

Abstract
Clinical whole-exome sequencing (WES) for identification of mutations leading to Mendelian disease has been offered to the medical community since 2011. Clinically undiagnosed neurological disorders are the most frequent basis for test referral, and currently, approximately 25% of such cases are diagnosed at the molecular level. To date, there are approximately 4,000 “known” disease-associated loci, and many are associated with striking dysmorphic features, making genotype-phenotype correlations relatively straightforward. A significant fraction of cases, however, lack characteristic dysmorphism or clinical pathognomonic traits and are dependent upon molecular tests for definitive diagnoses. Further, many molecular diagnoses are guided by recent gene-disease association discoveries. Hence, there is a critical interplay between clinical testing and research leading to gene-disease association discovery. Here, we describe four probands, all of whom presented with hypotonia, intellectual disability, global developmental delay, and mildly dysmorphic facial features. Three of the four also had sleep apnea. Each was a simplex case without a remarkable family history. Using WES, we identified AHDC1 de novo truncating mutations that most likely cause this genetic syndrome.
Main Text
De novo pathogenic mutations are a major cause of sporadic human genetic disease.1,2 Whole-exome sequencing (WES)3 using next-generation-sequencing methods has proven to be a powerful tool for molecular diagnosis of mutations in genes known to underlie Mendelian disease,1 as well as for the discovery of novel disease-associated loci.4 Despite the rapid development of these new molecular tools, the majority of individuals who are suspected to have a genetic disease remain undiagnosed. In part, this reflects the incomplete status of the catalog of characterized Mendelian-disease-associated genes; this catalog currently includes about 4,000 entries and represents less than one-quarter of the annotated genes (∼21,000) in the human genome.
We applied WES to identify de novo genetic changes in a parent-offspring trio in which the proband exhibited developmental delay, hypotonia, mild dysmorphic features, sleep apnea, and other symptoms (Figure 1; Table 1; Table S1, available online). A truncating de novo mutant allele was found in AT-hook, DNA-binding motif, containing 1 (AHDC1 [RefSeq accession number NM_001029882.2]). We subsequently identified an additional three independent simplex cases with similar phenotypes and de novo truncating events in the same gene. This pattern of de novo variation in AHDC1 is highly unlikely to have occurred by chance and most likely represents the underlying cause of the symptoms in these individuals.
Figure 1.

Pedigrees and Mutations of Four Affected Families
Table 1.
Major Clinical Features of Four Probands
|
Subject |
||||
|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |
| Gender | female | female | male | male |
| Age | 18 months | 4 years | 8 years | 11 years |
| Ethnicity | European descent | South Asian | European descent | European descent |
| Intellectual disability | NA | moderate | mild | moderate to severe |
| Speech delay | no words at 18 months of age | two words at 4 years of age | first words after 1 year of age, persistent speech therapy | no words, noncommunicating autism |
| Motor delay | no sitting at 18 months of age | sitting at 19 months of age, walking at 24 months of age | sitting at 9 months of age, walking at 18 months of age | sitting at 15 months of age, no independent ambulation |
| Hypotonia and failure to thrive | yes | yes | yes | yes |
| Dysmorphic facial features | low-set ears, esotropia, upslanting palpebral fissures, micrognathia, flat nasal bridge | protuberant ears, upslanting palpebral fissures, flat nasal bridge | protuberant low-set ears, small earlobes, hypertelorism, downslanting palpebral fissures, mild ptosis, micrognathia | upturned earlobes, hypertelorism, esotropia, flat nasal bridge |
| Anatomic upper-airway obstruction | laryngomalacia, obstructive sleep apnea | obstructive sleep apnea | laryngomalacia, obstructive sleep apnea | suspected tracheomalacia in infancy, history of snoring |
| Family history | negative, one healthy sibling | negative | negative, one healthy sibling | negative, two healthy siblings |
| Previous testing | MD, SMA, PWS, CMA, metabolic work-up | FX, CMA (18 Mb AOH on chromosome 5), metabolic work-up | CMA, FX, metabolic work-up | SMA, PWS, CMA, metabolic work-up |
See Table S1 for additional details of clinical presentations. Abbreviations are as follows: AOH, absence of heterozygosity; CMA, chromosome microarray; FX, fragile X chromosome; MD, myotonic dystrophy; NA, not available; PWS, Prader-Willi syndrome; and SMA, spinal muscular atrophy.
