36] or 10 μm (PM10) [37] size correlating with either reduced insulin sensitivity or the incidence of diabetes. Collectively, these studies suggest that a diverse array of environmental contaminants may play a central role in the pathophysiology of diabetes and its antecedent states in some individuals.
Similar to studies in diabetes, research evaluating the associations between cardiovascular risk factors and exposure to environmental toxins has provided intriguing preliminary evidence to suggest a potential contribution of EDCs to the pathogenesis of CVD. Atherosclerosis is a progressive metabolic and inflammatory disease of the vasculature that plays a central role in CVD, the leading cause of death in the United States [38]. In epidemiological studies, circulating levels of phthalates [39], BPA [26], and multiple PCB congeners [40] have all been linked to atherosclerosis or CVD in different populations. In addition, inorganic arsenic (iAs), the primary arsenic species in groundwater contamination, has been positively associated with carotid intimal medial thickness (cIMT), a clinical measure of atherosclerosis [41,42], as well as increased levels serum matrix metalloproteinase-9, a biomarker for CVD [43,44]. Interestingly, a recent analysis suggested that the relationship between arsenic exposure and cIMT may be potentiated by incomplete arsenic methylation [45], emphasizing the need to consider specific arsenic species in the analysis of EDC associations with CVD. Importantly, a number of risk factors associated with obesity, including diabetes and insulin resistance as well as dyslipidemia and hypertension are associated with the development of atherosclerosis [46]. As such, EDCs that promote these conditions would be predicted to enhance the development of CVD. In addition to those environmental toxins that promote metabolic dysregulation discussed above, other studies have shown positive associations between serum levels of seven different PCB congeners and hypertension, suggesting that elevated PCB levels may play a role in raising blood pressure [47]. Furthermore, serum organochlorine pesticide levels have been associated with increased triglyceride levels, an independent risk factor for CVD [15,48]. In a study of endemic arsenic exposure in Bangladesh, arsenic levels were associated with lower levels of high-density lipoprotein (HDL) and increases in atherogenic oxidized low-density lipoprotein (LDL) despite lower total levels of LDL and total cholesterol [49].
In addition to metabolic parameters, tobacco smoke has long been appreciated as a prominent risk factor for atherosclerosis and CVD [50] and is also a prominent source of local hazardous air pollution [51]. Tobacco smoke is comprised of a multitude of chemicals, many of which are toxic or carcinogenic and may also play a pathophysiological role in the development of atherosclerosis [52]. One component of environmental tobacco smoke of interest is particulate matter [53]. In the context of ambient air pollution, exposure to particulate matter has been linked to long-term cardiovascular effects including hypertension [36], markers of oxidative stress [54], and CVD-related mortality [55]. Cadmium, another pollutant metal found in tobacco, has also been associated with an increased prevalence of atherosclerotic plaque formation [56,57]. Taken as a whole, these population-based data support the hypothesis that various environmental contaminants have the potential capacity to promote the development of diabetes and other risk factors associated with atherosclerosis ultimately leading to the development of CVD.
Diabetes and Cardiovascular Disease in Animal Models of EDC Action
This epidemiological evidence provides important evidence of potential deleterious effects of EDCs; however, many of these studies are cross-sectional, making it difficult to draw conclusions regarding causality. The suggestions drawn from these studies are, however, supported by animal models of exposure that have examined the pathogenesis of diabetes and vascular disease. A number of compounds have been shown to promote glucose intolerance and frank hyperglycemia in animal models. This includes organic toxins, including the plasticizer diethylhexylphthalate (DEHP) [58], PCBs [59], and triphenyltin [60] as well as the inorganic contaminant arsenic [61]. In addition to overt disruption of glucose handling, several EDCs have been shown to promote hyperinsulinism and insulin resistance. Chronic exposure of mice to the PCB mixture Aroclor 1254 promoted insulin resistance and hyperinsulinemia [62]. In addition, BPA [63,64], the flame retardant polybrominated diphenyl ethers [65], POPs [66], atrazine [67], particulate air pollution [68], and arsenic [69] have all been shown to cause impairments in insulin action and glucose homeostasis. Interestingly, in one study, the insulin resistance induced by exposure to PM2.5 air pollution was only observed in the presence of a high fat diet, suggesting potential synergy between EDCs and dietary risk factors for metabolic disease [68].
Of particular interest in the field of endocrine disruption is the possibility that susceptibility to adverse effects varies across the lifespan. The Developmental Origins of Health and Disease hypothesis postulates that organisms have periods of unique sensitivity to environmental insults during development and that exposure during these sensitive windows predisposes to the development of disease later in life [70]. Furthermore, exposure during these periods may also result in heritable changes through epigenetic modifications that may promote development of disease in generations remote from the initial chemical exposure [71,72]. In a recent study, the effects of perinatal BPA exposure were found to be dose-, sex-and time-dependent, further suggesting that the perinatal period is a critical window of EDC susceptibility [64]. Multiple studies have shown that exposure to BPA during pregnancy can alter metabolic homeostasis in both the mother and in her adult offspring [63,73,74]. Similarly, gestational and lactational exposure to perfluorooctane sulfonate (PFOS), a worldwide industrial pollutant once commonly used in stain and water repellents [75], was shown to impair glucose and lipid homeostasis in adult rats [76,77]. Thus, consideration of not only the chemical but the timing of exposure across the lifespan is critical for assessing the effects of EDCs on energy metabolism.
In addition to studies demonstrating connections between EDCs and pro-atherogenic disruptions in glucose metabolism and insulin action, several studies have specifically interrogated the role of environmental toxicants in the development of atherosclerosis and other atherosclerosis-associated risk factors (Table 1). Apolipoprotein-E-deficient (ApoE−/−) mice are commonly used as a model of macrovascular disease since these mice are susceptible to the development of diet-induced atherosclerotic plaques [78]. Subchronic treatment of ApoE−/− mice with 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) demonstrated a trend toward earlier onset and greater severity of atherosclerotic lesions in one model of exposure [79], while a second study revealed that TCDD aggravated atherosclerosis progression, an effect enhanced by high fat diet feeding [80]. Polycyclic aromatic hydrocarbons (PAHs) (e.g. benzo[a]pyrene) have also been shown to increase the size of atherosclerotic plaques in ApoE−/− mice [81], and in a more recent study, exposure to ambient particular matter air pollution from Beijing increased aortic arch atherosclerotic plaque growth in ApoE−/− mice [82].
Table 1.
Models of EDC-mediated in vivo disruption of vascular and metabolic function
| Pollutant | Animal Model | Dose | Physiological Alterations | Ref. |
|---|---|---|---|---|
| Arsenic | FvB mice | 100 ppb, drinking water (6 months) |
|
[135] |
| Wistar Kyoto rats | 133 μg/ml, drinking water (20 weeks) |
|
[86] | |
| Atrazine | Sprague-Dawley rats | 30 or 300 μg/kg/day (5 months) |
|
[67] |
| Benzo[a]pyrene | ApoE−/− mice | 5 mg/kg, once/week, orally (24 weeks) |
|
[81] |
| BPA | Wistar rats (gestational/lactational exposure) | 50 μg/kg/day, offspring fed on HFD |
|
[74] |
| Wistar rats (gestational/lactational exposure) | 50 μg/kg/day, oral gavage |
|
[163] | |
| OF-1 mice (gestational exposure) | 10 or 100 μg/kg/day |
|
[63] | |
| CD-1 mice (gestational exposure) | 5 – 5×104 μg/kg/day |
|
[73] | |
| ICR mice (gestational/lactational exposure) | 1 or 10 μg/ml, in drinking water |
|
[87] | |
| Cadmium | ApoE−/− mice | 100 μg/L, in drinking water |
|
[57] |
| Male Sprague-Dawley rats | 2 mg/kg/day (4 days) |
|
[164] | |
| Male Wistar rats | 5 or 50 mg/l, in drinking water (6 months) |
|
[84] | |
| Male Albino rats | 1 mg/kg/day, i.p. (4 weeks) |
|
[85] | |
| Male Slc:ICR mice | 0, 5, 10, 20 μM/kg, subcutaneously (2 weeks) |
|
[165] | |
| Combustion emissions | Male ApoE−/− mice | 6 h/day inhalation exposure (50 days) |
|
[166] |
| Diazinon | Sprague-Dawley rats | 0.5 or 2 mg/kg/day, subcutaneously (4 days) |
|
[143] |
| Dioxins / TCDD | C57BL/6 and ApoE−/− mice | 5 μg/kg, i.p. (3 days) |
|
[167] |
| C57BL/6 mice | 10 μg/kg, i.p. |
|
[94] | |
| C57BL/6 mice | 300 ng/kg, oral gavage, 3× week (60 days) |
|
[168] | |
| ApoE−/− mice | 15 μg/kg, i.p. (once) |
|
[80] | |
| Malathion | Male Wistar rats | 100–400 ppm, orally (4 weeks) |
|
[169] |
| Nicotine | Wistar rats (transgenerational exposure) | 1 mg/kg/day, subcutaneous injection |
|
[91] |
| Sprague-Dawley rats (gestational exposure) | 4 μg/kg/min. via osmotic mini-pump |
|
[140] | |
| PCBs | ApoE−/− mice | 49 mg/kg, i.p. |
|
[133] |
| C57BL/6 mice | 50 mg/kg, oral gavage |
|
[59] | |
| C57BL/6 mice | 36 mg/kg/wk (20 wks) |
|
[62] | |
| AhR+/+ (mixed) mice | 170 μmol/kg, i.p. |
|
[170] | |
| ApoE−/− mice | 49 mg/kg, i.p. |
|
[129] | |
| Sprague-Dawley rats | 224 μg/kg, i.p. (total dosage) |
|
[171] | |
| PFOS | Wistar rats (gestational/lactational exposure) | 0.5 or 1.5 mg/kg/day |
|
[76] |
| PM2.5 | C57BL/6 mice | 10 weeks, whole-body exposure (inhalation) |
|
[110] |
| Various mouse strains | 6 h/day, 5 days/week (20 weeks) |
|
[172] | |
| Male ApoE−/− mice | 6 h/day, 5 days/week (2 months) |
|
[120] | |
| Male Sprague-Dawley rats | Once per week, intratracheal installation (3 weeks) |
|
[68] | |
| C57BL/6 mice | 6hrs/day, 5 days/week (128 days) |
|
[112] | |
| Tributyltin | C57BL/6 mice (gestational exposure) | 0.05 or 0.5 mg/kg/day |
|
[173] |
| Male KM mice | 0.5, 5, 50 μg/kg, once/3 days (45 days total) |
|
[174] | |
| Triflumizole | CD-1 mice (gestational exposure) | 0.1, 1, 10 μM, in drinking water |
|
[175] |
Disruptions in lipid metabolism can lead to the development of an atherogenic dyslipidemia, including an increase in small, dense LDL, elevated triglycerides, and reduced anti-atherogenic HDL. High concentrations of circulating atherogenic lipoproteins enhances lipid accumulation in the subendothelial space where oxidized-LDL (oxLDL) is taken up by macrophages, generating “foam cells” and triggering an inflammatory cascade resulting in formation and progression of atherosclerotic plaques [83]. Environmental pollutants that promote the dysregulation of lipid metabolism, therefore, are predicted to enhance the risk of macrovascular disease. In male rats, cadmium exposure was found to increase plasma free fatty acids and LDL while also decreasing HDL [84]. Similar effects of cadmium on LDL and HDL were observed in a second model of rat exposure that also demonstrated an increase in serum triglycerides [85]. Coupled with a high cholesterol diet, arsenic was shown to promote a pro-atherogenic reduction in the HDL-to-LDL cholesterol ratio without altering total cholesterol or triglyceride levels [86]. In ApoE−/− mice, TCDD exposure was shown to increase LDL levels [79]. The increased atherogenesis observed in ApoE−/− mice exposed to ambient particular matter was associated with an increase in serum total cholesterol and LDL-C [82]. Similar to models of diabetes, developmental exposure to BPA has been shown to increase total serum cholesterol levels [87], while in utero TCDD attenuated HDL-C increases in high-fat diet-fed ApoE−/− mice [88].
