

Abstract
Homozygosity for a recurrent 290 kb deletion of NPHP1 is the most frequent cause of isolated nephronophthisis (NPHP) in humans. A deletion of the same genomic interval has also been detected in individuals with Joubert syndrome (JBTS), and in the mouse, Nphp1 interacts genetically with Ahi1, a known JBTS locus. Given these observations, we investigated the contribution of NPHP1 in Bardet-Biedl syndrome (BBS), a ciliopathy of intermediate severity. By using a combination of array-comparative genomic hybridization, TaqMan copy number assays, and sequencing, we studied 200 families affected by BBS. We report a homozygous NPHP1 deletion CNV in a family with classical BBS that is transmitted with autosomal-recessive inheritance. Further, we identified heterozygous NPHP1 deletions in two more unrelated persons with BBS who bear primary mutations at another BBS locus. In parallel, we identified five families harboring an SNV in NPHP1 resulting in a conserved missense change, c.14G>T (p.Arg5Leu), that is enriched in our Hispanic pedigrees; in each case, affected individuals carried additional bona fide pathogenic alleles in another BBS gene. In vivo functional modeling in zebrafish embryos demonstrated that c.14G>T is a loss-of-function variant, and suppression of nphp1 in concert with each of the primary BBS loci found in our NPHP1-positive pedigrees exacerbated the severity of the phenotype. These results suggest that NPHP1 mutations are probably rare primary causes of BBS that contribute to the mutational burden of the disorder.
Main Text
Nephrocystin-1 is a ciliary protein encoded by NPHP1 on human chromosome 2q13 (MIM 607100; RefSeq accession number NM_000272.3; GI, 189491772). The genetic structure surrounding the gene and locus includes flanking 330 kb inverted low-copy repeats (LCR) that have 45 kb direct repeats embedded within them; this genomic architecture renders the locus susceptible to genomic instability mainly via nonallelic homologous recombination (NAHR).1 A common 290 kb recurrent microdeletion with breakpoints located within the smaller direct repeats is present in approximately 1/400 normal individuals of northern European descent (estimated from the Database of Genomic Variants [DGV]). Homozygous deletions have never been detected in controls, suggesting that loss of NPHP1 gives rise to penetrant pathology. However, the clinical expressivity of the homozygous deletion can be variable. In most cases reported, the deletion has been causally associated with isolated nephronophthisis (NPHP [MIM 256100])1,2 or NPHP and retinal degeneration (Senior-Loken syndrome [SLS] [MIM 266900]);3 however, rare deletion cases have also been identified with Joubert syndrome (JBTS [MIM 213300]), a severe ciliopathy hallmarked by developmental delay and central nervous system malformations.4 Multiple in vivo and in vitro studies have demonstrated physical interaction, protein colocalization, and genetic interaction between NPHP1 and other ciliary proteins that include nephrocystin-3, nephrocystin-4, and inversin.5–9 Moreover, a dosage-sensitive interaction between Nphp1 and Ahi1, for which the human ortholog is responsible for approximately 10% of JBTS,10 has also been shown in rodent models; mice deficient for both proteins have a more severe retinal phenotype than do the single-gene knockouts.11
These observations prompted us to ask whether NPHP1 harbors alleles that are contributory to other ciliopathies. To this end, we queried NPHP1 mutations in a cohort of individuals with Bardet-Biedl syndrome (BBS [MIM 209900]), a rare (1:13,500–1:160,000) multisystemic ciliopathy characterized by retinal degeneration, obesity, polydactyly, mental retardation, renal dysfunction, and hypogonadism; BBS is also underscored by extensive genetic heterogeneity, allelic overlap with other ciliopathies, and both recessive and oligogenic forms of inheritance.12–16
First, we tested the copy-number status of NPHP1 in 200 BBS-affected families (82.5% of individuals of European ancestry, 15% of Arab ancestry, 2.5% of Hispanic origin) and 229 control subjects of European descent. This analysis was part of a custom high-resolution oligonucleotide array comparative genomic hybridization (aCGH) scan of 772 genes prioritized from the ciliary proteome;17 at the NPHP1 locus, we placed 187 oligonucleotide probes, distributed across the intragenic coding and noncoding regions with an average resolution of 100 bp and 500 bp, respectively. The experiments were performed according to the manufacturer’s recommendations with minor modifications via whole-genome amplified DNA in BBS cases and genomic DNA in controls.18 Additional individuals from our pedigrees, as well as confirmatory analysis of candidate CNVs, were genotyped by TaqMan copy-number assays performed according to the manufacturer’s recommendations. We used three different assays, located in exon 1 (Hs00036264_cn), exon 10 (Hs00095856_cn), and exon 20 (Hs00002984_cn) of NPHP1 (RefSeq NM_000272.3; UCSC Genome Browser build hg19). Both the usage of human DNA samples and model organisms has been reviewed and approved by ethics board and animal care and use committees at Duke University. Informed consent was obtained before individuals were included in the study.
We identified three BBS-affected families in which the NPHP1 recurrent deletion segregated with disease: one consanguineous Hispanic pedigree with a homozygous deletion (RC2), one consanguineous Israeli family with a heterozygous deletion (family 44), and one pedigree of northern European descent (AR704) also bearing a heterozygous deletion (Table 1, Figures 1 and 2). The deletion was not detected in any of our control samples. Moreover, to obtain a more accurate assessment of the frequency of the NPHP1 deletion in the general population, and thus the power of the analysis, we combined our experimental control data with publicly available data from a genome-wide copy-number variant (CNV) screen by Itsara et al.19 In this data set, heterozygous deletions were detected in 2/1,607 European controls.
Table 1.