Subject 1 was an 18-month-old female (born to unrelated parents) who presented with hypotonia, delayed motor milestones, dysmorphic features, hepatomegaly, and laryngomalacia (Figure 2). Both the healthy parents and the proband were analyzed by WES.1 Informed consent was obtained, and all procedures were followed in accordance with the ethical standards prescribed and approved by the Baylor College of Medicine Institutional Review Board. The DNA from each of the three samples was sequenced at an average depth of coverage of greater than 120-fold, and greater than 95% of the targeted bases were covered at 20-fold or higher. The results identified de novo events, including single-nucleotide variants (SNVs) or small indel mutations in the proband, in five genes: c.415G>A (p.Glu139Lys) in CALY (MIM 604647; RefSeq NM_015722.3), c.1429G>A (p.Gly477Arg) in PTPRB (MIM 176882; RefSeq NM_001109754.2), c.1076C>A (p.Ala359Glu) in TBCK (RefSeq NM_033115.4), c.1093dup (p.Met365Asnfs∗4) in CCDC66 (RefSeq NM_001141947.1), and c.2373_2374delTG (p.Cys791Trpfs∗57) in AHDC1; all sequence coordinates are based on human reference genome hg19 (UCSC Genome Browser). A comparison of the minor allele frequencies between these variants and similar mutations in the NHLBI Exome Sequencing Project Exome Variant Server (EVS) and a local variant database (see below) eliminated three missense mutations and one putative frameshift mutation as likely disease-causing candidates because of their occurrence in individuals without suspected developmental disorders. The remaining variant, the de novo deletion mutation in AHDC1 (c.2373_2374delTG), results in a frameshift of the AHDC1 open reading frame, beginning at codon 791, and by conceptual translation is predicted to cause a premature termination codon (p.Cys791Trpfs∗57). Predicted truncating mutations in AHDC1 are absent from the 1000 Genomes database (healthy individuals), the EVS (about 6,500 individuals), and a database of exome data from the Atherosclerosis Risk in Communities study (approximately 8,000 community-based individuals). The absence of truncating mutations in AHDC1 in these databases suggests that such gene perturbations are not consistent with general good health, and therefore the observed AHDC1 mutation was considered likely to be pathogenic.
Figure 2.
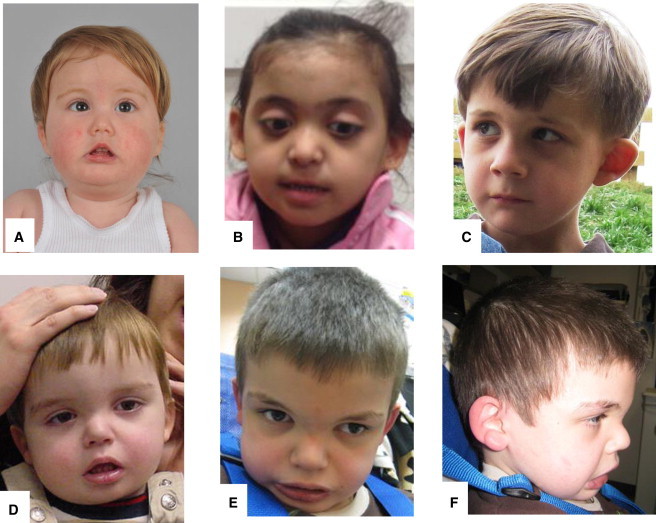
Facial Features of Probands
(A) Subject 1 (17 months old) with a round face, full cheeks, horizontal eyebrows, a depressed nasal bridge, anteverted nares, hypoplastic alae nasi, tented upper-lip vermillion, and microstomia.