Hypertension is a key risk factor in the development of CVD. Nicotine, highly prevalent in human populations mainly as a result of voluntary exposure, represents a potentially hazardous compound with potentially high levels of in utero exposure from smoking mothers [89,90]. Offspring of exposed mothers had elevated blood pressure, demonstrating cardiovascular abnormalities resulting from nicotine exposure [91]. A separate study found that nicotine exposure promoted atherosclerotic lesion growth in a mouse model of the disease [92]. This diverse set of data suggests that various environmental contaminants in a variety of experimental contexts have the capacity to promote dysregulation of energy metabolism while facilitating the development of atherosclerosis and its associated risk factors.
Mechanisms of EDC-Induced Metabolic Dysregulation and Cardiovascular Risk Factors
Studies at the population and animal levels have providing intriguing insights into the potential role of environmental toxicants in the pathogenesis of diabetes and macrovascular disease; however, they fail to fully characterize the molecular mechanisms by which EDCs exert their deleterious effects. In order to identify pathophysiological pathways, predict novel EDCs, and develop novel therapeutic targets, several studies have aimed to identify the molecular mechanisms responsible for pro-diabetic and pro-atherogenic environmental toxicants. These studies show that environmental pollutants indicated in the pathogenesis of T2DM and CVD in vivo can modulate important cellular events involved in insulin production and glucose homeostasis, and also disrupt processes crucial for regulating vascular health (Figure 1).
Figure 1. Contributions of environmental pollutants to cardiovascular disease pathology.
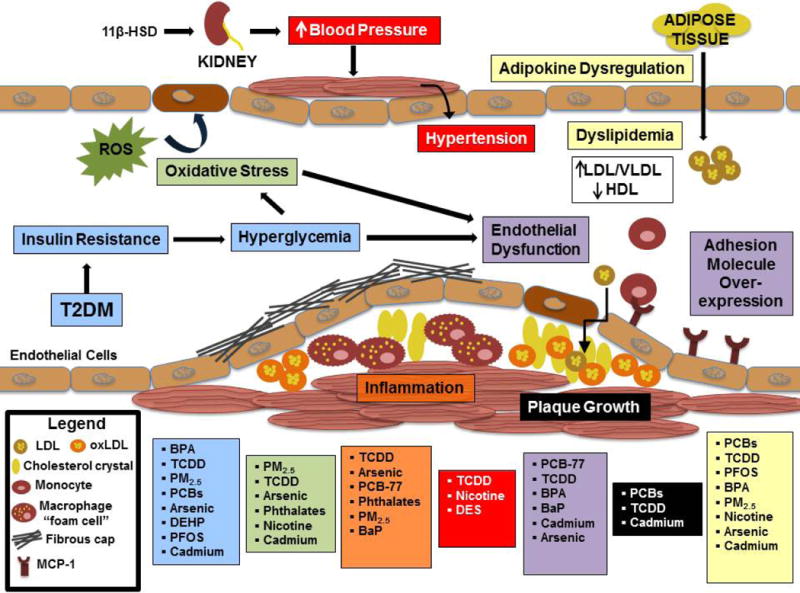
This figure represents mechanisms seen in either in vivo exposure studies or in vitro cellular models.
In healthy individuals glucose levels are maintained within a very tight range through an augmentation of insulin secretion from pancreatic β cells in response to increases in insulin resistance [93]. Under conditions of significant and sustained insulin resistance, however, β cells begin to lose their ability to adequately compensate at times of peak demand, and the individuals transitions to a state of impaired glucose tolerance. Ultimately, the persistent β-cell stress results in insufficient insulin secretion even during periods of fasting, and the patient enters a state of frank T2DM. Thus, EDCs that impair β-cell insulin secretion or interfere with peripheral insulin action can promote the development of T2DM. The effects of various environmental contaminants on β-cell physiology and insulin action have been examined (reviewed in ref. [Sargis, Diabetes & Metabolism Journal, in press]). Several compounds have been shown to disrupt β-cell function, promote β-cell death, or disrupt signal transduction pathways in β cells. These include organic compounds such as TCDD [94,95,96], PCBs [97], BPA [98,99] and triphenyltin [100]. Inorganic compounds have also been shown to modulate β-cell function as well, including cadmium [101] and mercury [102]. Furthermore, arsenic in both its inorganic and methylated forms has been shown to disrupt β-cell function [103,104]. Interestingly, BPA [98] and PCBs [105] have been shown to augment insulin secretion; however, this may still reflect a deleterious disruption in energy homeostasis, possibly through insulin-induced insulin resistance.
In addition to those compounds affecting insulin secretion, a number of compounds have been shown to antagonize cellular insulin action in a variety of experimental systems. TCDD [106], BPA [107] and DEHP [108] have also been shown to reduce insulin receptor levels in some studies, whereas the phenylsulfamide fungicide tolylfluanid [109], particulate matter [110], TCDD [106] and DEHP [108] have been shown to reduce levels of the insulin receptor substrate-1 (IRS-1), a key intermediate in the insulin signaling cascade. Downstream of IRS-1, the insulin-stimulated activating phosphorylation of Akt (protein kinase B) has been shown to be attenuated by a host of environmental toxicants, including arsenic [111], particular matter [112], PCB-77 [113], tolylfluanid [109] and BPA [63], while arsenic has also been shown to antagonize insulin action distal to Akt phosphorylation [114]. Finally, antagonism of cellular insulin action at the level of the facilitative glucose transporter, type 4 has been shown for DEHP [108], TCDD [106], and cadmium [115]. Thus, a host of environmental pollutants have the capacity to alter energy homeostasis through a variety of cellular mechanisms that are predicted to promote the development of diabetes and its associated complications, including vascular injury.
In addition to direct effects on insulin production or insulin action, EDCs may augment the risk of T2DM indirectly by altering the expression of the various secreted factors that modulate global insulin sensitivity. For example, adipose tissue plays a critical role in energy metabolism through the secretion of a number of adipokines. Adiponectin is one such secretory product that promotes insulin sensitivity while also exerting anti-inflammatory effects and promoting β-cell function [116]. Environmentally relevant doses of BPA suppress adiponectin release from adipose tissue ex vivo [117]. In addition, cadmium [118], tributyltin [119], and particulate matter [120] have also been shown to reduce adiponectin expression and/or release. In addition to promoting dysglycemia, EDC-induced reductions in adiponectin may also accelerate atherosclerosis, as this adipokine appears to play an important protective role in the vasculature by suppressing foam cell formation and promoting macrophage cholesterol efflux [121]. In contrast, tumor necrosis factor-α (TNFα) and IL-6 induce insulin resistance and are increased by TCDD [106], PCB-77 [59,113], and particulate matter [112]. EDC-induced changes in these pro-inflammatory mediators may also play a role in enhancing the development of atherosclerosis (reviewed in ref. [122]).
Atherosclerosis is a multifaceted disease resulting from multiple metabolic and inflammatory derangements. Atherogenic, apolipoprotein-B-containing lipoproteins infiltrate the subendothelial space where they can undergo oxidative modification. Oxidized lipoproteins cause alterations in endothelial function and protein expression that lead to the recruitment of monocytes to the subendothelial space. These infiltrating monocytes are activated, accumulate lipid, and transform into macrophage “foam cells.” This results in a cascade of inflammatory responses that recruits other cell types, including smooth muscle cells. As the atherosclerotic plaque enlarges and becomes necrotic, a fibrous cap forms, rupture of which promotes platelet adherence and initiates the coagulation cascade resulting in acute occlusion of the vessel lumen. EDCs that modulate any of these events or that alter risk factors associated with these processes are predicted to promote the development of macrovascular disease.
Several EDCs have been examined for their effects on mechanisms specific to the development of CVD (Table 2). Vascular inflammation, a hallmark of atherosclerosis, has been studied extensively in cellular models of exposure, including exposure to PCBs [123], and particulate matter [124]. PCB-77 [125,126] and the PAH benzo[a]pyrene [127] have been shown to up-regulate expression of monocyte chemoattractant protein-1 (MCP-1), an inducible cytokine responsible for attracting circulating monocytes to sites of inflammation (e.g. early atherosclerotic lesion). TCDD and other PCB congeners have also been shown to up-regulate MCP-1 expression in other tissues; however, whether these effects are also observed in the vascular tissue is incompletely known [128,129]. Similarly, endothelial dysfunction and inflammation have been shown in animal models of exposure to cadmium [130] and PCBs [126,131]. Endothelial dysfunction, in turn, is known to promote the progression of inflammatory vascular diseases, including atherosclerosis [132]. Activation of the aryl hydrocarbon receptor (AhR) induces vascular inflammation and promotes atherosclerosis in apoE−/− mice, and the AhR is also a common target for a number of diverse EDCs including TCDD [94], dioxin-like PCBs [133], and the fungicide cyprodinil [134]. Systemic inflammation has also been shown to be increased in mice exposed to aerosolized particulate matter [110] and arsenite [135], and TCDD exposure was associated with the induction of global inflammatory gene responses [80].
Table 2.