Detected Mutations and Clinical Findings in Affected Individuals with NPHP1 Mutations
| Subject; Origin | NPHP1 Mutation (RefSeq NM_000272.3) | BBS Locus (Primary Causal Locus unless Indicated Otherwise) | Zebrafish Dataa | RP | Polydactyly | Obesity | Hypogonadism | Renal Anomalies | Developmental Delay |
|---|---|---|---|---|---|---|---|---|---|
| RC2-03; Latino | NPHP1: del; hom | BBS2b (RefSeq NM_031885.3): c.367A>G (p.Ile123Val); hom | hypomorph | yes | yes | no | yes | nephronophthisis | yes |
| AR704-03; N. European | NPHP1: del; het | BBS10c (RefSeq NM_024685.3): c.145C>T (p.Arg49Trp); het | null | yes | yes | yes | NA | no | yes |
| AR704-04; N. European | NPHP1: del; het | BBS10c (RefSeq NM_024685.3): c.145C>T (p.Arg49Trp); het | null | yes | yes | yes | NA | no | yes |
| 44/3; Israel | NPHP1: del; het | BBS7 (RefSeq NM_176824.2): c.87_88delCA (p.His29Glnfs∗12); hom | NA | yes | yes | yes | yes | born with one kidney; chronic renal failure; kidney transplantation | yes |
| 44/4; Israel | NPHP1: del; het | BBS7 (RefSeq NM_176824.2): c.87_88delCA (p.His29Glnfs∗12); hom | NA | yes | yes | yes | NA | no | yes |
| AR888-03; Latino | NPHP1: c.14G>T (p.Arg5Leu); het | BBS1 (RefSeq NM_024649.4): c.1645G>T (p.Glu549∗); het | NA | yes | yes | yes | ND | impaired renal function; kidney cysts; enlarged kidney | yes |
| BBS1d (RefSeq NM_024649.4): del exon1_11; het | NA | ||||||||
| RC1-03; Latino | NPHP1: c.14G>T (p.Arg5Leu); het | BBS10 (RefSeq NM_024685.3): c.926T>C (p.Leu309Pro); het | ND | yes | yes | yes | yes | no | yes |
| BBS10 (RefSeq NM_024685.3): c.9_14delTTCTAT (p.Ser3_Met5delinsArg); het | ND | ||||||||
| DM012-003; N. European | NPHP1: c.14G>T (p.Arg5Leu); het | BBS10 (RefSeq NM_024685.3): c.271dupT (Cys91Leufs∗5); hom | NA | yes | yes | yes | no | no | yes |
| R1-04; Latino | NPHP1: c.14G>T (p.Arg5Leu); het | BBS9 (RefSeq NM_198428.2): c.1536A>G [p.(=)]; hom | ND | yes | ND | ND | ND | ND | ND |
| R1-05; Latino | NPHP1: c.14G>T (p.Arg5Leu); het | BBS9 (RefSeq NM_198428.2): c.1536A>G [p.(=)]; hom | yes | ND | ND | ND | ND | ND | |
| AR082-03; Latino | NPHP1: c.14G>T (p.Arg5Leu); het | BBS9 (RefSeq NM_198428.2): c.1789C>T (p.Gln597∗); hom | NA | yes | ND | ND | ND | ND | ND |
Abbreviations are as follows: RP, retinitis pigmentosa; del, deletion; het, heterozygous; hom, homozygous; NA, not applicable (affected individual is female); ND, not determined.
Data from Zaghloul et al.26
Putative second-site modulator
Heterozygous change, unclear whether causal locus
Publicly available in dbVar (study ID: nstd93)
Figure 1.
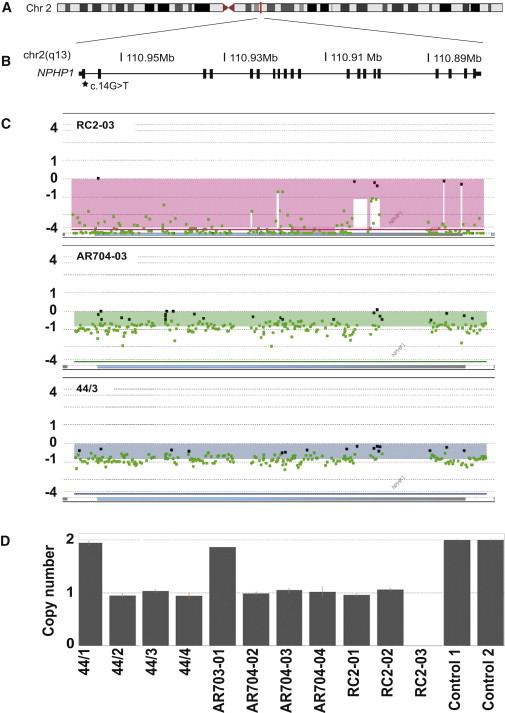
NPHP1 Deletions in Individuals with BBS
(A) Schematic of human chromosome 2. A vertical red line indicates the position of NPHP1 (RefSeq NM_000272.3) at 2q13.
(B) A zoomed depiction of the NPHP1 locus with a schematic illustration of exons (vertical black bars). The genomic location on chromosome 2 (in Mb) is shown. c.14G>T (p.Arg5Leu) is indicated with a black star.
(C) High-resolution results from the aCGH analyses in individuals RC2-03 (top), AR704-03 (middle), and 44/3 (bottom) are visualized as a plot. The horizontal blue bar represents NPHP1. Individual dots represent specific oligonucleotide probes and are indicated as black (normal copy number), red (copy number gain), and green (copy number loss) compared to a reference sample. The normalized log2 ratios of the Cy5/Cy3 intensity values for cases versus controls are shown on the y axis.
(D) Segregation of the NPHP1 deletion by Taqman copy number analysis using a probe (Hs00002984_cn) located within the NPHP1 locus; individuals 44/2, 44/3, 44/4, AR704-02, AR704-03, AR704-04, RC2-01, and RC2-02 harbor a heterozygous deletion and RC2-03 has a homozygous deletion. Error bars were calculated by the CopyCaller Software and represent the copy number range of triplicate reactions.
Figure 2.
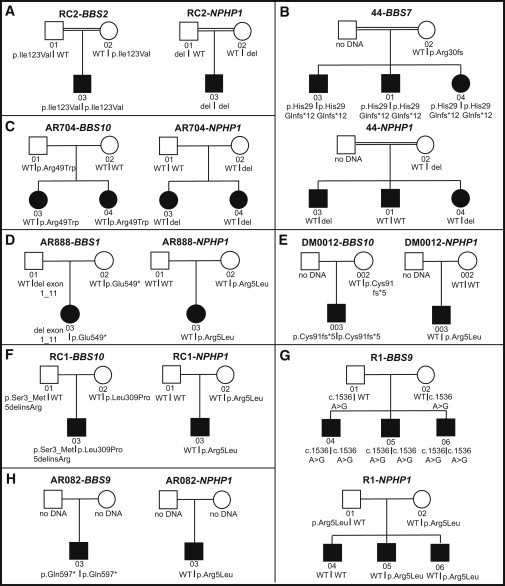
Pedigrees of the Families with NPHP1 Mutations
Pedigrees and BBS gene mutational analysis results in all families with NPHP1 mutations. Squares denote males and circles denote females. Blackened symbols represent the affected children and individual identification numbers are given below the symbols. Double lines indicate consanguinity.