(B) Subject 2 (4 years old) with thin eyebrows, a depressed nasal bridge, a bulbous nasal tip, and protuberant ears.
(C) Subject 3 (8 years old) with horizontal eyebrows, low-set ears, simple earlobes, and micrognathia.
(D) Subject 4 (21 months old) with a round face, full cheeks, horizontal eyebrows, a depressed nasal bridge, anteverted nares, tented upper-lip vermillion, and microstomia.
(E and F) Front (E) and side (F) views of subject 4 (9 years old) with a round face, full cheeks, horizontal eyebrows, an acute nasal angle, and fleshy pinna.
We next screened 2,000 entries of clinical WES data at the Whole Genome Laboratory at Baylor College of Medicine to identify possible additional AHDC1 mutations. Those data consist primarily of individuals who had a sample submitted by their physician for clinical WES, as previously described.1 The clinical WES test focuses on exome sequencing of the proband, and complete parental WES data are not routinely generated. Among 2,000 previously tested individuals, of whom 1,700 had developmental delay and/or intellectual disabilities, three were found to harbor frameshift alleles in AHDC1 (c.2898delC [p.Tyr967Thrfs∗175] at chr1: 27,875,729, c.2373_2374delTG [p.Cys791Trpfs∗57] at chr1: 27,876,253–27,876,254, and c.2547delC [p.Ser850Profs∗82] at chr1: 27,876,080; Figures 1 and 3). The mutations in the three individuals were further demonstrated by PCR and Sanger DNA sequencing to be absent in maternal and paternal DNA and were therefore interpreted as de novo events. Interestingly, subjects 1 and 3 had the same de novo mutation (c.2373_2374delTG [p.Cys791Trpfs∗57]). Except for the AHDC1 mutations, we did not find other molecular events that could potentially explain the conditions in these probands. Therefore, the de novo mutations observed here in AHDC1 are the most likely causes of the disease.
Figure 3.
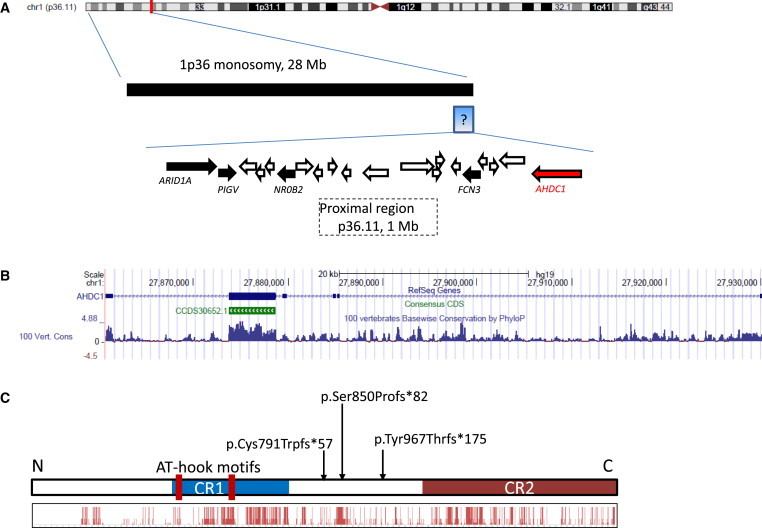
AHDC1 Genomic Organization
(A) Chromosomal location of AHDC1 and the genomic region surrounding it.
(B and C) Organization of AHDC1 (B) and the AT-hook DNA-binding region, conserved regions, and location of the three different truncating alterations in the disorder (C). The histogram shows evolutionary conservation (see Figure S3 for details).
Clinical review of the four probands with AHCD1 truncating mutations revealed that all had a history of congenital hypotonia and failure to thrive (Table 1; Table S1). The developmental histories were all remarkable for delayed speech, especially expressive language. All had mildly dysmorphic facial features that could be seen at a young age, and those of subject 4 persisted at an older age. Three probands also had a history of obstructive sleep apnea, potentially because of upper-airway structural abnormalities. All probands had prior brain MRI demonstrating hypoplasia of the corpus callosum. Simplification of the gyral pattern and delayed myelination were also observed. A retrocerebellar cyst was present in two of the four subjects (Figure S1).