Mechanisms linking environmental pollutants to disruption of vascular biology
| Pollutant | Mechanism of Disruption | Model System | Ref. |
|---|---|---|---|
| 2,4-D | Impaired glucose uptake and metabolism | Rat Sertoli cells | [176] |
| Arsenic / iAS | Activation of NFκB through increased C-reactive protein expression | HepG2 cells | [135] |
| Augmented uptake of ox-LDL | Mouse aortic endothelial cells | [139] | |
| Impaired glucose-stimulated insulin secretion by β cells | INS-1(832/13) cells | [104] | |
| Impaired insulin-stimulated glucose uptake, induction of inflammatory response genes | 3T3-L1 adipocytes | [111] | |
| Atrazine | Potentiated cAMP activity | Rat pituitary and testicular Leydig cells | [144] |
| Benzo[a]pyrene | Increased MCP-1 expression | HUVEC | [127] |
| BPA | Disrupted endothelial cell proliferation with mitotic abnormalities | HUVEC | [142] |
| Suppressed adiponectin release | Primary human adipose tissue | [117] | |
| Modulated metabolic and inflammatory gene expression | Human PBMCs | [177] | |
| Cadmium | Impaired glucose tolerance | Primary rat adipose tissue | [164] |
| Increased ROS production, increased oxidative stress | Isolated rat aortas | [130] | |
| Increased vascular endothelial permeability, inhibited cell proliferation | HUVE cells | [57] | |
| Cyprodinil | Induced nuclear translocation and transcriptional activity of the AhR | HO-23 cells | [134] |
| DEHP | Increased H2O2 and OH− radicals, decreased glucose uptake | Primary rat adipose tissue | [108] |
| Nonylphenol | Enhanced ischemia/reperfusion injury | Guinea pig heart | [178] |
| PM2.5 | Pro-inflammatory cytokine induction, C-reactive protein induction | U937 cells | [179] |
| PCB-77 | Increased MCP-1 expression | Primary mouse endothelial cells | [126] |
| Pro-inflammatory cytokine expression and release | 3T3-L1 adipocytes | [129] | |
| PCB-126 | Altered metabolic gene expression | Human PBMCs | [177] |
| TCDD (dioxin) | Decreased nuclear ER levels through aryl hydrocarbon receptor (AhR) | MCF-7 cells | [180] |
| Impaired insulin-stimulated glucose uptake, induction of inflammatory response (TNF-α) genes | 3T3-L1 adipocytes | [106] | |
| Impaired glucose-stimulated insulin secretion by β cells | Primary mouse pancreatic islets | [94] | |
| Impaired glucose-stimulated insulin secretion by β cells | INS-1E β-cell line | [95] | |
| Altered gene expression associated with cell growth and cell death | Mouse VSMCs | [181] |
AhR, aryl hydrocarbon receptor; iAS, inorganic arsenic species; 2,4-D, 2,4-dichlorophenoxyacetic acid; HUVEC, human umbilical vascular endothelial cells; PBMCs, peripheral blood mononuclear cells; ROS, reactive oxygen species,
Oxidative stress is an important pathway in the initiation and progression of the atherosclerotic lesion through the oxidative modification of lipoproteins and the induction of cellular dysfunction [136,137]. Persistent hyperglycemia, as seen in uncontrolled diabetes, induces endothelial dysfunction, likely through increased oxidative stress [138]. Thus, EDCs that promote the development of diabetes may also aggravate the development of atherosclerosis. A recent epidemiological study showed that urinary phthalate (e.g. DEHP) levels correlated with levels of the oxidative stress marker malondialdehyde [32], further suggesting this mechanism as relevant for EDC-mediated atherosclerotic disease. Uptake of oxidized LDL by endothelial cells has been shown to be up-regulated by arsenic [139]; whether arsenic has similar effects in promoting macrophage foam cell formation, however, requires further study. In utero exposure to nicotine has been shown to promote vascular dysfunction mediated by oxidative stress [90,140]. Acute cadmium exposure in isolated rat aortic rings induced vascular injury by increasing endothelial cell oxidative stress [130], and co-planar PCBs have been shown to induce oxidative stress in vascular endothelial cells through the NF-κB pathway [131].
In addition to their effects on oxidative stress, selected PCB congeners were also found to decrease the angiogenic capacity human umbilical vein endothelial cells (HUVEC) [141]. A follow-up study showed that the endothelial toxicity of co-planar PCBs could be inhibited by metabolites produced from the oxidation of omega-3 fatty acids, providing a possible mechanism to explain the anti-inflammatory properties of these lipid species [123]. A recent study also showed that BPA at environmentally relevant doses interferes with endothelial cell proliferation and mitotic division [142]. Together, these studies suggest that some EDCs may drive atherosclerotic lesion progression by inducing endothelial cell dysfunction while also antagonizing reparative mechanisms critical for restoring blood flow.
Hypertension is a clear risk factor for micro- and macrovascular disease, and promotes atherosclerosis by enhancing sheer stresses and endothelial inflammation mediated by oxidative stress. Blood pressure is regulated by a host of local and systemic signaling molecules, some of which have been shown to be modulated by EDCs. Developmental exposure of rats to nicotine resulted in an enhanced blood pressure response to angiotensin II in adulthood [90]. Catecholamines play a critical role in regulating blood pressure through their action on adrenergic G-protein coupled receptors, some of which signal through adenylate cyclase. Organophosphorus pesticides, including diazinon and parathion, have been shown to up-regulate adenylate cyclase activity in a model of neonatal rat exposure [143]. Additionally, exposure to the herbicide atrazine has been shown to potentiate cAMP signaling in cultured rat pituitary cells [144]. Thus, EDC-induced sensitization of the cAMP pathway to catecholamines may potentiate their effects on blood pressure. In the kidneys, aldosterone acting on mineralocorticoid receptors plays a critical role in regulating blood pressure. Because cortisol circulates at much higher concentrations than aldosterone and has affinity for mineralocorticoid receptors, cortisol-induced activation of these receptors is prevented physiologically through the local inactivation of glucocorticoids by 11β-hydroxysteroid dehydrogenase, type 2 (11β-HSD-2). Several studies have examined the capacity for EDCs to inhibit this enzyme. The dithiocarbamate pesticide Thiram [145], organotin compounds [146], and cadmium [147] all exhibit the capacity to inhibit this enzyme. Since antagonism of 11β-HSD-2 would be predicted to elevate blood pressure, determining whether these or other EDCs enhance the development of macrovascular disease through the promotion of hypertension is worthy of further investigation. Finally, because insulin exerts vasodilatory effects on the vasculature, compounds discussed that induce insulin resistance may also raise blood pressure through antagonism of insulin action on vascular smooth muscle.
Disruption of nuclear hormone signaling may play a critical role in mediating the negative vascular effects of EDCs. Sex steroids play a critical role in lipoprotein metabolism, and disruption of sex steroid signaling has been a central area of interest in the field of endocrine disruption. A variety of compounds have been shown to modulate estrogenic and/or androgenic signaling (reviewed in ref. [20,148,149]), and some of these compounds may promote the development of an atherogenic lipid profile. Similarly, thyroid hormone is known to play a critical role in lipoprotein metabolism [150]. Disruption of thyroid hormone action has also been described for a number of different EDCs, including hydroxylated PCBs [151,152]. Whether these or other compounds have the capacity to augment cardiovascular risk through the modulation of lipoprotein metabolism requires further study. Finally, states of glucocorticoid excess (i.e. Cushing’s Syndrome) are characterized by hypertension, dyslipidemia, and hyperglycemia, in addition to other abnormalities. Agents that promote glucocorticoid signaling may therefore contribute to elevated CVD risk through multiple mechanisms. Tolylfluanid was shown to stimulate glucocorticoid action in adipose tissue [153], while studies in rat EDR3 cells showed that low doses of arsenic stimulated GR-mediated gene transcription [154]. These studies suggest a potential role for EDC disruption of glucocorticoid signaling as a potential mediator of metabolic and vascular disease. Finally, other nuclear hormones that play a role in pathways regulating energy and lipid metabolism [e.g. liver X receptor and steroid and xenobiotic receptor (SXR)] may also be important sites of endocrine disruption of vascular health [155].
In addition to further clarification of these mechanisms, a greater understanding is required of how EDCs modulate lipoprotein metabolism, including lipid synthesis, transport, enzymatic and non-enzymatic modification, and receptor-mediated uptake. Because many EDCs are highly lipophilic, they are transported in the circulation incorporated into lipoproteins. As such, they are enriched at sites of lipid metabolism and may have unique potent effects on cellular processes governing lipid handling. For example, it has been shown that the fatty acid and phospholipid composition of lipid membranes and lipoproteins as well as membrane fluidity modulate lipid oxidation, a key step in atherogenesis [156,157]. Likewise, the physical properties of membranes govern cholesterol efflux [158], and intercalation of lipophilic EDCs into membranes may modulate this process. Enzymatic action on lipoproteins is also governed by lipid composition, which may influence delivery of critical fatty acids to tissues such as the brain [159]. Likewise, it will be important to determine whether EDCs modulate activity of proteins mediating lipid uptake and efflux in order to fully appreciate their role in contributing to vascular disease. Whether lipid soluble environmental contaminants alter metabolic function by interfering with lipid handling remains largely unresolved. Studies correlating EDC exposure with alterations in circulating levels of the CVD biomarker serum matrix metalloproteinase-9 levels [43,44], suggest that specific investigations examining EDC effects on the extracellular matrix of the atherosclerotic plaque as well as plaque stability are warranted. Finally, as effects are often concentration-dependent and non-monotonic [73,160,161], better assessment of human exposure to EDCs is required to inform these mechanistic studies as well as to improve risk assessment.
Potential Clinical Implications and Conclusions
The current state of scientific evidence supports a potential role for EDCs in the pathogenesis of metabolic diseases and atherosclerotic disease. Whether any specific individual can tie their disease to a particular exposure, however, is much more complex given the immense heterogeneity of chemicals, concentrations, combinations, timing, and duration of exposure. However, as improvements are made in identifying risk to specific individuals, current data may suggest how an individual’s EDC exposure may influence both metabolic disease development and response to specific therapies. For example, SXR regulates expression of cytochrome P450 enzymes [162], and this nuclear receptor is disrupted by multiple EDCs, including DDT and nonylphenol [155]. This suggests that individuals with exposure to these EDCs may experience differential efficacy or enhanced side effects from drugs metabolized by these enzymes, including key CVD drugs such as statins. Evidence that atrazine and organophosphate pesticides can potentiate the action of adenylate cyclase [143,144] may suggest enhanced efficacy for adrenergic blockers in the treatment of hypertension or the use of agents with anti-glucagon effects in the treatment of diabetes in patients exposed to these agents. Patients with known ongoing exposure to EDCs that inhibit 11β-HSD-2 may experience enhanced benefit with agents targeting the mineralocorticoid receptor (e.g. spironolactone). Similarly, those with exposures contributing to the development of their diabetes may receive preferential benefit with either insulin replacement or an insulinsensitizing agent depending on the mechanisms of action of the EDCs to which they are exposed. Given the fact that there are over 150,000 chemicals registered with the European Chemicals Agency [22], most exposure is polychemical, many chemicals have multiple mechanisms of action, dose-effect relationships are sometimes characterized as nonmonotonic, and phenotypes are influenced by the timing of exposure; it may be impossible to achieve the level of granularity that would lead to specific conclusions regarding the efficacy of any particular treatment. However, in instances of known exposures (e.g. occupational, recreational, or accidental), our burgeoning knowledge base may ultimately provide us with the tools necessary to improve the care of specific patient groups.
The last two decades has witnessed an important transformation in our knowledge of toxicity with the recognition that environmental pollutants have the capacity to modulate endocrine and metabolic signaling pathways, opening the door to a greater appreciation of the myriad factors contributing to the burgeoning global metabolic disease epidemic. Epidemiological studies support a role of a variety of organic and inorganic pollutants in the development of insulin resistance, obesity, diabetes, and cardiovascular disease; and these studies are supported by preclinical studies associating individual exposures with specific mechanisms of disease development. While many challenges remain, the current level of evidence supports the hypothesis that deleterious health consequences, at least in part, arise from our exposure to a milieu of environmental toxicants. It is hoped that scientific advances at the population and basic science levels will permit us to better address this important issue through environmental remediation, targeted therapies, and sound public policy.
Acknowledgments
Due to reference constraints the authors were unable to include all the important work performed in the field of endocrine disruption of metabolism and cardiovascular disease. The current manuscript was meant to emphasize important aspects of environmental disruption of energy homeostasis and cardiovascular risk; any omissions were not meant to exclude important work contributing to the hypothesis that environmental contaminants play an important pathogenic role in the global epidemic of metabolic and cardiovascular disease. This work was supported by grants from the National Institutes of Health (K08-ES019176, R21-ES021354, and the Diabetes Research and Training Center [P60-DK020595]).