Overall, we observed a deletion incidence of 1.5% of BBS-affected pedigrees (3/200). Although a rare event, data from the total BBS cohort versus the combined control data sets (our 229 controls plus the Itsara et al.19 set) showed a significant enrichment of the NPHP1 deletion allele in BBS (p = 0.008, Fisher’s exact test). These data were likewise significant when compared to the entire DGV mixed population control data from 20,086 individuals (p = 0.011, chi-square with Yates correction). These findings suggest that the NPHP1 deletion may contribute to BBS.
Next, we turned our attention to point mutations. We selected probands from 96 BBS-affected families without preselection for known mutations or CNVs, we PCR amplified all NPHP1 (RefSeq NM_000272.3, hg19) coding exons and splice junctions, and we sequenced them according to standard Sanger methodology (primers and PCR conditions are available on request). We identified a single heterozygous variant that encoded a missense change, c.14G>T (p.Arg5Leu; rs190983114; RefSeq NM_000272.3; hg19), in three BBS-affected families (Table 1, Figure 2) of Hispanic (n = 2; RC1, AR888) or northern European (n = 1; DM012) origin. The variant position is evolutionarily invariant in all 39 species with a detectable NPHP1 ortholog and is present in ultrarare frequencies in the Exome Variant Server database (EVS) (6/13,000 with an equal MAF of 0.04% in either European American [EA] or African American [AA] samples). To replicate this finding, we assessed the prevalence of the c.14G>T change in the second half of our BBS cohort by Sanger sequencing of NPHP1 exon 1; we detected two additional BBS-affected families (AR082 and R1) that were heterozygous for the c.14G>T allele; both families were of Hispanic origin. Combined, we found that 2.5% of our overall cohort (5/200 families) carried the c.14G>T allele, which, similar to the deletion burden in our cohort, represents an apparent enrichment compared to controls.
We were struck by the fact that 4/5 of families with the c.14G>T allele were of Hispanic origin, a subset that is heavily underrepresented in our cohort, with only 14/200 families (n = 4/14 Hispanics with NPHP1 c.14G>T; MAF 28%). This finding represents a significant enrichment of this allele compared to EA and AA controls (MAF < 0.04%). Given that EVS is bereft of Hispanic control data, and conscious of the fact that we might be observing a population-specific variant, we sequenced 277 control individuals of Hispanic origin; although the MAF of this allele was elevated in this population, the enrichment in our BBS cohort remained significant (28% versus 4.9% MAF%; p = 0.04; Fisher’s exact test).
Our findings are reminiscent of the genetic architecture of other BBS and ciliopathy loci, in which low-frequency variants can contribute both causal and contributory alleles to the disease burden13,20–26 but for which the genetic data alone suggest association but are insufficient to provide robust arguments for causality. We therefore turned to functional assays. We reasoned that (1) if NPHP1 is relevant to BBS pathology, then suppression of the zebrafish ortholog of this gene should mimic the previously established phenotypes of bbs morphants at midsomitic stages26,27 and also give rise to renal defects; and (2) if the c.14G>T allele is relevant to the function of the protein and the clinical phenotype of the affected individuals, this allele should fail to complement suppression of nphp1 in vivo.
The Danio rerio genome has a single zebrafish NPHP1 ortholog (nphp1 [RefSeq NM_001077170]; 56% protein sequence identity with human NPHP1). We obtained a previously published nphp1 translation-blocking (tb) morpholino (MO)6 as well as a splice-blocking (sb) MO targeting the junction of nphp1 exon 2/intron 2–3 (5′-ACAGATACAAGTCCTCTACCTCTGC-3′; Gene Tools). We injected progressively increasing sb-MO or tb-MO concentrations (3–9 ng, sb; 2–6 ng, tb) into wild-type (WT) zebrafish embryos at the 1–4 cell stage and we scored embryo clutches for shortened body axes, broader somites, and broad and kinked notochords at the 8–10 somite stage according to previously established objective criteria.26 For each MO independently, we observed a dose-dependent increase in both the number of affected morphants and also the severity of the defects (Figure S1 available online) that phenocopied the phenotypes observed in numerous ciliopathy morphants, including all tested BBS genes.20,22,23,27–30 To test sb-MO efficiency, we harvested whole embryos in Trizol (Invitrogen) for total RNA extraction and reverse transcribed oligo-dT primed cDNA (Superscript III, Invitrogen) for PCR. An aberrant splicing product was detected in sb-MO-injected embryos (Figure S1).
To test for MO specificity, we coinjected MO with capped human WT NPHP1 mRNA. In brief, to generate human NPHP1 message, we cloned the full-length NPHP1 open reading frame into the pCS2+ plasmid, linearized with NotI, and performed in vitro transcription with the SP6 mMessage mMachine kit (Ambion). Coinjection of 200 pg of WT human NPHP1 mRNA with MO (3 ng sb-MO; 2 ng tb-MO) resulted in a significant rescue of morphant phenotypes (55% versus 39% abnormal for sb-MO versus sb-MO+WT; p = 0.003; Figure 3; n = 60–62 embryos/injection and 69% versus 45% abnormal for tb-MO versus tb-MO+WT; p < 0.0001; Figure S1; n = 31–41 embryos/injection). We then used the gastrulation phenotype to test the pathogenicity of the heterozygous c.14G>T NPHP1 variant by comparing the ability of equivalent doses of NPHP1 WT or c.14G>U mRNA to rescue the nphp1 sb-MO-induced defects. Coinjection of nphp1 sb-MO with human NPHP1 mRNA harboring the c.14G>U mutation was significantly worse from embryo clutches injected with sb-MO coinjected with WT mRNA, suggesting that the mutation impedes NPHP1 function (Figures 3A and 3B). Injection of mutant NPHP1 mRNA was indistinguishable from wild-type injection (p = 0.79), but injection of decreasing doses of c.14G>U mRNA alone resulted in a dose-dependent decrease of affected embryos (Figure S2), which indicates indirectly the decreased function conferred by the c.14G>T mutation.