The independent occurrence of four de novo mutational events at this locus in individuals with similar phenotypes is highly unlikely5 (discussed in Bainbridge et al.6) and can be asserted as extremely strong evidence that these mutations in ACHD1 cause this simplex disorder. To the best of our knowledge, the overall clinical presentations of these probands do not precisely match any previously known disease and, together with the statistical and molecular data, suggest a genetic syndrome defined by the mutations in AHDC1.
In subject 4, we also identified a de novo missense change, c.2006A>C (p.Asp669Ala), in ANKRD11 (MIM 611192; RefSeq NM_013275.5). Haploinsufficiency of ANKRD11 has been associated with KBG syndrome (MIM 148050), characterized by macrodontia, variable facial dysmorphic features, mild skeletal anomalies, seizures in some individuals, and mild to moderate intellectual disability. However, the proband reported here did not have macrodontia, skeletal defects, or other features of KGB phenotypes and therefore did not meet the KBG diagnostic criteria proposed by Skjei et al.7 Additionally, the well-characterized pathogenic mutations in ANKRD11 are truncating.8 Thus, the significance of ANKRD11 missense variant c.2006A>C (p.Asp669Ala) is unclear.
AHDC1 is located on the short arm of chromosome 1 within the cytogenetic band 1p36.11, but it is more proximal than the regions identified from partial or complete monosomy of 1p36,9 other small interstitial deletions,10,11 and the nearby ARID1A, mutations in which cause autosomal-dominant Coffin-Siris syndrome12 (MIM 135900). In the RefSeq and CCDS databases, the structural organization of AHDC1 includes five untranslated exons upstream of a single 4,929 bp coding exon followed by a single downstream exon. This intronless coding structure is a common feature for newly evolved genes created by RNA-based retroposition.13 Indeed, orthologs of AHDC1 can only be found in vertebrate animals. Gaining intron structures during evolution is correlated with higher expression levels.14 The expression level and patterns of AHDC1 are more similar to those of the multi-intron ARID1A than to those of the intronless FOXG1 (MIM 164874), two other genes associated with severe developmental disorders in humans (Figure S2). Therefore, it can be postulated that the introns of the UTRs of AHDC1 affect and/or enhance the expression levels in various human tissues. On the nucleotide level, the single coding exon of AHDC1 is well conserved among vertebrates (Figure 3). The 3′ untranslated exon also shows conservation levels similar to those of the coding region, suggesting a potential functional significance of this exon.
Human AHDC1 encodes a protein of 1,603 amino acids. By aligning human AHDC1 against the protein sequences of AHDC1 orthologs in mouse, zebrafish, and western clawed frog, we found that the conserved amino acids are clustered into two regions (Figure 3C; Figure S3), suggesting two functional units. AHDC1 has two AT-hook DNA-binding motifs located at codons 396–408 and 544–556, contained in conserved region 1. AT-hook domains are DNA-binding motifs that act to fasten proteins to AT-rich sequences in DNA.15 Although conserved, region 2 contains no known functional domains. In vitro protein-interaction assays have shown that AHDC1 interacts with a number of other nuclear proteins.16–21 Therefore, conserved regions 1 and 2 of AHDC1 might interact with the DNA elements or protein partners. Interestingly, all of the de novo mutations found in these four probands might truncate conserved region 2 but preserve region 1. Given that each mutation identified here occurs in a single coding exon, the modified mRNA might escape nonsense-mediated decay,22 suggesting that the autosomal-dominant mode of inheritance of these mutations is possibly due to the formation of dominant-negative proteins rather than haploinsufficiency.
Future research to better delineate the functional domains of AHDC1 is now enhanced by the phenotypic association with the truncating mutations reported here. Also, the phenotypes of the four probands are clearly similar in retrospect. However, speech delay and obstructive sleep apnea are sufficiently common conditions that it is unlikely that this syndrome would have been identified if the de novo mutations had not been uncovered first.