Abbreviations
- AhR
aryl hydrocarbon receptor
- ApoE
apolipoprotein E
- BaP
benzo[a]pyrene
- BPA
bisphenol A
- cIMT
carotid intima-media thickness
- CVD
cardiovascular disease
- DDE
dichlorodiphenyldichloroethylene
- DEHP
diethylhexylphthalate
- DDT
dichlorodiphenyltrichloroethane
- EDCs
endocrine disrupting chemicals
- GLUT4
glucose transporter type 4
- HDL
high density lipoprotein
- 11β-HSD-2
11β-hydroxysteroid dehydrogenase, type 2
- HUVEC
human umbilical vein endothelial cells
- iAs
inorganic arsenic
- IRS-1
insulin receptor substrate-1
- LDL
low-density lipoprotein
- MCP-1
monocyte chemoattractant protein-1
- NTP
National Toxicology Program
- oxLDL
oxidized low density lipoprotein
- PAHs
polycyclic aromatic hydrocarbons
- PCBs
polychlorinated biphenyls
- PFOS
perfluorooctane sulfonate
- PM
particulate matter
- POPs
persistent organic pollutants
- ROS
reactive oxygen species
- SXR
steroid and xenobiotic receptor
- T2DM
type 2 diabetes mellitus
- TCDD
2,3,7,8-tetrachlorodibenzo-p-dioxin
- TNFα
tumor necrosis factor α
- VLDL
very low density lipoprotein
Footnotes
Compliance with Ethics Guidelines
Conflict of Interest
Andrew G. Kirkley declares that he has no conflict of interest.
Robert M. Sargis has received honoraria from the Korean Diabetes Association.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
References
- 1.Centers for Disease Control and Prevention. National diabetes fact sheet: national estimates and general information on diabetes and prediabetes in the United States, 2011. US Department of Health and Human Services, Centers for Disease Control and Prevention; 2011. [Google Scholar]
- 2.ADA. Economic costs of diabetes in the U.S. in 2012. Diabetes Care. 2013;36:1033–1046. doi: 10.2337/dc12-2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Imperatore G, Boyle JP, Thompson TJ, Case D. Projections of Type 1 and Type 2 Diabetes Burden in the U.S. Population Aged <20 Years Through 2050. Diabetes Care. 2012;35:2515–2520. doi: 10.2337/dc12-0669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gregg EW, Boyle JP, Thompson TJ, Barker LE. Modeling the impact of prevention policies on future diabetes prevalence in the United States: 2010–2030. Population Health Metrics. 2013;11 doi: 10.1186/1478-7954-11-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.IDF. Diabetes Atlas. 2013. 6th. [Google Scholar]
- 6.Matheus AS, Tannus LR, Cobas RA, Palma CC, Negrato CA, et al. Impact of diabetes on cardiovascular disease: an update. Int J Hypertens. 2013;2013:653789. doi: 10.1155/2013/653789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cherian B ea. Therapeutic implications of diabetes in cardiovascular disease. American Journal of Therapeutics. 2009;16:e51–e59. doi: 10.1097/MJT.0b013e31815db924. [DOI] [PubMed] [Google Scholar]
- 8.Libby P, Nathan DM, Abraham K, Brunzell JD, Fradkin JE, et al. Report of the National Heart, Lung, and Blood Institute-National Institute of Diabetes and Digestive and Kidney Diseases Working Group on Cardiovascular Complications of Type 1 Diabetes Mellitus. Circulation. 2005;111:3489–3493. doi: 10.1161/CIRCULATIONAHA.104.529651. [DOI] [PubMed] [Google Scholar]
- 9.Grundy SM, Benjamin IJ, Burke GL, Chait A, Eckel RH, et al. Diabetes and Cardiovascular Disease : A Statement for Healthcare Professionals From the American Heart Association. Circulation. 1999;100:1134–1146. doi: 10.1161/01.cir.100.10.1134. [DOI] [PubMed] [Google Scholar]
- 10.Carnethon MR, Biggs ML, Barzilay J, Kuller LH, Mozaffarian D, et al. Diabetes and coronary heart disease as risk factors for mortality in older adults. Am J Med. 2010;123:556, e551–559. doi: 10.1016/j.amjmed.2009.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hill JO. Environmental Contributions to the Obesity Epidemic. Science. 1998;280:1371–1374. doi: 10.1126/science.280.5368.1371. [DOI] [PubMed] [Google Scholar]
- 12.Baillie-Hamilton PF. Chemical Toxins: A Hypothesis to Explain the Global Obesity Epidemic. THE JOURNAL OF ALTERNATIVE AND COMPLEMENTARY MEDICINE. 2002;8:185–192. doi: 10.1089/107555302317371479. [DOI] [PubMed] [Google Scholar]
- 13•.Thayer KA, Heindel JJ, Bucher JR, Gallo MA. Role of environmental chemicals in diabetes and obesity: a National Toxicology Program workshop review. Environ Health Perspect. 2012;120:779–789. doi: 10.1289/ehp.1104597. An important overview of the work performed by the National Toxicology Program in reviewing the evidence linking various pollutants with the development of metabolic disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diamanti-Kandarakis E, Bourguignon JP, Giudice LC, Hauser R, Prins GS, et al. Endocrine-disrupting chemicals: an Endocrine Society scientific statement. Endocr Rev. 2009;30:293–342. doi: 10.1210/er.2009-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee DH, Lee IK, Porta M, Steffes M, Jacobs DR., Jr Relationship between serum concentrations of persistent organic pollutants and the prevalence of metabolic syndrome among non-diabetic adults: results from the National Health and Nutrition Examination Survey 1999–2002. Diabetologia. 2007;50:1841–1851. doi: 10.1007/s00125-007-0755-4. [DOI] [PubMed] [Google Scholar]
- 16.Finkelstein EA, Trogdon JG, Cohen JW, Dietz W. Annual medical spending attributable to obesity: payer-and service-specific estimates. Health Aff (Millwood) 2009;28:w822–831. doi: 10.1377/hlthaff.28.5.w822. [DOI] [PubMed] [Google Scholar]
- 17.Kuo CC, Moon K, Thayer KA, Navas-Acien A. Environmental chemicals and type 2 diabetes: an updated systematic review of the epidemiologic evidence. Curr Diab Rep. 2013;13:831–849. doi: 10.1007/s11892-013-0432-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alonso-Magdalena P, Quesada I, Nadal A. Endocrine disruptors in the etiology of type 2 diabetes mellitus. Nat Rev Endocrinol. 2011;7:346–353. doi: 10.1038/nrendo.2011.56. [DOI] [PubMed] [Google Scholar]
- 19.Lee DH, Steffes MW, Sjodin A, Jones RS, Needham LL, et al. Low dose of some persistent organic pollutants predicts type 2 diabetes: a nested case-control study. Environ Health Perspect. 2010;118:1235–1242. doi: 10.1289/ehp.0901480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Coster S, van Larebeke N. Endocrine-disrupting chemicals: associated disorders and mechanisms of action. J Environ Public Health. 2012;2012:713696. doi: 10.1155/2012/713696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Regnier SM, Sargis RM. Adipocytes under assault: Environmental disruption of adipose physiology. Biochim Biophys Acta. 2013 doi: 10.1016/j.bbadis.2013.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22•.Lind L, Lind PM. Can persistent organic pollutants and plastic-associated chemicals cause cardiovascular disease? J Intern Med. 2012;271:537–553. doi: 10.1111/j.1365-2796.2012.02536.x. An excellent review suggesting that environmental contaminants may play a role in cardiovascular disease. This review also discusses the sources of exposure to several chemicals linked to cardiovascular disease. [DOI] [PubMed] [Google Scholar]
- 23••.Taylor KW, Novak RF, Anderson HA, Birnbaum LS, Blystone C, et al. Evaluation of the association between persistent organic pollutants (POPs) and diabetes in epidemiological studies: a national toxicology program workshop review. Environ Health Perspect. 2013;121:774–783. doi: 10.1289/ehp.1205502. A comprehensive review of the literature linking persistent organic pollutants with the development of diabetes. Based on this analysis, the authors concluded that there is sufficient evidence to suggest a link between persistent organic pollutants and diabetes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24•.Maull EA, Ahsan H, Edwards J, Longnecker MP, Navas-Acien A, et al. Evaluation of the association between arsenic and diabetes: a National Toxicology Program workshop review. Environ Health Perspect. 2012;120:1658–1670. doi: 10.1289/ehp.1104579. A comprehensive review of the evidence linking arsenic to diabetes. While the authors note that the connection is not firm, there is some evidence supporting a potential role for arsenic in the development of diabetes in certain populations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vandenberg LN, Colborn T, Hayes TB, Heindel JJ, Jacobs DR, Jr, et al. Hormones and endocrine-disrupting chemicals: low-dose effects and nonmonotonic dose responses. Endocr Rev. 2012;33:378–455. doi: 10.1210/er.2011-1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lang IA, Galloway TS, Scarlett A. Association of Urinary BisphenolA Concentration With Medical Disorders and Laboratory Abnormalities in Adults. JAMA. 2008;300:1303–1310. doi: 10.1001/jama.300.11.1303. [DOI] [PubMed] [Google Scholar]
- 27.Gasull M, Pumarega J, Tellez-Plaza M, Castell C, Tresserras R, et al. Blood concentrations of persistent organic pollutants and prediabetes and diabetes in the general population of Catalonia. Environ Sci Technol. 2012;46:7799–7810. doi: 10.1021/es300712g. [DOI] [PubMed] [Google Scholar]
- 28.Hoogduijn MJ, Rakonczay Z, Genever PG. The effects of anticholinergic insecticides on human mesenchymal stem cells. Toxicol Sci. 2006;94:342–350. doi: 10.1093/toxsci/kfl101. [DOI] [PubMed] [Google Scholar]
- 29.Roos V, Rönn M, Salihovic S, Lind L, Bavel Bv, et al. Circulating Levels of Persistent Organic Pollutants in Relation to Visceral and Subcutaneous Adipose Tissue by Abdominal MRI. Obesity. 2012 doi: 10.1002/oby.20267. [DOI] [PubMed] [Google Scholar]
- 30.Frederiksen H, Skakkebaek NE, Andersson AM. Metabolism of phthalates in humans. Mol Nutr Food Res. 2007;51:899–911. doi: 10.1002/mnfr.200600243. [DOI] [PubMed] [Google Scholar]
- 31.Stahlhut RW, van Wijngaarden E, Dye TD, Cook S, Swan SH. Concentrations of urinary phthalate metabolites are associated with increased waist circumference and insulin resistance in adult U.S. males. Environ Health Perspect. 2007;115:876–882. doi: 10.1289/ehp.9882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim JH, Park HY, Bae S, Lim YH, Hong YC. Diethylhexyl phthalates is associated with insulin resistance via oxidative stress in the elderly: a panel study. PLoS One. 2013;8:e71392. doi: 10.1371/journal.pone.0071392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang T, Li M, Chen B, Xu M, Xu Y, et al. Urinary bisphenol A (BPA) concentration associates with obesity and insulin resistance. J Clin Endocrinol Metab. 2012;97:E223–227. doi: 10.1210/jc.2011-1989. [DOI] [PubMed] [Google Scholar]
