

Abstract
Purpose
Multiple-gene sequencing is entering practice, but its clinical value is unknown. We evaluated the performance of a customized germline-DNA sequencing panel for cancer-risk assessment in a representative clinical sample.
Methods
Patients referred for clinical BRCA1/2 testing from 2002 to 2012 were invited to donate a research blood sample. Samples were frozen at −80° C, and DNA was extracted from them after 1 to 10 years. The entire coding region, exon-intron boundaries, and all known pathogenic variants in other regions were sequenced for 42 genes that had cancer risk associations. Potentially actionable results were disclosed to participants.
Results
In total, 198 women participated in the study: 174 had breast cancer and 57 carried germline BRCA1/2 mutations. BRCA1/2 analysis was fully concordant with prior testing. Sixteen pathogenic variants were identified in ATM, BLM, CDH1, CDKN2A, MUTYH, MLH1, NBN, PRSS1, and SLX4 among 141 women without BRCA1/2 mutations. Fourteen participants carried 15 pathogenic variants, warranting a possible change in care; they were invited for targeted screening recommendations, enabling early detection and removal of a tubular adenoma by colonoscopy. Participants carried an average of 2.1 variants of uncertain significance among 42 genes.
Conclusion
Among women testing negative for BRCA1/2 mutations, multiple-gene sequencing identified 16 potentially pathogenic mutations in other genes (11.4%; 95% CI, 7.0% to 17.7%), of which 15 (10.6%; 95% CI, 6.5% to 16.9%) prompted consideration of a change in care, enabling early detection of a precancerous colon polyp. Additional studies are required to quantify the penetrance of identified mutations and determine clinical utility. However, these results suggest that multiple-gene sequencing may benefit appropriately selected patients.
INTRODUCTION
Clinical genetic testing for cancer-risk assessment has become widespread over the last two decades, with evidence-based testing guidelines for hereditary breast and ovarian cancer (BRCA1 and BRCA2; BRCA1/2), Lynch syndrome (MLH1, MSH2, MSH6, PMS2, and EPCAM), familial adenomatous polyposis (APC), hereditary diffuse gastric cancer (CDH1), Li-Fraumeni syndrome (TP53), Cowden's syndrome (PTEN), and a few other conditions.1–4 Cancer genetic counseling and risk-reducing interventions have accordingly been developed for high penetrance, autosomal dominant conditions. Most of these interventions, especially prophylactic surgery, are excessive for carriers of mutations that have uncertain pathogenicity.5–7 Recently, next-generation technology has enabled massively parallel sequencing at low cost, and panels of multiple cancer-associated genes are newly available for clinical use.8,9
Despite these advances in technology, a critical knowledge deficit remains about the clinical value of multiple-gene panels for cancer susceptibility. Major questions include how many and which genes to sequence, whether results are sufficiently understood to guide intervention, and how best to counsel patients about variants of low or moderate penetrance.8,10–12 We designed a customized germline sequencing panel of 42 cancer-associated genes and evaluated its information yield among women referred for clinical evaluation of hereditary breast and ovarian cancer. Specifically, we aimed to assess the concordance of results with prior clinical sequencing, the prevalence of potentially actionable results, and the downstream effects on cancer screening and risk-reduction.
METHODS
Participant Accrual
Patients were eligible to participate if they were female, at least 18 years old, and had undergone clinical BRCA1/2 testing at the Stanford University Clinical Cancer Genetics program from 2002 to 2012. The criteria for clinical genetic counseling and BRCA1/2 testing were those accepted by insurance carriers, based on guidelines of the National Comprehensive Cancer Network at the time of evaluation. Some patients had a personal history of breast and/or ovarian cancer; some had a family history of breast, ovarian, pancreatic, and/or prostate cancers; and some had both personal and family history of such cancers.2,3 Since 2002, all patients undergoing clinical BRCA1/2 testing (for an identified familial mutation; the Ashkenazi Jewish three-founder mutations panel; full sequencing with analysis of five common BRCA1 rearrangements; and/or full sequencing with comprehensive rearrangement analysis, depending on the indication and year) were offered participation in a study approved by the Stanford University institutional review board (IRB). Patients were informed that participation would consist of donating two 5-mL tubes of blood for clinical BRCA1/2 testing and that they might be contacted about participation in future research. Blood specimens were frozen at −80° C and were linked to demographic, clinical, and genetic data stored in a secure research database.13–15
Gene Selection
Genes were selected through a review of published literature. Most genes had a reported breast cancer association but some were associated with other cancer syndromes or DNA repair pathways, rendering a breast cancer–risk association plausible (Table 1).