Acknowledgments
We thank the families and clinical staff at all locations for participation in this study. This work was supported in part by NIH grants to R.A.G. (NHGRI 5 U54 HG003273), J.R.L. (NHGRI RO1NS058529 and U54HG006542), and S.E.P. (NHGRI 5 U01 HG006485). M.W. received funding from the National Institute of Neurological Disorders and Stroke (NS076547) and the Simons Foundation. T.Y.T. is partially supported by the National Health and Medical Research Council of Australia (fellowship 607431). Baylor College of Medicine performs genetic testing as a service for a fee. M.N.B. is a founder of Codified Genomics, and J.R.L. is a consultant for Athena Diagnostics, 23andMe, and Ion Torrent Systems Inc. and holds multiple US and European patents for DNA diagnostics.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Atherosclerosis Risk in Communities Study, http://www2.cscc.unc.edu/aric/
Baylor College of Medicine Whole Genome Laboratory, https://www.bcm.edu/research/medical-genetics-labs/wholegenomelab
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://omim.org/
Accession Numbers
The ClinVar accession numbers for the DNA variant data reported in this paper are SCV000148377, SCV000148378, and SCV000148379.
References
- 1.Yang Y., Muzny D.M., Reid J.G., Bainbridge M.N., Willis A., Ward P.A., Braxton A., Beuten J., Xia F., Niu Z. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med. 2013;369:1502–1511. doi: 10.1056/NEJMoa1306555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Veltman J.A., Brunner H.G. De novo mutations in human genetic disease. Nat. Rev. Genet. 2012;13:565–575. doi: 10.1038/nrg3241. [DOI] [PubMed] [Google Scholar]
- 3.Bainbridge M.N., Wang M., Burgess D.L., Kovar C., Rodesch M.J., D’Ascenzo M., Kitzman J., Wu Y.Q., Newsham I., Richmond T.A. Whole exome capture in solution with 3 Gbp of data. Genome Biol. 2010;11:R62. doi: 10.1186/gb-2010-11-6-r62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gonzaga-Jauregui C., Lupski J.R., Gibbs R.A. Human genome sequencing in health and disease. Annu. Rev. Med. 2012;63:35–61. doi: 10.1146/annurev-med-051010-162644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neale B.M., Kou Y., Liu L., Ma’ayan A., Samocha K.E., Sabo A., Lin C.F., Stevens C., Wang L.S., Makarov V. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bainbridge M.N., Hu H., Muzny D.M., Musante L., Lupski J.R., Graham B.H., Chen W., Gripp K.W., Jenny K., Wienker T.F. De novo truncating mutations in ASXL3 are associated with a novel clinical phenotype with similarities to Bohring-Opitz syndrome. Genome Med. 2013;5:11. doi: 10.1186/gm415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Skjei K.L., Martin M.M., Slavotinek A.M. KBG syndrome: report of twins, neurological characteristics, and delineation of diagnostic criteria. Am. J. Med. Genet. A. 2007;143:292–300. doi: 10.1002/ajmg.a.31597. [DOI] [PubMed] [Google Scholar]
- 8.Sirmaci A., Spiliopoulos M., Brancati F., Powell E., Duman D., Abrams A., Bademci G., Agolini E., Guo S., Konuk B. Mutations in ANKRD11 cause KBG syndrome, characterized by intellectual disability, skeletal malformations, and macrodontia. Am. J. Hum. Genet. 2011;89:289–294. doi: 10.1016/j.ajhg.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gajecka M., Mackay K.L., Shaffer L.G. Monosomy 1p36 deletion syndrome. Am. J. Med. Genet. C. Semin. Med. Genet. 2007;145C:346–356. doi: 10.1002/ajmg.c.30154. [DOI] [PubMed] [Google Scholar]