- 34.Chang JW, Chen HL, Su HJ, Liao PC, Guo HR, et al. Dioxin exposure and insulin resistance in Taiwanese living near a highly contaminated area. Epidemiology. 2010;21:56–61. doi: 10.1097/EDE.0b013e3181c2fc6e. [DOI] [PubMed] [Google Scholar]
- 35.Brook RD, Xu X, Bard RL, Dvonch JT, Morishita M, et al. Reduced metabolic insulin sensitivity following sub-acute exposures to low levels of ambient fine particulate matter air pollution. Sci Total Environ. 2013;448:66–71. doi: 10.1016/j.scitotenv.2012.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coogan PF, White LF, Jerrett M, Brook RD, Su JG, et al. Air pollution and incidence of hypertension and diabetes mellitus in black women living in Los Angeles. Circulation. 2012;125:767–772. doi: 10.1161/CIRCULATIONAHA.111.052753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim JH, Hong YC. GSTM1, GSTT1, and GSTP1 polymorphisms and associations between air pollutants and markers of insulin resistance in elderly Koreans. Environ Health Perspect. 2012;120:1378–1384. doi: 10.1289/ehp.1104406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Greenberg CC, Danos AM, Brady MJ. Central role for protein targeting to glycogen in the maintenance of cellular glycogen stores in 3T3-L1 adipocytes. Mol Cell Biol. 2006;26:334–342. doi: 10.1128/MCB.26.1.334-342.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lind PM, Lind L. Circulating levels of bisphenol A and phthalates are related to carotid atherosclerosis in the elderly. Atherosclerosis. 2011;218:207–213. doi: 10.1016/j.atherosclerosis.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 40.Lind PM, van Bavel B, Salihovic S, Lind L. Circulating levels of persistent organic pollutants (POPs) and carotid atherosclerosis in the elderly. Environ Health Perspect. 2012;120:38–43. doi: 10.1289/ehp.1103563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Epstein LHD, K K, Roba LG, Finkelstein E. The Influence of Taxes and Subsidies on Energy Purchased in an Experimental Purchasing Study. Psychological Science. 2010 doi: 10.1177/0956797610361446. [DOI] [PubMed] [Google Scholar]
- 42.Osorio-Yanez C, Ayllon-Vergara JC, Aguilar-Madrid G, Arreola-Mendoza L, Hernandez-Castellanos E, et al. Carotid intima-media thickness and plasma asymmetric dimethylarginine in mexican children exposed to inorganic arsenic. Environ Health Perspect. 2013;121:1090–1096. doi: 10.1289/ehp.1205994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fukuda D, Shimada K, Tanaka A, Kusuyama T, Yamashita H, et al. Comparison of levels of serum matrix metalloproteinase-9 in patients with acute myocardial infarction versus unstable angina pectoris versus stable angina pectoris. Am J Cardiol. 2006;97:175–180. doi: 10.1016/j.amjcard.2005.08.020. [DOI] [PubMed] [Google Scholar]
- 44.Burgess JL, Kurzius-Spencer M, O’Rourke MK, Littau SR, Roberge J, et al. Environmental arsenic exposure and serum matrix metalloproteinase-9. J Expo Sci Environ Epidemiol. 2013;23:163–169. doi: 10.1038/jes.2012.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen Y, Wu F, Graziano JH, Parvez F, Liu M, et al. Arsenic exposure from drinking water, arsenic methylation capacity, and carotid intima-media thickness in Bangladesh. Am J Epidemiol. 2013;178:372–381. doi: 10.1093/aje/kwt001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fruchart JC, Nierman MC, Stroes ES, Kastelein JJ, Duriez P. New risk factors for atherosclerosis and patient risk assessment. Circulation. 2004;109:III15–19. doi: 10.1161/01.CIR.0000131513.33892.5b. [DOI] [PubMed] [Google Scholar]
- 47.Everett CJ, Mainous AG, 3rd, Frithsen IL, Player MS, Matheson EM. Association of polychlorinated biphenyls with hypertension in the 1999–2002 National Health and Nutrition Examination Survey. Environ Res. 2008;108:94–97. doi: 10.1016/j.envres.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 48.Bannenberg G, Martin HJ, Belai I, Maser E. 11beta-Hydroxysteroid dehydrogenase type 1: tissue-specific expression and reductive metabolism of some anti-insect agent azole analogues of metyrapone. Chem Biol Interact. 2003;143–144:449–457. doi: 10.1016/s0009-2797(02)00183-7. [DOI] [PubMed] [Google Scholar]
- 49.Karim MR, Rahman M, Islam K, Mamun AA, Hossain S, et al. Increases in oxidized low-density lipoprotein and other inflammatory and adhesion molecules with a concomitant decrease in high-density lipoprotein in the individuals exposed to arsenic in Bangladesh. Toxicol Sci. 2013;135:17–25. doi: 10.1093/toxsci/kft130. [DOI] [PubMed] [Google Scholar]
- 50.Auerbach O, Hammond EC, Garfinkel L. Smoking in relation to atherosclerosis of the coronary arteries. The New England Journal of Medicine. 1965;273:775–779. doi: 10.1056/NEJM196510072731501. [DOI] [PubMed] [Google Scholar]
- 51.Nazaroff WW, Singer BC. Inhalation of hazardous air pollutants from environmental tobacco smoke in US residences. J Expo Anal Environ Epidemiol. 2004;14(Suppl 1):S71–77. doi: 10.1038/sj.jea.7500361. [DOI] [PubMed] [Google Scholar]
- 52.Ding YS, Zhang L, Jain RB, Jain N, Wang RY, et al. Levels of tobacco-specific nitrosamines and polycyclic aromatic hydrocarbons in mainstream smoke from different tobacco varieties. Cancer Epidemiol Biomarkers Prev. 2008;17:3366–3371. doi: 10.1158/1055-9965.EPI-08-0320. [DOI] [PubMed] [Google Scholar]
- 53.Invernizzi G, Ruprecht A, Mazza R, Rossetti E, Sasco A, et al. Particulate matter from tobacco versus diesel car exhaust: an educational perspective. Tob Control. 2004;13:219–221. doi: 10.1136/tc.2003.005975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bae S, Pan XC, Kim SY, Park K, Kim YH, et al. Exposures to particulate matter and polycyclic aromatic hydrocarbons and oxidative stress in schoolchildren. Environ Health Perspect. 2010;118:579–583. doi: 10.1289/ehp.0901077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pope CA, 3rd, Burnett RT, Thurston GD, Thun MJ, Calle EE, et al. Cardiovascular mortality and long-term exposure to particulate air pollution: epidemiological evidence of general pathophysiological pathways of disease. Circulation. 2004;109:71–77. doi: 10.1161/01.CIR.0000108927.80044.7F. [DOI] [PubMed] [Google Scholar]
- 56.Fagerberg B, Bergstrom G, Boren J, Barregard L. Cadmium exposure is accompanied by increased prevalence and future growth of atherosclerotic plaques in 64-year-old women. J Intern Med. 2012;272:601–610. doi: 10.1111/j.1365-2796.2012.02578.x. [DOI] [PubMed] [Google Scholar]
- 57.Messner B, Knoflach M, Seubert A, Ritsch A, Pfaller K, et al. Cadmium is a novel and independent risk factor for early atherosclerosis mechanisms and in vivo relevance. Arterioscler Thromb Vasc Biol. 2009;29:1392–1398. doi: 10.1161/ATVBAHA.109.190082. [DOI] [PubMed] [Google Scholar]
- 58.Richardson VM, Staskal DF, Ross DG, Diliberto JJ, DeVito MJ, et al. Possible mechanisms of thyroid hormone disruption in mice by BDE 47, a major polybrominated diphenyl ether congener. Toxicol Appl Pharmacol. 2008;226:244–250. doi: 10.1016/j.taap.2007.09.015. [DOI] [PubMed] [Google Scholar]
- 59.Baker NA, Karounos M, English V, Fang J, Wei Y, et al. Coplanar polychlorinated biphenyls impair glucose homeostasis in lean C57BL/6 mice and mitigate beneficial effects of weight loss on glucose homeostasis in obese mice. Environ Health Perspect. 2013;121:105–110. doi: 10.1289/ehp.1205421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ogino K, Inukai T, Miura Y, Matsui H, Y T. Triphenyltin chloride induces glucose intolerance by the suppression of insulin release from hamster pancreatic beta-cells. Exp Clin Endocrinol Diabetes. 1996;104:409–411. doi: 10.1055/s-0029-1211476. [DOI] [PubMed] [Google Scholar]
- 61.Hill DS, Wlodarczyk BJ, Mitchell LE, RH F. Arsenate-induced maternal glucose intolerance and neural tube defects in a mouse model. Toxicol Appl Pharmacol. 2009;239:29–36. doi: 10.1016/j.taap.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gray SL, Shaw AC, Gagne AX, Chan HM. Chronic Exposure to PCBs (Aroclor 1254) Exacerbates Obesity-Induced Insulin Resistance and Hyperinsulinemia in Mice. J Toxicol Environ Health A. 2013;76:701–715. doi: 10.1080/15287394.2013.796503. [DOI] [PubMed] [Google Scholar]
- 63•.Alonso-Magdalena P, Vieira E, Soriano S, Menes L, Burks D, et al. Bisphenol A exposure during pregnancy disrupts glucose homeostasis in mothers and adult male offspring. Environ Health Perspect. 2010;118:1243–1250. doi: 10.1289/ehp.1001993. An important manuscript from a productive group specifically linking developmental endocrine disruptor exposure to metabolic derangements. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu J, Yu P, Qian W, Li Y, Zhao J, et al. Perinatal bisphenol A exposure and adult glucose homeostasis: identifying critical windows of exposure. PLoS One. 2013;8:e64143. doi: 10.1371/journal.pone.0064143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.JT N, DT S, GB C. Polybrominated diphenyl ethers alter hepatic phosphoenolpyruvate carboxykinase enzyme kinetics in male Wistar rats: implications for lipid and glucose metabolism. J Toxicol Environ Health A. 2013;76:142–156. doi: 10.1080/15287394.2012.738457. [DOI] [PubMed] [Google Scholar]
- 66.Ruzzin J, Petersen R, Meugnier E, Madsen L, Lock EJ, et al. Persistent organic pollutant exposure leads to insulin resistance syndrome. Environ Health Perspect. 2010;118:465–471. doi: 10.1289/ehp.0901321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lim S, Ahn SY, Song IC, Chung MH, Jang HC, et al. Chronic exposure to the herbicide, atrazine, causes mitochondrial dysfunction and insulin resistance. PLoS One. 2009;4:e5186. doi: 10.1371/journal.pone.0005186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yan YH, Chou CC, Lee CT, Liu JY, Cheng TJ. Enhanced insulin resistance in dietinduced obese rats exposed to fine particles by instillation. Inhal Toxicol. 2011;23:507–519. doi: 10.3109/08958378.2011.587472. [DOI] [PubMed] [Google Scholar]