Table 1.
Sequenced Genes and Criteria for Their Inclusion
| Fully Sequenced Genes (n = 42) | Breast Cancer Relative Risk (or other inclusion criterion) | References |
|---|---|---|
| APC | Unknown (causes Familial Adenomatous Polyposis) | Redston et al16 |
| ATM | 1.5-3.8 | Renwick et al17 |
| BLM | 1.2-3.3 | Broberg et al18 |
| BMPR1A | 1.3 | Saetrom et al19 |
| BRCA1 | 4.0-7.0 | Chen et al20 |
| BRCA2 | 4.0-7.0 | Chen et al21 |
| BRIP1 | 1.2-3.2 | Seal et al22 |
| CDH1 | 5.9-7.3 | Kaurah et al,23 Pharoah et al24 |
| CDK4 | Unknown (functions as DNA repair gene) | Dean et al25 |
| CDKN2A | 1.1-1.7 | Debniak et al26 |
| EPCAM | 1.2-1.6 | Jiang et al27 |
| FANCA | 0.9-1.0 | Barroso et al28 |
| FANCB | 0.8-1.2 | Barroso et al28 |
| FANCC | 1.0-1.4 | Barroso et al28 |
| FANCD2 | 1.1-1.7 | Barroso et al28 |
| FANCE | 0.9-1.2 | Barroso et al28 |
| FANCF | 0.9-1.4 | Barroso et al28 |
| FANCG | 0.8-1.0 | Barroso et al28 |
| FANCI | 0.8-1.1 | Barroso et al28 |
| FANCL | 1.0-1.1 | Barroso et al28 |
| LIG4 | 0.4-1.0 | Kuschel et al29 |
| MEN1 | Unknown (causes multiple endocrine neoplasia 1) | Lemmens et al30 |
| MET | Unknown (associated with papillary renal cell carcinoma risk) | Neklason et al31 |
| MLH1 | 0.2-2.0 | Win et al32 |
| MSH2 | 1.2-3.7 | Win et al32 |
| MSH6 | 0-13 | Win et al32 |
| MUTYH | 1.0-3.4 | Rennert et al33 |
| NBN | 1.4-6.6 | Bogdanova et al,34 Seemanová et al,35 Zhang et al36 |
| PALB2 | 1.4-3.9 | Rahman et al37 |
| PALLD | Unknown (associated with pancreatic cancer risk) | Pogue-Geile et al38 |
| PMS2 | Unknown (causes Lynch syndrome) | Win32 |
| PRSS1 | 0.7-1.6 | Wagner et al39 |
| PTCH1 | Unknown (associated with basal cell nevus syndrome and glioblastoma) | Lee et al40 |
| PTEN | 2.0-5.0 | Pilarski et al,41 Tan et al42 |
| RAD51C | 1.5-7.8 | Meindl et al43 |
| RET | Unknown (causes multiple endocrine neoplasia 2) | Machens et al44 |
| SLX4 | 1.0-2.0 | Landwehr et al,45 Shah et al46 |
| SMAD4 | Unknown (mutated in breast tumors) | Tram et al47 |
| SPINK1 | Unknown (mutated in breast tumors) | Soon et al48 |
| STK11 | 2.0-4.0 | Hearle et al,49 Lim et al50 |
| TP53 | 4.3-9.3 | Gonzalez et al51 |
| VHL | Unknown (mutated in breast tumors) | Kong et al52 |
Sequencing
Sequencing was performed at InVitae (San Francisco, CA), a clinical laboratory improvement amendments (CLIA) –approved laboratory. Two micrograms of genomic DNA per patient were sheared on a Covaris E220 sonicator (Woburn, MA) to 250 base pair (bp) mean fragment size. Genomic DNA was quantified and assessed for quality using the Life Technologies Quant-iT PicoGreen double-strand DNA assay kit (Carlsbad, CA). The entire coding region, exon-intron boundaries (± 10 bp), and other regions containing known pathogenic variants were targeted and captured using Agilent SureSelect custom RNA probes (Santa Clara, CA) and Integrated DNA Technologies xGen Lockdown custom DNA probes (Coral, IL). Sequencing libraries were constructed using the Agilent SureSelectXT protocol and were quantified with the KAPA Biosystems Library Quantification Kit (Woburn, MA). These steps were performed in an automated fashion using the Agilent Bravo automated liquid-handling platform. Quantified libraries were sequenced on the Illumina MiSeq platform (San Diego, CA) using the 2 × 151 bp configuration to at least 400× average coverage. Bioinformatics and data quality control followed the Genome Analysis Toolkit best-practices (Broad Institute, Cambridge, MA), with additional algorithms to detect larger insertions, deletions, and duplications.53 PMS2 exons 12 to 15 were excluded from analysis because of high homology to the known pseudogene. Deletion/duplication data were not available for 54 samples sequenced before the development of calibration standards used by the algorithm.