Figure 3.
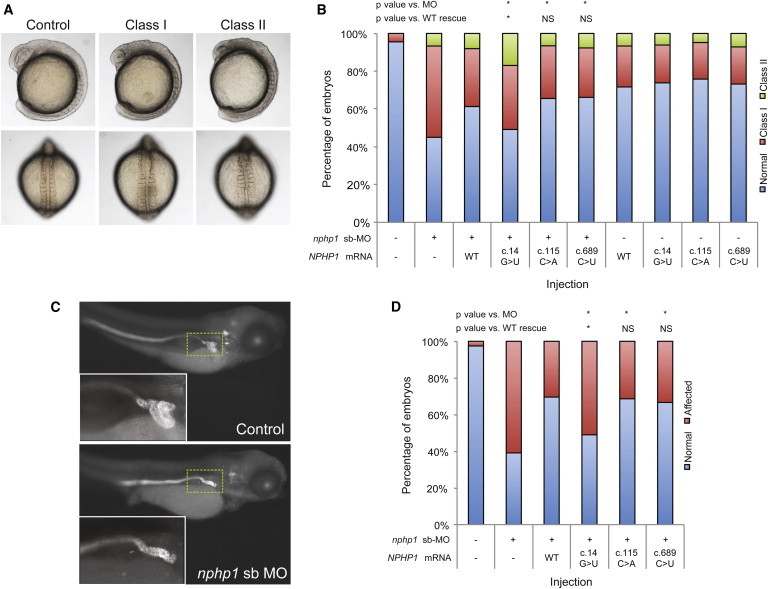
Morpholino-Induced Suppression of nphp1 in Zebrafish Embryos Generates Gastrulation Defects and Abnormal Renal Morphology
(A) Live dorsal and lateral views of normal embryos and class I and class II morphants are shown. Live images of embryos were acquired on a Nikon AZ100 microscope at 8× magnification, with image capture facilitated by NIS Elements software (Nikon).
(B) Quantification of live scoring of nphp1 MO and human mRNA (co)injections. Embryos were injected with the indicated dose (2 ng sb-MO and/or 200 pg NPHP1 mRNA) and scored live according to criteria shown. n = 48–65 embryos/injection, repeated at least twice; with masked scoring; all comparisons were made using χ2 tests; ∗p < 0.002; NS, not significant; see Table S1 for p values. c.115C>A (rs33958626) and c.689C>T (rs113450177) have similar minor allele frequencies (MAF; 4% in European Americans and 3.3% in African Americans) to that of c.14G>T (rs190983114) in our overall BBS cohort (2.5%), but result in rescue not significantly different from WT mRNA.
(C) Lateral views of representative larva fluorescently stained at 4 days postfertilization (dpf) with anti-NaK ATPase antibody (a6F, Developmental Studies Hybridoma Bank) to demarcate renal tubules. In comparison to controls, embryos injected with nphp1 sb-MO have atrophy and reduced convolution of the proximal tubule. Inset area indicated by dashed green line.
(D) Quantification of renal phenotyping of nphp1 MO and human mRNA (co)injections. Embryos were injected with the indicated dose (2 ng sb-MO and/or 200 pg NPHP1 mRNA) and immunostained with anti-NaK ATPase antibody and scored as normal or affected (as shown in panel C), n = 46–51 embryos/injection, repeated at least twice; with masked scoring; all comparisons were made with χ2 tests; ∗p < 0.05; NS, not significant; see Table S1 for p values.
In parallel, we tested two missense variants mined from EVS that had both a modest allele frequency (>1% MAF) and an apparent ethnicity-specific enrichment similar to c.14G>T (c.115C>A, rs33958626, MAF 0.6% in AA versus 4% in EA; and c.689C>T, rs113450177, MAF 3.3% AA versus 0% EA; RefSeq NM_000272.3; hg19). The efficiency of both variants to improve the nphp1 MO gastrulation defects was not significantly different than that of WT mRNA, lending further evidence in favor of the specificity of the c.14G>T pathogenicity (Figure 3B, Table S1).
Next, we corroborated these findings in 4 day postfertilization (dpf) zebrafish larva by using a phenotypic readout directly relevant to NPHP. In brief, we reinjected sb-MO and/or mRNA at identical doses to those used for the gastrulation scoring and fixed embryos for whole-mount immunostaining with anti-NaK ATPase antibody (a6F, Developmental Studies Hybridoma Bank) as described.31 Although we did not observe significant cyst formation in nphp1 morphant batches (<1%), we did observe atrophy and diminished convolution of the proximal tubule, which is an orthologous renal feature consistent with NPHP in humans32 (Figure 3C). Coinjection of sb-MO with WT mRNA improved significantly this defect (61% versus 30% affected for sb-MO versus WT+sb-MO, p < 0.0001, n = 46 embryos/injection with masked scoring), whereas embryo clutches injected with MO and c.14G>U mRNA were significantly worse than WT rescue (51% affected, p < 0.0001). These data corroborated our CE studies and suggested further that c.14G>T is pathogenic. Moreover, negative control variants c.115C>A and c.689C>T both scored benign in this assay, once again similar to the CE studies (Figures 3C and 3D; Table S1).
In aggregate, our data indicate that eight BBS-affected families bear loss-of-function mutations in NPHP1 from either deletion CNVs or from a deleterious point mutation (SNV). We therefore asked (1) whether any of these alleles might be genetically necessary to drive the phenotype under a recessive paradigm and thus represent a Penetrant Mendelizing Variant (PMV); and (2) similar to other ciliopathy loci, whether the phenotype could be explained by the amalgam of mutations at multiple loci.
We first segregated all pathogenic variants in our families. In the family with the homozygous NPHP1 deletion, segregation showed autosomal-recessive inheritance, with each of the parents being heterozygous carriers, as confirmed by a Taqman copy number assay (Figures 1D and 2A). By contrast, in the other two families with a heterozygous NPHP1 deletion, there was no evidence of a second CNV in NPHP1, and sequencing of all exons was likewise negative for candidate pathogenic changes. These data suggested that this lesion might be coincidental in these families or be contributory to the genetic load of these pedigrees similar to what has been observed for other BBS loci.13,14,20–26 We made similar observations for the five families bearing the c.14G>T allele, none of which carried additional NPHP1 pathogenic alleles that were detectable within the constraints of our methodology.