- 69.Paul DS, Walton FS, Saunders RJ, M S. Characterization of the impaired glucose homeostasis produced in C57BL/6 mice by chronic exposure to arsenic and high-fat diet. Environ Health Perspect. 2011;119:1104–1109. doi: 10.1289/ehp.1003324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McMillen IC, Robinson JS. Developmental origins of the metabolic syndrome: prediction, plasticity, and programming. Physiol Rev. 2005;85:571–633. doi: 10.1152/physrev.00053.2003. [DOI] [PubMed] [Google Scholar]
- 71.Newbold RR, Padilla-Banks E, Jefferson WN. Adverse effects of the model environmental estrogen diethylstilbestrol are transmitted to subsequent generations. Endocrinology. 2006;147:S11–17. doi: 10.1210/en.2005-1164. [DOI] [PubMed] [Google Scholar]
- 72.Manikkam M, Guerrero-Bosagna C, Tracey R, Haque MM, Skinner MK. Transgenerational actions of environmental compounds on reproductive disease and identification of epigenetic biomarkers of ancestral exposures. PLoS One. 2012;7:e31901. doi: 10.1371/journal.pone.0031901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Angle BM, Do RP, Ponzi D, Stahlhut RW, Drury BE, et al. Metabolic disruption in male mice due to fetal exposure to low but not high doses of bisphenol A (BPA): Evidence for effects on body weight, food intake, adipocytes, leptin, adiponectin, insulin and glucose regulation. Reprod Toxicol. 2013 doi: 10.1016/j.reprotox.2013.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wei J, Lin Y, Li Y, Ying C, Chen J, et al. Perinatal exposure to bisphenol A at reference dose predisposes offspring to metabolic syndrome in adult rats on a high-fat diet. Endocrinology. 2011;152:3049–3061. doi: 10.1210/en.2011-0045. [DOI] [PubMed] [Google Scholar]
- 75.Leibel RL. Molecular physiology of weight regulation in mice and humans. Int J Obes (Lond) 2008;32(Suppl 7):S98–108. doi: 10.1038/ijo.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lv Z, Li G, Li Y, Ying C, Chen J, et al. Glucose and lipid homeostasis in adult rat is impaired by early-life exposure to perfluorooctane sulfonate. Environ Toxicol. 2013;28:532–542. doi: 10.1002/tox.20747. [DOI] [PubMed] [Google Scholar]
- 77.White SS, Fenton SE, Hines EP. Endocrine disrupting properties of perfluorooctanoic acid. J Steroid Biochem Mol Biol. 2011;127:16–26. doi: 10.1016/j.jsbmb.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zadelaar S, Kleemann R, Verschuren L, de Vries-Van der Weij J, van der Hoorn J, et al. Mouse models for atherosclerosis and pharmaceutical modifiers. Arterioscler Thromb Vasc Biol. 2007;27:1706–1721. doi: 10.1161/ATVBAHA.107.142570. [DOI] [PubMed] [Google Scholar]
- 79.TP D, JK K, B W. Dioxin Exposure Is an Environmental Risk Factor for Ischemic Heart Disease. Cardiovascular Toxicology. 2001;01:285–298. doi: 10.1385/ct:1:4:285. [DOI] [PubMed] [Google Scholar]
- 80.Wu D, Nishimura N, Kuo V, Fiehn O, Shahbaz S, et al. Activation of aryl hydrocarbon receptor induces vascular inflammation and promotes atherosclerosis in apolipoprotein E−/− mice. Arterioscler Thromb Vasc Biol. 2011;31:1260–1267. doi: 10.1161/ATVBAHA.110.220202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Curfs DM, Knaapen AD, Pachen DM. Polycyclic aromatic hydrocarbons induce an inflammatory atherosclerotic plaque phenotype irrespective of their DNA binding properties. The FASEB Journal. 2005 doi: 10.1096/fj.04-2269fje. [DOI] [PubMed] [Google Scholar]
- 82.Chen T, Jia G, Wei Y, Li J. Beijing ambient particle exposure accelerates atherosclerosis in ApoE knockout mice. Toxicol Lett. 2013;223:146–153. doi: 10.1016/j.toxlet.2013.09.004. [DOI] [PubMed] [Google Scholar]
- 83.Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–355. doi: 10.1016/j.cell.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rogalska J, Brzoska MM, Roszczenko A, Moniuszko-Jakoniuk J. Enhanced zinc consumption prevents cadmium-induced alterations in lipid metabolism in male rats. Chem Biol Interact. 2009;177:142–152. doi: 10.1016/j.cbi.2008.09.011. [DOI] [PubMed] [Google Scholar]
- 85.Olisekodiaka MJ, Igbeneghu CA, Onuegbu AJ, Oduru R, Lawal AO. Lipid, lipoproteins, total antioxidant status and organ changes in rats administered high doses of cadmium chloride. Med Princ Pract. 2012;21:156–159. doi: 10.1159/000333385. [DOI] [PubMed] [Google Scholar]
- 86.Cheng TJ, Chuu JJ, Chang CY, Tsai WC, Chen KJ, et al. Atherosclerosis induced by arsenic in drinking water in rats through altering lipid metabolism. Toxicol Appl Pharmacol. 2011;256:146–153. doi: 10.1016/j.taap.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 87.Miyawaki J, Sakayama K, Kato H. Perinatal and postnatal exposure to bisphenol A increases adipose tissue mass and serum cholesterol level in mice. Journal of Atherosclerosis and Thrombosis. 2007;14 doi: 10.5551/jat.e486. [DOI] [PubMed] [Google Scholar]
- 88.Sugai E, Yoshioka W, Kakeyama M, Ohsako S, Tohyama C. In utero and lactational exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin modulates dysregulation of the lipid metabolism in mouse offspring fed a high-calorie diet. J Appl Toxicol. 2013 doi: 10.1002/jat.2881. [DOI] [PubMed] [Google Scholar]
- 89.Bruin JE, Gerstein HC, Holloway AC. Long-term consequences of fetal and neonatal nicotine exposure: a critical review. Toxicol Sci. 2010;116:364–374. doi: 10.1093/toxsci/kfq103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xiao D, Huang X, Yang S, Zhang L. Estrogen normalizes perinatal nicotine-induced hypertensive responses in adult female rat offspring. Hypertension. 2013;61:1246–1254. doi: 10.1161/HYPERTENSIONAHA.113.01152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Holloway AC, Cuu DQ, Morrison KM, Gerstein HC, Tarnopolsky MA. Transgenerational effects of fetal and neonatal exposure to nicotine. Endocrine. 2007;31:254–259. doi: 10.1007/s12020-007-0043-6. [DOI] [PubMed] [Google Scholar]
- 92.Heeschen C, Jang JJ, Weis M. Nicotine stimulates angiogenesis and promotes tumor growth and atherosclerosis. Nature Medicine. 2001;7:833–839. doi: 10.1038/89961. [DOI] [PubMed] [Google Scholar]
- 93.Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444:840–846. doi: 10.1038/nature05482. [DOI] [PubMed] [Google Scholar]
- 94.Kurita H, Yoshioka W, Nishimura N, Kubota N, Kadowaki T, et al. Aryl hydrocarbon receptor-mediated effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on glucose-stimulated insulin secretion in mice. J Appl Toxicol. 2009;29:689–694. doi: 10.1002/jat.1459. [DOI] [PubMed] [Google Scholar]
- 95.Piaggi S, Novelli M, Martino L, Masini M, Raggi C, et al. Cell death and impairment of glucose-stimulated insulin secretion induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in the beta-cell line INS-1E. Toxicol Appl Pharmacol. 2007;220:333–340. doi: 10.1016/j.taap.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 96.Novelli M, Piaggi S, De Tata V. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-induced impairment of glucose-stimulated insulin secretion in isolated rat pancreatic islets. Toxicol Lett. 2005;156:307–314. doi: 10.1016/j.toxlet.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 97.D W, M W, C L. Ultrastructure of beta-cells of the endocrine pancreas in rats receiving polychlorinated biphenyls. Environ Physiol Biochem. 1975;5:322–340. [PubMed] [Google Scholar]
- 98.Alonso-Magdalena P, Laribi O, Ropero AB, Fuentes E, Ripoll C, et al. Low doses of bisphenol A and diethylstilbestrol impair Ca2+ signals in pancreatic alpha-cells through a nonclassical membrane estrogen receptor within intact islets of Langerhans. Environ Health Perspect. 2005;113:969–977. doi: 10.1289/ehp.8002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Soriano S, Alonso-Magdalena P, Garcia-Arevalo M, Novials A, Muhammed SJ, et al. Rapid insulinotropic action of low doses of bisphenol-A on mouse and human islets of Langerhans: role of estrogen receptor beta. PLoS One. 2012;7:e31109. doi: 10.1371/journal.pone.0031109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Miura Y, Matsui H. Triphenyltin impairs a protein kinase A (PKA)-dependent increase of cytosolic Na+ and Ca2+ and PKA-independent increase of cytosolic Ca2+ associated with insulin secretion in hamster pancreatic beta-cells. Toxicol Appl Pharmacol. 2006;216:363–372. doi: 10.1016/j.taap.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 101.Chang KC, Hsu CC, Liu SH, Su CC, Yen CC, et al. Cadmium induces apoptosis in pancreatic beta-cells through a mitochondria-dependent pathway: the role of oxidative stress-mediated c-Jun N-terminal kinase activation. PLoS One. 2013;8:e54374. doi: 10.1371/journal.pone.0054374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen YW, Huang CF, Yang CY, Yen CC, Tsai KS, et al. Inorganic mercury causes pancreatic beta-cell death via the oxidative stress-induced apoptotic and necrotic pathways. Toxicol Appl Pharmacol. 2010;243:323–331. doi: 10.1016/j.taap.2009.11.024. [DOI] [PubMed] [Google Scholar]
- 103.Douillet C, Currier J, Saunders J, Bodnar WM, Matousek T, et al. Methylated trivalent arsenicals are potent inhibitors of glucose stimulated insulin secretion by murine pancreatic islets. Toxicol Appl Pharmacol. 2013;267:11–15. doi: 10.1016/j.taap.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fu J, Woods CG, Yehuda-Shnaidman E, Zhang Q, Wong V, et al. Low-level arsenic impairs glucose-stimulated insulin secretion in pancreatic beta cells: involvement of cellular adaptive response to oxidative stress. Environ Health Perspect. 2010;118:864–870. doi: 10.1289/ehp.0901608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fischer LJ, Zhou HR, MA W. Polychlorinated biphenyls release insulin from RINm5F cells. Life Sci. 1996;59:2041–2049. doi: 10.1016/s0024-3205(96)00557-7. [DOI] [PubMed] [Google Scholar]
- 106.Nishiumi S, Yoshida M, Azuma T, Yoshida K, Ashida H. 2,3,7,8-tetrachlorodibenzo-p-dioxin impairs an insulin signaling pathway through the induction of tumor necrosis factor-alpha in adipocytes. Toxicol Sci. 2010;115:482–491. doi: 10.1093/toxsci/kfq052. [DOI] [PubMed] [Google Scholar]