Variant Classification
Sequence variants and large insertions and deletions were classified according to the American College of Medical Genetics (ACMG) guidelines for variant interpretation.54 Variants were classified as pathogenic or likely pathogenic (collectively termed, pathogenic) if they conferred a truncating, initiation codon or splice donor/acceptor effect, if functional data demonstrated an effect on protein function relevant to disease phenotype, or if pathogenicity was otherwise demonstrated in published literature. If no functional data were available, missense, silent, and intronic variants were classified as variants of uncertain significance (VUS), benign or likely benign based on allele frequency in the 1,000 Genomes Study,55 dbSNP,56 or the Exome Variant Server.57 Also, published literature on BRCA1/2 VUS was reviewed to classify those variants further as benign or likely benign following the ACMG guidelines. For pathogenic variants, we then reviewed published literature and practice guidelines to assign a clinical status of potentially actionable versus not actionable. We defined potentially actionable results as pathogenic variants that either cause recognized hereditary cancer syndromes, such as Lynch syndrome or hereditary diffuse gastric cancer,58 or have a published association with a two-fold or greater relative risk of breast cancer compared with that of an average woman, under which circumstance guidelines recommend annual screening with breast magnetic resonance imaging (MRI) and mammograms.59–61 Participants with potentially actionable results were contacted by telephone, were invited to a genetic counseling visit, and were offered a CLIA-approved test to confirm research findings. Cancer screening and prevention recommendations were consistent with clinical practice guidelines focused on the estimated magnitude of cancer risk, given the absence of gene-specific guidelines for many of the sequenced genes.2,59,60,62,63 Participants were not notified about results that do not currently affect care recommendations, including absence of sequence abnormalities, pathogenic variants considered not actionable, or variants of uncertain significance.2,58,59,60,63
Statistical Analysis
Participant characteristics and sequencing results were tabulated, with descriptive statistics including medians, means, and standard deviations for continuous data and proportions with 95% CI for categoric data. Proportions were compared using the χ2 statistic. All P values are two-tailed.
RESULTS
Participant Characteristics
From January 1, 2001 to June 30, 2013, 1,805 patients underwent clinical BRCA1/2 testing in the Stanford Clinical Cancer Genetics program. Six hundred fifty-four patients (36.2%) donated a research blood sample during clinical testing. From 654 research samples, 198 were randomly selected for study participation; 174 participants had breast cancer, and 57 carried a BRCA1/2 mutation. Participants were diverse in age and race/ethnicity and were generally representative of the clinical population from which they were recruited (Table 2).
Table 2.