Given these observations, and the fact that the BBS phenotype is more severe than what would be expected typically from a homozygous NPHP1 deletion, we asked whether our NPHP1-positive pedigrees might bear additional pathogenic lesions in the known BBS loci; therefore, we performed Sanger-based sequencing analysis of the BBS1-16 exons and splice junctions, which together account for ∼75% of the disease burden in BBS.15,33
Consistent with the NPHP1 deletion being the primary driver of BBS, sequencing of DNA from individuals in family RC2 yielded no biallelic mutations under a recessive model. These data, together with the fact that the NPHP1 deletion has never been observed in homozygosity in control individuals, indicates that this lesion is the most likely primary driver of the disease in the family, or PMV, potentially defining a new BBS locus. Consistent with this notion, phenotypic analysis of the affected individual (RC2-03) that included a renal biopsy indicated that the proband, in addition to his classical BBS features (retinal degeneration, polydactyly, developmental delay, hypogonadism), was notable for tubular interstitial and glomerular injury, findings uncommon in BBS34 but typical of nephronophthisis (Figure 4).
Figure 4.
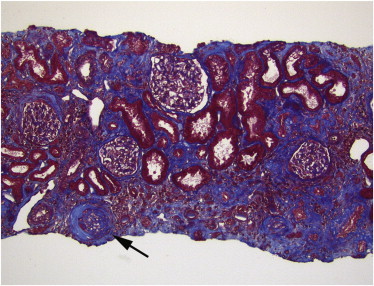
Renal Biopsy on Individual RC2-03
Renal biopsy on RC2-03, showing moderate tubular atrophy and interstitial fibrosis, with one obsolescent glomerulus (arrow). In the context of this individual, the findings are consistent with juvenile nephronophthisis. (Trichrome stain, original magnification ×100.)
Of note, individual RC2-03 harbors a homozygous BBS2 variant, c.367A>G (p.Ile123Val). This allele cannot be causal to BBS, because it is present in homozygosity in ∼4% of EA and 8% of Hispanic control subjects (rs11373; dbSNP; RefSeq NM_031885.3; hg19). However, this change has been shown previously to be a functional hypomorph that is enriched significantly and overtransmitted in individuals with BBS,26 raising the possibility that NPHP1 and BBS2 might interact genetically.
We similarly analyzed the coding regions of BBS1-1616 by Sanger sequencing in the other seven families. In five of them, we identified a primary BBS driver locus. In the other two families (AR888 and AR704), we evaluated BBS1 and BBS10, respectively, for CNVs that could unmask the second recessive allele. We identified a paternally inherited 17.7 kb deletion of BBS1 (RefSeq NM_024649.4; hg19) exons 1–11 (in AR888 in trans with a truncating point mutation), thereby adding a sixth pedigree with a characterized primary driver locus (Table 1, Figures 2 and S3). The causal locus in AR704 remains unclear; we cannot rule out the possibility that AR704 harbors a second BBS10 allele in a regulatory region, or that BBS10 functions as a second-site modulator to a hitherto unidentified BBS gene.
Together, our CNV and sequencing data showed one pedigree in which homozygous loss of NPHP1 is coincident with a homozygous hypomorph in BBS2; in the remaining families, heterozygous NPHP1 loss-of-function alleles are coinherited with mutations in each of BBS1, BBS7, BBS9, and BBS10. We note that these predominant causal loci in the NPHP1-mutation-bearing pedigrees differ somewhat from the reported relative contributions of BBS genes to the disorder,15,33 which may either be a chance event or may be suggestive of a unique biochemical relationship between NPHP1 and these BBS proteins.
As a first step toward investigating these possibilities, we asked whether genetic interaction between NPHP1 and the primary BBS driver loci can exacerbate the severity of disease in a manner reminiscent of the reported NPHP1-AHI1 interaction,11 as well as the interaction reported previously for various binary combinations of ciliopathy13,20,22–26,35–38 and nonciliopathy21 loci. We therefore coinjected subeffective doses of the nphp1 sb-MO (0.5 ng) with each of bbs1 (3 ng), bbs2 (1 ng), bbs7 (3.5 ng), bbs9 (2 ng), and bbs10 (4 ng)26 (Figure 5, Table S1). In each pairwise combination, gastrulation phenotypes were significantly more severe in the double MO injected embryos compared to the single MO injections alone (n = 61–255 embryos/injection; masked scoring). Similarly, the presence of proximal renal tubule atrophy in 4 dpf larva showed that for each pairwise combination, renal phenotypes were exacerbated significantly in comparison to single MO injections alone (n = 16–95 embryos/injection; masked scoring). Together, these data indicate that suppression of NPHP1 activity, in combination with loss of function of any tested BBS locus, can interact to exacerbate phenotypic severity. We do not have the resolution to determine whether these effects are additive or multiplicative, although our functional studies are more consistent with an additive model.
Figure 5.
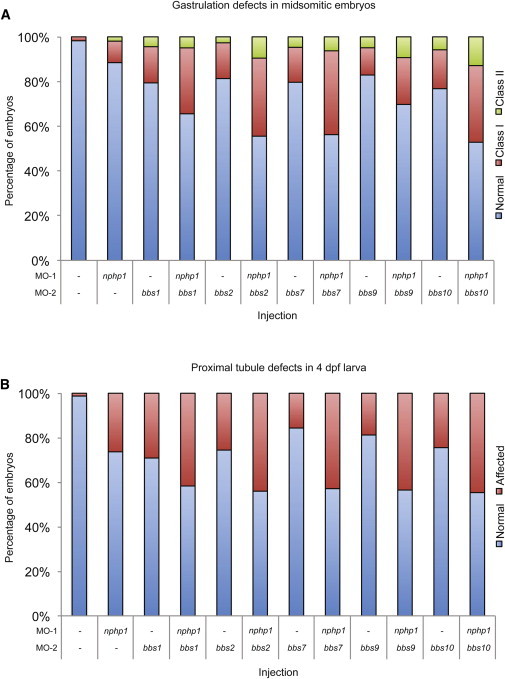
nphp1 Interacts Genetically with All Primary BBS Loci Identified in BBS Cases with NPHP1 Mutations
(A) Gastrulation defects in midsomitic embryos. Coinjection of a subeffective dose of nphp1 sb-MO together with each of bbs1, bbs2, bbs7, bbs9, and bbs10 MOs significantly increase the gastrulation phenotype compared to the same dose injected alone. n = 61–255 embryos/injection, 0.5 ng of nphp1 sb-MO, 3 ng of bbs1-MO, 1 ng of bbs2-MO, 3.5 ng of bbs7-MO, 2 ng of bbs9-MO, and 4 ng of bbs10-MO was used, scoring as in Figure 3A.