- 107.Jayashree S, Indumathi D, Akilavalli N, Sathish S, Selvaraj J, et al. Effect of Bisphenol-A on insulin signal transduction and glucose oxidation in liver of adult male albino rat. Environ Toxicol Pharmacol. 2013;35:300–310. doi: 10.1016/j.etap.2012.12.016. [DOI] [PubMed] [Google Scholar]
- 108.Rajesh P, Sathish S, Srinivasan C, Selvaraj J, Balasubramanian K. Phthalate is associated with insulin resistance in adipose tissue of male rat: role of antioxidant vitamins. J Cell Biochem. 2013;114:558–569. doi: 10.1002/jcb.24399. [DOI] [PubMed] [Google Scholar]
- 109.Sargis RM, Neel BA, Brock CO, Lin Y, Hickey AT, et al. The novel endocrine disruptor tolylfluanid impairs insulin signaling in primary rodent and human adipocytes through a reduction in insulin receptor substrate-1 levels. Biochim Biophys Acta. 2012;1822:952–960. doi: 10.1016/j.bbadis.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zheng Z, Xu X, Zhang X, Wang A, Zhang C, et al. Exposure to ambient particulate matter induces a NASH-like phenotype and impairs hepatic glucose metabolism in an animal model. J Hepatol. 2013;58:148–154. doi: 10.1016/j.jhep.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Xue P, Hou Y, Zhang Q, Woods CG, Yarborough K, et al. Prolonged inorganic arsenite exposure suppresses insulin-stimulated AKT S473 phosphorylation and glucose uptake in 3T3-L1 adipocytes: involvement of the adaptive antioxidant response. Biochem Biophys Res Commun. 2011;407:360–365. doi: 10.1016/j.bbrc.2011.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sun Q, Yue P, Deiuliis JA, Lumeng CN, Kampfrath T, et al. Ambient air pollution exaggerates adipose inflammation and insulin resistance in a mouse model of dietinduced obesity. Circulation. 2009;119:538–546. doi: 10.1161/CIRCULATIONAHA.108.799015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wang J, Lv X, Y D. Inflammatory response and insulin signaling alteration induced by PCB77. J Environ Sci (China) 2010;22:1086–1090. doi: 10.1016/s1001-0742(09)60221-7. [DOI] [PubMed] [Google Scholar]
- 114.Yen YP, Tsai KS, Chen YW, Huang CF, Yang RS, et al. Arsenic inhibits myogenic differentiation and muscle regeneration. Environ Health Perspect. 2010;118:949–956. doi: 10.1289/ehp.0901525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Han JC, Park SY, Hah BG, Choi GH, Kim YK, et al. Cadmium induces impaired glucose tolerance in rat by down-regulating GLUT4 expression in adipocytes. Archives of Biochemistry and Biophysics. 2003;413:213–220. doi: 10.1016/s0003-9861(03)00120-6. [DOI] [PubMed] [Google Scholar]
- 116.AT T, PE S. Adiponectin: mechanistic insights and clinical implications. Diabetologia. 2012;55:2319–2326. doi: 10.1007/s00125-012-2598-x. [DOI] [PubMed] [Google Scholar]
- 117.Hugo ER, Brandebourg TD, Woo JG, Loftus J, Alexander JW, et al. Bisphenol A at environmentally relevant doses inhibits adiponectin release from human adipose tissue explants and adipocytes. Environ Health Perspect. 2008;116:1642–1647. doi: 10.1289/ehp.11537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kawakami T, Sugimoto H, Furuichi R, Kadota Y, Inoue M, et al. Cadmium reduces adipocyte size and expression levels of adiponectin and Peg1/Mest in adipose tissue. Toxicology. 2010;267:20–26. doi: 10.1016/j.tox.2009.07.022. [DOI] [PubMed] [Google Scholar]
- 119.Zuo Z, Chen S, Wu T, Zhang J, Su Y, et al. Tributyltin causes obesity and hepatic steatosis in male mice. Environ Toxicol. 26:79–85. doi: 10.1002/tox.20531. [DOI] [PubMed] [Google Scholar]
- 120.Xu Z, Xu X, Zhong M, Hotchkiss IP, Lewandowski RP, et al. Ambient particulate air pollution induces oxidative stress and alterations of mitochondria and gene expression in brown and white adipose tissues. Part Fibre Toxicol. 2011;8:20. doi: 10.1186/1743-8977-8-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wang M, Wang D, Zhang Y, Wang X, Liu Y, et al. Adiponectin increases macrophages cholesterol efflux and suppresses foam cell formation in patients with type 2 diabetes mellitus. Atherosclerosis. 2013;229:62–70. doi: 10.1016/j.atherosclerosis.2013.01.017. [DOI] [PubMed] [Google Scholar]
- 122.Kleemann R, Zadelaar S, Kooistra T. Cytokines and atherosclerosis: a comprehensive review of studies in mice. Cardiovasc Res. 2008;79:360–376. doi: 10.1093/cvr/cvn120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Majkova Z, Layne J, Sunkara M, Morris AJ, Toborek M, et al. Omega-3 fatty acid oxidation products prevent vascular endothelial cell activation by coplanar polychlorinated biphenyls. Toxicol Appl Pharmacol. 2011;251:41–49. doi: 10.1016/j.taap.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Vogel CFA, Sciullo E, Wong P, Kuzmicky P, Kado N, et al. Induction of Proinflammatory Cytokines and C-Reactive Protein in Human Macrophage Cell Line U937 Exposed to Air Pollution Particulates. Environmental Health Perspectives. 2005;113:1536–1541. doi: 10.1289/ehp.8094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Libby P. Inflammation and Atherosclerosis. Circulation. 2002;105:1135–1143. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- 126.Majkova Z, Smart E, Toborek M, Hennig B. Up-regulation of endothelial monocyte chemoattractant protein-1 by coplanar PCB77 is caveolin-1-dependent. Toxicol Appl Pharmacol. 2009;237:1–7. doi: 10.1016/j.taap.2009.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Knaapen AM, Curfs DM, Pachen DM, Gottschalk RW, de Winther MP, et al. The environmental carcinogen benzo[a]pyrene induces expression of monocytechemoattractant protein-1 in vascular tissue: a possible role in atherogenesis. Mutat Res. 2007;621:31–41. doi: 10.1016/j.mrfmmm.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 128.Vogel CF, Nishimura N, Sciullo E, Wong P, Li W, et al. Modulation of the chemokines KC and MCP-1 by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in mice. Arch Biochem Biophys. 2007;461:169–175. doi: 10.1016/j.abb.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 129•.Arsenescu V, Arsenescu RI, King V, Swanson H, Cassis LA. Polychlorinated biphenyl-77 induces adipocyte differentiation and proinflammatory adipokines and promotes obesity and atherosclerosis. Environ Health Perspect. 2008;116:761–768. doi: 10.1289/ehp.10554. An excellent basic science studying examining the effects of the a specific PCB congener on the development of metabolic and cardiovascular disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Angeli JK, Cruz Pereira CA, de Oliveira Faria T, Stefanon I, Padilha AS, et al. Cadmium exposure induces vascular injury due to endothelial oxidative stress: the role of local angiotensin II and COX-2. Free Radic Biol Med. 2013;65C:838–848. doi: 10.1016/j.freeradbiomed.2013.08.167. [DOI] [PubMed] [Google Scholar]
- 131.Hennig B. Proinflammatory Properties of Coplanar PCBs: In Vitro and in Vivo Evidence. Toxicology and Applied Pharmacology. 2002;181:174–183. doi: 10.1006/taap.2002.9408. [DOI] [PubMed] [Google Scholar]
- 132.Birch LL, Deysher M. Caloric compensation and sensory specific satiety: evidence for self regulation of food intake by young children. Appetite. 1986;7:323–331. doi: 10.1016/s0195-6663(86)80001-0. [DOI] [PubMed] [Google Scholar]
- 133.Arsenescu V, Arsenescu R, Parulkar M, Karounos M, Zhang X, et al. Polychlorinated biphenyl 77 augments angiotensin II-induced atherosclerosis and abdominal aortic aneurysms in male apolipoprotein E deficient mice. Toxicol Appl Pharmacol. 2011;257:148–154. doi: 10.1016/j.taap.2011.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Fang CC, Chen FY, Chen CR, Liu CC, Wong LC, et al. Cyprodinil as an activator of aryl hydrocarbon receptor. Toxicology. 2013;304:32–40. doi: 10.1016/j.tox.2012.11.018. [DOI] [PubMed] [Google Scholar]
- 135.Druwe IL, Sollome JJ, Sanchez-Soria P, Hardwick RN, Camenisch TD, et al. Arsenite activates NFkappaB through induction of C-reactive protein. Toxicol Appl Pharmacol. 2012;261:263–270. doi: 10.1016/j.taap.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wolin MS. Interactions of Oxidants With Vascular Signaling Systems. Arteriosclerosis, Thrombosis, and Vascular Biology. 2000;20:1430–1442. doi: 10.1161/01.atv.20.6.1430. [DOI] [PubMed] [Google Scholar]
- 137.Dikalov SI, Nazarewicz RR. Angiotensin II-induced production of mitochondrial reactive oxygen species: potential mechanisms and relevance for cardiovascular disease. Antioxid Redox Signal. 2013;19:1085–1094. doi: 10.1089/ars.2012.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ceriello A. Evidence for an Independent and Cumulative Effect of Postprandial Hypertriglyceridemia and Hyperglycemia on Endothelial Dysfunction and Oxidative Stress Generation: Effects of Short- and Long-Term Simvastatin Treatment. Circulation. 2002;106:1211–1218. doi: 10.1161/01.cir.0000027569.76671.a8. [DOI] [PubMed] [Google Scholar]
- 139.Hossain E, Ota A, Karnan S, Damdindorj L, Takahashi M, et al. Arsenic augments the uptake of oxidized LDL by upregulating the expression of lectin-like oxidized LDL receptor in mouse aortic endothelial cells. Toxicol Appl Pharmacol. 2013 doi: 10.1016/j.taap.2013.10.012. [DOI] [PubMed] [Google Scholar]
- 140.Xiao D, Huang X, Lawrence J, Yang S, Zhang L. Fetal and neonatal nicotine exposure differentially regulates vascular contractility in adult male and female offspring. J Pharmacol Exp Ther. 2007;320:654–661. doi: 10.1124/jpet.106.113332. [DOI] [PubMed] [Google Scholar]
- 141.Tavolari S, Bucci L, Tomasi V, Guarnieri T. Selected polychlorobiphenyls congeners bind to estrogen receptor alpha in human umbilical vascular endothelial (HUVE) cells modulating angiogenesis. Toxicology. 2006;218:67–74. doi: 10.1016/j.tox.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 142.Ribeiro-Varandas E, Viegas W, Sofia Pereira H, Delgado M. Bisphenol A at concentrations found in human serum induces aneugenic effects in endothelial cells. Mutat Res. 2013;751:27–33. doi: 10.1016/j.mrgentox.2012.10.007. [DOI] [PubMed] [Google Scholar]