Characteristics of the Study Participants Compared With the Larger Clinical Source Population
| Characteristic | Study Participants (n = 198) |
BRCA1/2 Test at Stanford, 2001-2013 (n = 1,805) |
||
|---|---|---|---|---|
| No. of Participants | % | No. of Participants | % | |
| Age, years | ||||
| Median | 48 | 46 | ||
| Race/ethnicity | ||||
| NH white, Ashkenazi Jew | 7 | 3.5 | 102 | 5.6 |
| NH white, not Ashkenazi Jew | 140 | 70.7 | 1,205 | 66.7 |
| NH black | 3 | 1.5 | 30 | 1.7 |
| NH Asian/Pacific Islander | 39 | 19.7 | 292 | 16.2 |
| Hispanic | 7 | 3.5 | 92 | 5.1 |
| Unknown/other race or ethnicity* | 2 | 1.0 | 84 | 4.6 |
| Personal history of breast cancer | ||||
| Unilateral | 138 | 69.7 | 1,139 | 63.1 |
| Bilateral† | 36 | 18.1 | 139 | 7.7 |
| Age at first breast cancer diagnosis, years | ||||
| Median | 44 | 44 | ||
| Personal history of ovarian cancer† | 1 | 0.5 | 136 | 7.5 |
| Age at ovarian cancer diagnosis, median | 72 | 52 | ||
| Personal history of other cancer‡ | 18 | 9.1 | 175 | 9.7 |
| Family history of breast cancer | 148 | 74.7 | 1,285 | 71.2 |
| Family history of ovarian cancer | 52 | 26.3 | 444 | 24.6 |
| BRCA1 mutation§ | 35 | 17.7 | 213 | 11.8 |
| BRCA2 mutation | 24 | 12.1 | 194 | 10.7 |
| Year of first clinic visit | ||||
| Median | 2008 | 2009 | ||
| Range | 2005-2011 | 2001-2013 | ||
Abbreviation: NH, non-Hispanic.
For comparison between study participants and source population, P = .01 (χ2 two-tailed test).
For comparison between study participants and source population, P < .001 (χ2 two-tailed test).
Other cancers were nonmelanoma skin cancer (n = 6), endometrial cancer (n = 3), melanoma (n = 2), colon cancer (n = 1), leukemia (n = 1), lymphoma (n = 1), pancreatic cancer (n = 1), salivary gland cancer (n = 1), thyroid cancer (n = 1), and unknown primary cancer (n = 1).
For comparison between study participants and source population, P = .02 (χ2 two-tailed test).
Pathogenic Variants in BRCA1 and BRCA2
Before study enrollment, 57 participants were known to carry 59 pathogenic variants in BRCA1/2 as determined by standard clinical testing (Myriad Genetics, Salt Lake City, UT). The remaining 141 participants had tested negative for BRCA1/2 mutations. Fifty-seven of these 59 mutations in BRCA1/2 were confirmed by the gene panel. Of the two others, one was detected but interpreted as a VUS because the information in the literature did not meet ACMG criteria for pathogenicity.54 The other was a large insertion in one of the 54 samples for which del/dup analysis was not performed in the panel test. Considering the assays performed, BRCA1/2 analysis was concordant with prior testing for 197 of 197 participants, and pathogenicity interpretation was concordant for 196 (99.5%) of 197 participants (95% CI, 96.9% to 100%).
Pathogenic Variants in Other Genes
Sixteen pathogenic variants were detected in women who tested negative for BRCA1/2 mutations, for a prevalence of 11.4% (95% CI, 7.0% to 17.7%). The affected genes were ATM (two women), BLM (one woman), CDH1 (one woman), CDKN2A (one woman), MLH1 (one woman), MUTYH (five women), NBN (two women), PRSS1 (one woman), and SLX4 (two women). Eleven variants were previously reported in the literature, and five were novel. Consistent with the larger study sample and source population, most of the women carrying pathogenic variants (80%) had a personal history of breast cancer; 67% were non-Hispanic white, 20% were non-Hispanic Asian, and 13% were Hispanic (Table 3).
Table 3.