(B) Proximal tubule defects in 4 dpf larva. Coinjection of a subeffective dose of nphp1 sb-MO together with each of the five bbs MOs (same doses as in A) increase significantly the renal phenotype at 4 dpf compared to the same dose injected alone. n = 16–95 embryos/injection, scoring as in Figure 3B. See Table S1 for p values.
Given that we discovered a single BBS-affected family with recessive NPHP1 deletions, it will be necessary to identify additional individuals who fulfill BBS clinical criteria but have causal molecular lesions at the NPHP1 locus. Browsing the Database of Chromosome Imbalance and Phenotype in Humans using Ensembl Resources (DECIPHER) identified one additional case with the homozygous NPHP1 deletion. The phenotypic information available (oculomotor apraxia; delayed speech and language development; intellectual disability) partially overlaps with BBS (but also with JBTS) but only for neurocognitive features that are typical triggers for ordering clinical aCGH testing. However, our data potentially extend the phenotypic spectrum of NPHP1 loss of function, wherein homozygous deletion of the locus can give rise to ciliopathy phenotypes in the intermediate continuum between classical isolated NPHP and the severe JBTS phenotype. These data predict that individuals affected by other ciliopathies might also bear similar lesions and suggest that extending the diagnostic analysis of ciliary disease cases to include this NPHP1 CNV, and potential CNVs at other ciliopathy loci,17 might inform further the genetics of ciliopathies and other complex traits.
More broadly, our data also suggest that NPHP1 mirrors other ciliopathy loci, contributing both causal and/or contributory alleles to the genetic burden of the disease. The observed deletion removes NPHP1 and a fragment of the neighboring gene MALL; however, previous studies have detected the heterozygous deletion in trans with a coding change in NPHP1 in persons with NPHP, but they did not detect mutations in MALL.2 As such, although we cannot formally exclude the contribution of MALL to BBS, we consider that unlikely.
Our observations, together with previous studies, suggest that the NPHP1 lesion might not be deterministic for renal disease. First, mouse breeding experiments have shown that the Nphp1-Ahi1 interaction can exacerbate retinal pathology.11 Second, although the individual (RC2-03) with the homozygous NPHP1 deletion does manifest hallmarks of NPHP-like renal pathology, only two of our other ten BBS-affected individuals with heterozygous NPHP1 lesions had renal defects. This is not surprising, given that we interrogated only a small fraction of the ciliary proteome (or the genome) and that, as emerging data suggest, the primary cilium is sensitive to trans-epistasis that can either exacerbate or ameliorate the phenotype, as exemplified by the protective effect of loss-of-function Bbs6 mutations on Cep290 and the likewise protective effect of loss-of-function axonemal mutations on a genetic background sensitized by PKD mutations in mice.36,38
In the present work, the challenges of interpreting the contribution of heterozygous rare alleles in the context of primary causal lesions are exacerbated by the identification of alleles with ethnic-specific enrichment. The c.14G>T allele is ultrarare in northern European and African American control subjects (MAF < 0.04%) but is elevated in Hispanics, a population that has been undersampled for BBS. In these instances, statistical arguments alone remain constrained by the availability of cases and appropriate controls. Thus, the synthesis of population data with rigorous functional studies of allele effect may aid interpretation of the potential functional consequences of the burden of rare alleles and their impact on the penetrance and expression of the disease in an ethnicity-specific manner.39
Acknowledgments
We are grateful to the BBS-affected families for their continued enthusiasm in our work. We thank John Belmont at Baylor College of Medicine and Shelby Strickland, Yutao Liu, Rand Allingham, and Michael Hauser at Duke University for Hispanic control DNAs. This work was funded by NIH grants DK072301, DK075972-09, and HD042601 to N.K., NS058529 to J.R.L., and EY021872 to E.E.D. and F32DK094578 to I.-C. T.; by funding from EU 7th FP under GA nr. 241955, project SYSCILIA to N.K. and E.E.D.; and by the Swedish Research Council grant 2010-978 to A.L. N.K. is a distinguished Jean and George Brumley Professor.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Database of Genomic Variants (DGV), http://dgv.tcag.ca/dgv/app/home
DECIPHER, http://decipher.sanger.ac.uk/
Ensembl Genome Browser, http://www.ensembl.org/index.html
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
UCSC Genome Browser, http://genome.ucsc.edu
Accession Numbers
The dbVar accession number for the study that identified the BBS1 copy-number variant reported in this paper is nstd93.