- 143.Adigun AA, Wrench N, Seidler FJ, Slotkin TA. Neonatal organophosphorus pesticide exposure alters the developmental trajectory of cell-signaling cascades controlling metabolism: differential effects of diazinon and parathion. Environ Health Perspect. 2010;118:210–215. doi: 10.1289/ehp.0901237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kucka M, Pogrmic-Majkic K, Fa S, Stojilkovic SS, Kovacevic R. Atrazine acts as an endocrine disrupter by inhibiting cAMP-specific phosphodiesterase-4. Toxicol Appl Pharmacol. 2012;265:19–26. doi: 10.1016/j.taap.2012.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Atanasov AG, Tam S, Röcken JM, Baker ME, Odermatt A. Inhibition of 11β-hydroxysteroid dehydrogenase type 2 by dithiocarbamates. Biochemical and Biophysical Research Communications. 2003;308:257–262. doi: 10.1016/s0006-291x(03)01359-7. [DOI] [PubMed] [Google Scholar]
- 146.Atanasov AG, Nashev LG, Tam S, Baker ME, Odermatt A. Organotins Disrupt the 11β-Hydroxysteroid Dehydrogenase Type 2–Dependent Local Inactivation of Glucocorticoids. Environmental Health Perspectives. 2005;113:1600–1606. doi: 10.1289/ehp.8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Yang K, Julan L, Rubio F, Sharma A, Guan H. Cadmium reduces 11 betahydroxysteroid dehydrogenase type 2 activity and expression in human placental trophoblast cells. Am J Physiol Endocrinol Metab. 2006;290:E135–E142. doi: 10.1152/ajpendo.00356.2005. [DOI] [PubMed] [Google Scholar]
- 148.Newbold RR, Padilla-Banks E, Jefferson WN. Environmental estrogens and obesity. Mol Cell Endocrinol. 2009;304:84–89. doi: 10.1016/j.mce.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Shanle EK, Xu W. Endocrine disrupting chemicals targeting estrogen receptor signaling: identification and mechanisms of action. Chem Res Toxicol. 2011;24:6–19. doi: 10.1021/tx100231n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Joharapurkar AA, Dhote VV, Jain MR. Selective thyromimetics using receptor and tissue selectivity approaches: prospects for dyslipidemia. J Med Chem. 2012;55:5649–5675. doi: 10.1021/jm2004706. [DOI] [PubMed] [Google Scholar]
- 151.Kitamura S, Jinno N, Suzuki T, Sugihara K, Ohta S, et al. Thyroid hormone-like and estrogenic activity of hydroxylated PCBs in cell culture. Toxicology. 2005;208:377–387. doi: 10.1016/j.tox.2004.11.037. [DOI] [PubMed] [Google Scholar]
- 152.Mastorakos G, Karoutsou EI, Mizamtsidi M, Creatsas G. The menace of endocrine disruptors on thyroid hormone physiology and their impact on intrauterine development. Endocrine. 2007;31:219–237. doi: 10.1007/s12020-007-0030-y. [DOI] [PubMed] [Google Scholar]
- 153.Neel BA, Brady MJ, Sargis RM. The endocrine disrupting chemical tolylfluanid alters adipocyte metabolism via glucocorticoid receptor activation. Mol Endocrinol. 2013;27:394–406. doi: 10.1210/me.2012-1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bodwell JE, Kingsley LA. Arsenic at Very Low Concentrations Alters Glucocorticoid Receptor (GR)-Mediated Gene Activation but Not GR-Mediated Gene Repression: Complex Dose-Response Effects Are Closely Correlated with Levels of Activated GR and Require a Functional GR DNA Binding Domain. Chem Res Toxicol. 2004;17:1064–1076. doi: 10.1021/tx0499113. [DOI] [PubMed] [Google Scholar]
- 155.Zhou T, Cong S, Sun S, Sun H, Zou R, et al. Identification of endocrine disrupting chemicals activating SXR-mediated transactivation of CYP3A and CYP7A1. Mol Cell Endocrinol. 2013;365:36–43. doi: 10.1016/j.mce.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 156.Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27:1047–1053. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- 157.Sargis RM, Subbaiah PV. Protection of membrane cholesterol by sphingomyelin against free radical-mediated oxidation. Free Radic Biol Med. 2006;40:2092–2102. doi: 10.1016/j.freeradbiomed.2006.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ratajczak MK, Ko YT, Lange Y, Steck TL, Lee KY. Cholesterol displacement from membrane phospholipids by hexadecanol. Biophys J. 2007;93:2038–2047. doi: 10.1529/biophysj.107.109553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Chen S, Subbaiah PV. Regioisomers of phosphatidylcholine containing DHA and their potential to deliver DHA to the brain: role of phospholipase specificities. Lipids. 2013;48:675–686. doi: 10.1007/s11745-013-3791-5. [DOI] [PubMed] [Google Scholar]
- 160.Marmugi A, Ducheix S, Lasserre F, Polizzi A, Paris A, et al. Low doses of bisphenol A induce gene expression related to lipid synthesis and trigger triglyceride accumulation in adult mouse liver. Hepatology. 2012;55:395–407. doi: 10.1002/hep.24685. [DOI] [PubMed] [Google Scholar]
- 161.Beausoleil C, Ormsby JN, Gies A, Hass U, Heindel JJ, et al. Low dose effects and non-monotonic dose responses for endocrine active chemicals: Science to practice workshop: Workshop summary. Chemosphere. 2013;93:847–856. doi: 10.1016/j.chemosphere.2013.06.043. [DOI] [PubMed] [Google Scholar]
- 162.Miki Y, Suzuki T, Tazawa C, Blumberg B, Sasano H. Steroid and xenobiotic receptor (SXR), cytochrome P450 3A4 and multidrug resistance gene 1 in human adult and fetal tissues. Mol Cell Endocrinol. 2005;231:75–85. doi: 10.1016/j.mce.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 163.Ma Y, Xia W, Wang DQ, Wan YJ, Xu B, et al. Hepatic DNA methylation modifications in early development of rats resulting from perinatal BPA exposure contribute to insulin resistance in adulthood. Diabetologia. 2013;56:2059–2067. doi: 10.1007/s00125-013-2944-7. [DOI] [PubMed] [Google Scholar]
- 164.Han JC, Park SY, Hah BG, Choi GH, Kim YK, et al. Cadmium induces impaired glucose tolerance in rat by down-regulating GLUT4 expression in adipocytes. Arch Biochem Biophys. 2003;413:213–220. doi: 10.1016/s0003-9861(03)00120-6. [DOI] [PubMed] [Google Scholar]
- 165.Kawakami T, Sugimoto H, Furuichi R, Kadota Y, Inoue M, et al. Cadmium reduces adipocyte size and expression levels of adiponectin and Peg1/Mest in adipose tissue. Toxicology. 2010;267:20–26. doi: 10.1016/j.tox.2009.07.022. [DOI] [PubMed] [Google Scholar]
- 166.Seilkop SK, Campen MJ, Lund AK, McDonald JD, Mauderly JL. Identification of chemical components of combustion emissions that affect pro-atherosclerotic vascular responses in mice. Inhal Toxicol. 2012;24:270–287. doi: 10.3109/08958378.2012.667455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Dalton TP, Kerzee JK, Wang B. Dioxin Exposure Is an Environmental Risk Factor for Ischemic Heart Disease. Cardiovasc Toxicol. 2001;01:285–298. doi: 10.1385/ct:1:4:285. [DOI] [PubMed] [Google Scholar]
- 168.Kopf PG, Huwe JK, Walker MK. Hypertension, cardiac hypertrophy, and impaired vascular relaxation induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin are associated with increased superoxide. Cardiovasc Toxicol. 2008;8:181–193. doi: 10.1007/s12012-008-9027-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Panahi P, Vosough-Ghanbari S, Pournourmohammadi S, Ostad SN, Nikfar S, et al. Stimulatory effects of malathion on the key enzymes activities of insulin secretion in langerhans islets, glutamate dehydrogenase and glucokinase. Toxicol Mech Methods. 2006;16:161–167. doi: 10.1080/15376520500191623. [DOI] [PubMed] [Google Scholar]
- 170.Hennig B. Proinflammatory Properties of Coplanar PCBs: In Vitro and in Vivo Evidence. Toxicol Appl Pharmacol. 2002;181:174–183. doi: 10.1006/taap.2002.9408. [DOI] [PubMed] [Google Scholar]
- 171.Lind PM, Orberg J, Edlund UB, Sjoblom L, Lind L. The dioxin-like pollutant PCB 126 (3,3′,4,4′,5-pentachlorobiphenyl) affects risk factors for cardiovascular disease in female rats. Toxicol Lett. 2004;150:293–299. doi: 10.1016/j.toxlet.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 172.Kampfrath T, Maiseyeu A, Ying Z, Shah Z, Deiuliis JA, et al. Chronic fine particulate matter exposure induces systemic vascular dysfunction via NADPH oxidase and TLR4 pathways. Circ Res. 2011;108:716–726. doi: 10.1161/CIRCRESAHA.110.237560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Grun F, Watanabe H, Zamanian Z, Maeda L, Arima K, et al. Endocrine-disrupting organotin compounds are potent inducers of adipogenesis in vertebrates. Mol Endocrinol. 2006;20:2141–2155. doi: 10.1210/me.2005-0367. [DOI] [PubMed] [Google Scholar]
- 174.Zuo Z, Chen S, Wu T, Zhang J, Su Y, et al. Tributyltin causes obesity and hepatic steatosis in male mice. Environ Toxicol. 2011;26:79–85. doi: 10.1002/tox.20531. [DOI] [PubMed] [Google Scholar]
- 175.Li X, Pham HT, Janesick AS, Blumberg B. Triflumizole is an obesogen in mice that acts through peroxisome proliferator activated receptor gamma (PPARgamma) Environ Health Perspect. 2012;120:1720–1726. doi: 10.1289/ehp.1205383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Alves MG, Neuhaus-Oliveira A, Moreira PI, Socorro S, Oliveira PF. Exposure to 2,4-dichlorophenoxyacetic acid alters glucose metabolism in immature rat Sertoli cells. Reprod Toxicol. 2013;38:81–88. doi: 10.1016/j.reprotox.2013.03.005. [DOI] [PubMed] [Google Scholar]
- 177.Wens B, De Boever P, Verbeke M, Hollanders K, Schoeters G. Cultured human peripheral blood mononuclear cells alter their gene expression when challenged with endocrine-disrupting chemicals. Toxicology. 2013;303:17–24. doi: 10.1016/j.tox.2012.10.019. [DOI] [PubMed] [Google Scholar]
- 178.Wang Y, Hu H, Zhao M, Zhao J, Yin D, et al. Nonylphenol disrupts the cardioprotective effects of 17beta-estradiol on ischemia/reperfusion injury in isolated hearts of guinea pig. J Toxicol Sci. 2013;38:731–740. doi: 10.2131/jts.38.731. [DOI] [PubMed] [Google Scholar]
- 179.Vogel CFA, Sciullo E, Wong P, Kuzmicky P, Kado N, et al. Induction of Proinflammatory Cytokines and C-Reactive Protein in Human Macrophage Cell Line U937 Exposed to Air Pollution Particulates. Environ Health Perspect. 2005;113:1536–1541. doi: 10.1289/ehp.8094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Wang X, Porter W, Krishnan V, Narasimhan TR, Safe S. Mechanism of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-mediated decrease of the nuclear estrogen receptor in MCF-7 human breast cancer cells. Mol Cell Endocrinol. 1993;96:159–166. doi: 10.1016/0303-7207(93)90106-t. [DOI] [PubMed] [Google Scholar]
- 181.Puga A, Sartor MA, Huang MY, Kerzee JK, Wei YD, et al. Gene expression profiles of mouse aorta and cultured vascular smooth muscle cells differ widely, yet show common responses to dioxin exposure. Cardiovasc Toxicol. 2004;4:385–404. doi: 10.1385/ct:4:4:385. [DOI] [PubMed] [Google Scholar]