- 10.Rosenfeld J.A., Crolla J.A., Tomkins S., Bader P., Morrow B., Gorski J., Troxell R., Forster-Gibson C., Cilliers D., Hislop R.G. Refinement of causative genes in monosomy 1p36 through clinical and molecular cytogenetic characterization of small interstitial deletions. Am. J. Med. Genet. A. 2010;152A:1951–1959. doi: 10.1002/ajmg.a.33516. [DOI] [PubMed] [Google Scholar]
- 11.Kang S.H., Scheffer A., Ou Z., Li J., Scaglia F., Belmont J., Lalani S.R., Roeder E., Enciso V., Braddock S. Identification of proximal 1p36 deletions using array-CGH: a possible new syndrome. Clin. Genet. 2007;72:329–338. doi: 10.1111/j.1399-0004.2007.00876.x. [DOI] [PubMed] [Google Scholar]
- 12.Tsurusaki Y., Okamoto N., Ohashi H., Kosho T., Imai Y., Hibi-Ko Y., Kaname T., Naritomi K., Kawame H., Wakui K. Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat. Genet. 2012;44:376–378. doi: 10.1038/ng.2219. [DOI] [PubMed] [Google Scholar]
- 13.Long M., VanKuren N.W., Chen S., Vibranovski M.D. New gene evolution: little did we know. Annu. Rev. Genet. 2013;47:307–333. doi: 10.1146/annurev-genet-111212-133301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shabalina S.A., Ogurtsov A.Y., Spiridonov A.N., Novichkov P.S., Spiridonov N.A., Koonin E.V. Distinct patterns of expression and evolution of intronless and intron-containing mammalian genes. Mol. Biol. Evol. 2010;27:1745–1749. doi: 10.1093/molbev/msq086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karlson J.R., Mørk E., Holtlund J., Laland S.G., Lund T. The amino acid sequence of the chromosomal protein HMG-Y, its relation to HMG-I and possible domains for the preferential binding of the proteins to stretches of A-T base pairs. Biochem. Biophys. Res. Commun. 1989;158:646–651. doi: 10.1016/0006-291x(89)92770-8. [DOI] [PubMed] [Google Scholar]
- 16.Eberl H.C., Spruijt C.G., Kelstrup C.D., Vermeulen M., Mann M. A map of general and specialized chromatin readers in mouse tissues generated by label-free interaction proteomics. Mol. Cell. 2013;49:368–378. doi: 10.1016/j.molcel.2012.10.026. [DOI] [PubMed] [Google Scholar]
- 17.Lim J., Hao T., Shaw C., Patel A.J., Szabó G., Rual J.F., Fisk C.J., Li N., Smolyar A., Hill D.E. A protein-protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration. Cell. 2006;125:801–814. doi: 10.1016/j.cell.2006.03.032. [DOI] [PubMed] [Google Scholar]
- 18.Nozawa R.S., Nagao K., Masuda H.T., Iwasaki O., Hirota T., Nozaki N., Kimura H., Obuse C. Human POGZ modulates dissociation of HP1alpha from mitotic chromosome arms through Aurora B activation. Nat. Cell Biol. 2010;12:719–727. doi: 10.1038/ncb2075. [DOI] [PubMed] [Google Scholar]
- 19.Shalaby M.A., Hampson L., Oliver A., Hampson I. Identification of PlexinD1 and AHDC1 as a putative interactors for Tip-1 protein. Genes & Genomics. 2011;33:399–405. [Google Scholar]
- 20.Wong K.A., Wilson J., Russo A., Wang L., Okur M.N., Wang X., Martin N.P., Scappini E., Carnegie G.K., O’Bryan J.P. Intersectin (ITSN) family of scaffolds function as molecular hubs in protein interaction networks. PLoS ONE. 2012;7:e36023. doi: 10.1371/journal.pone.0036023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vandamme J., Völkel P., Rosnoblet C., Le Faou P., Angrand P.O. Interaction proteomics analysis of polycomb proteins defines distinct PRC1 complexes in mammalian cells. Mol. Cell. Proteomics. 2011;10:002642. doi: 10.1074/mcp.M110.002642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schoenberg D.R., Maquat L.E. Regulation of cytoplasmic mRNA decay. Nat. Rev. Genet. 2012;13:246–259. doi: 10.1038/nrg3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.