References
- 1.Saunier S., Calado J., Benessy F., Silbermann F., Heilig R., Weissenbach J., Antignac C. Characterization of the NPHP1 locus: mutational mechanism involved in deletions in familial juvenile nephronophthisis. Am. J. Hum. Genet. 2000;66:778–789. doi: 10.1086/302819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hildebrandt F., Otto E., Rensing C., Nothwang H.G., Vollmer M., Adolphs J., Hanusch H., Brandis M. A novel gene encoding an SH3 domain protein is mutated in nephronophthisis type 1. Nat. Genet. 1997;17:149–153. doi: 10.1038/ng1097-149. [DOI] [PubMed] [Google Scholar]
- 3.Caridi G., Murer L., Bellantuono R., Sorino P., Caringella D.A., Gusmano R., Ghiggeri G.M. Renal-retinal syndromes: association of retinal anomalies and recessive nephronophthisis in patients with homozygous deletion of the NPH1 locus. Am. J. Kidney Dis. 1998;32:1059–1062. doi: 10.1016/s0272-6386(98)70083-6. [DOI] [PubMed] [Google Scholar]
- 4.Parisi M.A., Bennett C.L., Eckert M.L., Dobyns W.B., Gleeson J.G., Shaw D.W., McDonald R., Eddy A., Chance P.F., Glass I.A. The NPHP1 gene deletion associated with juvenile nephronophthisis is present in a subset of individuals with Joubert syndrome. Am. J. Hum. Genet. 2004;75:82–91. doi: 10.1086/421846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Delous M., Hellman N.E., Gaudé H.M., Silbermann F., Le Bivic A., Salomon R., Antignac C., Saunier S. Nephrocystin-1 and nephrocystin-4 are required for epithelial morphogenesis and associate with PALS1/PATJ and Par6. Hum. Mol. Genet. 2009;18:4711–4723. doi: 10.1093/hmg/ddp434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Slanchev K., Pütz M., Schmitt A., Kramer-Zucker A., Walz G. Nephrocystin-4 is required for pronephric duct-dependent cloaca formation in zebrafish. Hum. Mol. Genet. 2011;20:3119–3128. doi: 10.1093/hmg/ddr214. [DOI] [PubMed] [Google Scholar]
- 7.Otto E.A., Schermer B., Obara T., O’Toole J.F., Hiller K.S., Mueller A.M., Ruf R.G., Hoefele J., Beekmann F., Landau D. Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nat. Genet. 2003;34:413–420. doi: 10.1038/ng1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mollet G., Salomon R., Gribouval O., Silbermann F., Bacq D., Landthaler G., Milford D., Nayir A., Rizzoni G., Antignac C., Saunier S. The gene mutated in juvenile nephronophthisis type 4 encodes a novel protein that interacts with nephrocystin. Nat. Genet. 2002;32:300–305. doi: 10.1038/ng996. [DOI] [PubMed] [Google Scholar]
- 9.Olbrich H., Fliegauf M., Hoefele J., Kispert A., Otto E., Volz A., Wolf M.T., Sasmaz G., Trauer U., Reinhardt R. Mutations in a novel gene, NPHP3, cause adolescent nephronophthisis, tapeto-retinal degeneration and hepatic fibrosis. Nat. Genet. 2003;34:455–459. doi: 10.1038/ng1216. [DOI] [PubMed] [Google Scholar]
- 10.Ferland R.J., Eyaid W., Collura R.V., Tully L.D., Hill R.S., Al-Nouri D., Al-Rumayyan A., Topcu M., Gascon G., Bodell A. Abnormal cerebellar development and axonal decussation due to mutations in AHI1 in Joubert syndrome. Nat. Genet. 2004;36:1008–1013. doi: 10.1038/ng1419. [DOI] [PubMed] [Google Scholar]
- 11.Louie C.M., Caridi G., Lopes V.S., Brancati F., Kispert A., Lancaster M.A., Schlossman A.M., Otto E.A., Leitges M., Gröne H.J. AHI1 is required for photoreceptor outer segment development and is a modifier for retinal degeneration in nephronophthisis. Nat. Genet. 2010;42:175–180. doi: 10.1038/ng.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Badano J.L., Kim J.C., Hoskins B.E., Lewis R.A., Ansley S.J., Cutler D.J., Castellan C., Beales P.L., Leroux M.R., Katsanis N. Heterozygous mutations in BBS1, BBS2 and BBS6 have a potential epistatic effect on Bardet-Biedl patients with two mutations at a second BBS locus. Hum. Mol. Genet. 2003;12:1651–1659. doi: 10.1093/hmg/ddg188. [DOI] [PubMed] [Google Scholar]
- 13.Badano J.L., Leitch C.C., Ansley S.J., May-Simera H., Lawson S., Lewis R.A., Beales P.L., Dietz H.C., Fisher S., Katsanis N. Dissection of epistasis in oligogenic Bardet-Biedl syndrome. Nature. 2006;439:326–330. doi: 10.1038/nature04370. [DOI] [PubMed] [Google Scholar]
- 14.Katsanis N., Ansley S.J., Badano J.L., Eichers E.R., Lewis R.A., Hoskins B.E., Scambler P.J., Davidson W.S., Beales P.L., Lupski J.R. Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder. Science. 2001;293:2256–2259. doi: 10.1126/science.1063525. [DOI] [PubMed] [Google Scholar]
- 15.Zaghloul N.A., Katsanis N. Mechanistic insights into Bardet-Biedl syndrome, a model ciliopathy. J. Clin. Invest. 2009;119:428–437. doi: 10.1172/JCI37041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davis E.E., Katsanis N. The ciliopathies: a transitional model into systems biology of human genetic disease. Curr. Opin. Genet. Dev. 2012;22:290–303. doi: 10.1016/j.gde.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hjeij R., Lindstrand A., Francis R., Zariwala M.A., Liu X., Li Y., Damerla R., Dougherty G.W., Abouhamed M., Olbrich H. ARMC4 mutations cause primary ciliary dyskinesia with randomization of left/right body asymmetry. Am. J. Hum. Genet. 2013;93:357–367. doi: 10.1016/j.ajhg.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carvalho C.M., Zhang F., Liu P., Patel A., Sahoo T., Bacino C.A., Shaw C., Peacock S., Pursley A., Tavyev Y.J. Complex rearrangements in patients with duplications of MECP2 can occur by fork stalling and template switching. Hum. Mol. Genet. 2009;18:2188–2203. doi: 10.1093/hmg/ddp151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Itsara A., Cooper G.M., Baker C., Girirajan S., Li J., Absher D., Krauss R.M., Myers R.M., Ridker P.M., Chasman D.I. Population analysis of large copy number variants and hotspots of human genetic disease. Am. J. Hum. Genet. 2009;84:148–161. doi: 10.1016/j.ajhg.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davis E.E., Zhang Q., Liu Q., Diplas B.H., Davey L.M., Hartley J., Stoetzel C., Szymanska K., Ramaswami G., Logan C.V., NISC Comparative Sequencing Program TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat. Genet. 2011;43:189–196. doi: 10.1038/ng.756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Pontual L., Zaghloul N.A., Thomas S., Davis E.E., McGaughey D.M., Dollfus H., Baumann C., Bessling S.L., Babarit C., Pelet A. Epistasis between RET and BBS mutations modulates enteric innervation and causes syndromic Hirschsprung disease. Proc. Natl. Acad. Sci. USA. 2009;106:13921–13926. doi: 10.1073/pnas.0901219106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khanna H., Davis E.E., Murga-Zamalloa C.A., Estrada-Cuzcano A., Lopez I., den Hollander A.I., Zonneveld M.N., Othman M.I., Waseem N., Chakarova C.F. A common allele in RPGRIP1L is a modifier of retinal degeneration in ciliopathies. Nat. Genet. 2009;41:739–745. doi: 10.1038/ng.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leitch C.C., Zaghloul N.A., Davis E.E., Stoetzel C., Diaz-Font A., Rix S., Alfadhel M., Lewis R.A., Eyaid W., Banin E. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat. Genet. 2008;40:443–448. doi: 10.1038/ng.97. [DOI] [PubMed] [Google Scholar]
- 24.Putoux A., Thomas S., Coene K.L., Davis E.E., Alanay Y., Ogur G., Uz E., Buzas D., Gomes C., Patrier S. KIF7 mutations cause fetal hydrolethalus and acrocallosal syndromes. Nat. Genet. 2011;43:601–606. doi: 10.1038/ng.826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stoetzel C., Laurier V., Davis E.E., Muller J., Rix S., Badano J.L., Leitch C.C., Salem N., Chouery E., Corbani S. BBS10 encodes a vertebrate-specific chaperonin-like protein and is a major BBS locus. Nat. Genet. 2006;38:521–524. doi: 10.1038/ng1771. [DOI] [PubMed] [Google Scholar]
- 26.Zaghloul N.A., Liu Y., Gerdes J.M., Gascue C., Oh E.C., Leitch C.C., Bromberg Y., Binkley J., Leibel R.L., Sidow A. Functional analyses of variants reveal a significant role for dominant negative and common alleles in oligogenic Bardet-Biedl syndrome. Proc. Natl. Acad. Sci. USA. 2010;107:10602–10607. doi: 10.1073/pnas.1000219107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gerdes J.M., Liu Y., Zaghloul N.A., Leitch C.C., Lawson S.S., Kato M., Beachy P.A., Beales P.L., DeMartino G.N., Fisher S. Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nat. Genet. 2007;39:1350–1360. doi: 10.1038/ng.2007.12. [DOI] [PubMed] [Google Scholar]
- 28.Li C., Inglis P.N., Leitch C.C., Efimenko E., Zaghloul N.A., Mok C.A., Davis E.E., Bialas N.J., Healey M.P., Héon E. An essential role for DYF-11/MIP-T3 in assembling functional intraflagellar transport complexes. PLoS Genet. 2008;4:e1000044. doi: 10.1371/journal.pgen.1000044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Valente E.M., Logan C.V., Mougou-Zerelli S., Lee J.H., Silhavy J.L., Brancati F., Iannicelli M., Travaglini L., Romani S., Illi B. Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes. Nat. Genet. 2010;42:619–625. doi: 10.1038/ng.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McIntyre J.C., Davis E.E., Joiner A., Williams C.L., Tsai I.C., Jenkins P.M., McEwen D.P., Zhang L., Escobado J., Thomas S., NISC Comparative Sequencing Program Gene therapy rescues cilia defects and restores olfactory function in a mammalian ciliopathy model. Nat. Med. 2012;18:1423–1428. doi: 10.1038/nm.2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Drummond I.A., Davidson A.J. Zebrafish kidney development. Methods Cell Biol. 2010;100:233–260. doi: 10.1016/B978-0-12-384892-5.00009-8. [DOI] [PubMed] [Google Scholar]
- 32.Hildebrandt F., Attanasio M., Otto E. Nephronophthisis: disease mechanisms of a ciliopathy. J. Am. Soc. Nephrol. 2009;20:23–35. doi: 10.1681/ASN.2008050456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Redin C., Le Gras S., Mhamdi O., Geoffroy V., Stoetzel C., Vincent M.C., Chiurazzi P., Lacombe D., Ouertani I., Petit F. Targeted high-throughput sequencing for diagnosis of genetically heterogeneous diseases: efficient mutation detection in Bardet-Biedl and Alström syndromes. J. Med. Genet. 2012;49:502–512. doi: 10.1136/jmedgenet-2012-100875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beales P.L., Elcioglu N., Woolf A.S., Parker D., Flinter F.A. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J. Med. Genet. 1999;36:437–446. [PMC free article] [PubMed] [Google Scholar]
- 35.Huang L., Szymanska K., Jensen V.L., Janecke A.R., Innes A.M., Davis E.E., Frosk P., Li C., Willer J.R., Chodirker B.N. TMEM237 is mutated in individuals with a Joubert syndrome related disorder and expands the role of the TMEM family at the ciliary transition zone. Am. J. Hum. Genet. 2011;89:713–730. doi: 10.1016/j.ajhg.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma M., Tian X., Igarashi P., Pazour G.J., Somlo S. Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nat. Genet. 2013;45:1004–1012. doi: 10.1038/ng.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O’Toole J.F., Liu Y., Davis E.E., Westlake C.J., Attanasio M., Otto E.A., Seelow D., Nurnberg G., Becker C., Nuutinen M. Individuals with mutations in XPNPEP3, which encodes a mitochondrial protein, develop a nephronophthisis-like nephropathy. J. Clin. Invest. 2010;120:791–802. doi: 10.1172/JCI40076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rachel R.A., May-Simera H.L., Veleri S., Gotoh N., Choi B.Y., Murga-Zamalloa C., McIntyre J.C., Marek J., Lopez I., Hackett A.N. Combining Cep290 and Mkks ciliopathy alleles in mice rescues sensory defects and restores ciliogenesis. J. Clin. Invest. 2012;122:1233–1245. doi: 10.1172/JCI60981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wiszniewski W., Hunter J.V., Hanchard N.A., Willer J.R., Shaw C., Tian Q., Illner A., Wang X., Cheung S.W., Patel A. TM4SF20 ancestral deletion and susceptibility to a pediatric disorder of early language delay and cerebral white matter hyperintensities. Am. J. Hum. Genet. 2013;93:197–210. doi: 10.1016/j.ajhg.2013.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.