Pathogenic Variants in Genes Other Than BRCA1 or BRCA2, With Participant Characteristics and Clinical Decisions (potentially actionable or not)
| Study ID | Race/Ethnicity | Cancer Site/Subtype | Age at Diagnosis (years) | BRCA1/2 Mutation | Affected Gene | Variant Name | Variant Effect | Protein Change | Family Cancer History* | Clinical Decision | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| LS221 | NH white | Breast, bilateral: ER/PR+, HER2− | 44, 55 | None | ATM | NM_000051.3:c.1402_1403delAA | Frameshift | Lys468GlufsX18 | Breast (45); prostate (n = 3; 60s) | PA | Buzin et al64 |
| LS294 | NH white | Breast: ER−/PR−/HER2−; endometrial | 35, 46 | None | MLH1 | NM_000249.3:c.2190delT | Frameshift | Pro731LeufsX52 | Breast (70); neuroblastoma (10); ovary (55); pancreas (55) | PA | Rouleau et al65 |
| LS305 | Filipino | Breast: ER/PR+, HER2− | 43 | None | PRSS1 | NM_002769.4:c.346C>T | Missense | Arg116Cys | Breast (n = 3; 30s-70s); lymphoma (74); prostate (65) | PA | Szmola and Sahin-Tóth66 |
| LS306 | NH white | Breast, bilateral: ER/PR+, HER2− | 57, 63 | None | BLM | NM_000057.2:c.3558 + 1G>T | Splice donor | Not applicable | Breast, ovary (40s); colon (n = 3; 40s-70s); prostate (70) | PA | Novel |
| LS324 | Filipino | Breast, bilateral: ER/PR+, HER2− | 47, 55 | BRCA1 | MUTYH | NM_001048171.1:c.892-2A>G | Splice acceptor | Not applicable | Breast (57); ovarian (47); prostate (70) | PA | Novel |
| LS347 | NH white | No personal cancer history | None | None | SLX4 | NM_032444.2:c.5233_5234delGC | Frameshift | Ala1745SerfsX32 | Bladder (75); breast (n = 3; 40s); colon (85) | PA | Novel |
| LS358 | NH white | No personal cancer history | None | None | MUTYH | NM_001048171.1:c.1145G>A | Missense | Gly382Asp | Breast (45); colon (n = 2; 45, 65); ovarian (60) | PA | Ali et al,67 Eliason et al68 |
| LS373 | Hispanic | Breast: ER/PR+, HER2− | 36 | None | CDKN2A | NM_000077.4:c.146T>C | Missense | Ile49Thr | Lung (55); unknown primary | NA | Lal et al69 |
| LS380 | Hispanic | Breast, bilateral: ER/PR+, ER/PR− | 27, 33 | None | MUTYH | NM_001048171.1:c.694G>T | Missense | Val232Phe | Cervical (70); lung, larynx (65) | PA | Bai et al,70 Sieber et al71 |
| LS384 | Japanese | Breast: ER/PR+, HER2− | 48 | None | MUTYH | NM_001048171.1:c.892-2A>G | Splice acceptor | Not applicable | Breast (40s); gastric (90); ovarian (40s) | PA | Novel |
| LS400 | NH white | Breast: ER/PR+, HER2+ | 50 | None | NBN | NM_002485.4:c.643C>T | Missense | Arg215Trp | Breast (70s); melanoma (49) | PA | Seemanová et al72 |
| LS430 | NH white | Breast: ER/PR+, HER2− | 61 | None | SLX4 | NM_032444.2:c.5229insG | Frameshift | Gln1744AlafsX34 | Breast (n = 3; 40s); colon (n = 2, 39, 73); melanoma (27) | PA | Novel |
| LS443 | NH white | No personal cancer history | None | None | CDH1; NBN | NM_004360.3:c.532-18C>T; NM_002485.4:c.643C>T | Intronic; Missense | Not applicable; Arg215Trp | Breast (n = 4; 50s-70s); colon (70s); ovary (60s) | PA; PA | Seemanová et al,72 Suriano et al73 |
| LS456 | NH white | Breast: ER/PR+, HER2− | 49 | None | ATM | NM_000051.3:c.7271T>G | Missense | Val2424Gly | Breast (n = 2; 40s-60s); colon (n = 2; 30s-40s); melanoma (n = 2; 40, 58) | PA | McConville et al,74 Mitui et al75 |
| LS462 | NH white | Breast: ER/PR+, HER2− | 42 | None | MUTYH | NM_001048171.1:c.494A>G | Missense | Tyr165Cys | Breast (56); lung (54, 76); pancreas (44) | PA | Ali et al,67 Eliason et al68 |
Abbreviations: +, positive; −, negative; DCIS, ductal carcinoma in situ; ER, estrogen receptor; HER2, human epidermal growth factor receptor 2; NA, not actionable; NH, non-Hispanic; PA, potentially actionable; PR, progesterone receptor.
Family history of cancer in first- and second-degree relatives; parenthetical information includes patient age/decade of diagnosis (for multiple affected relatives, the number affected is reported, eg, n = 3; followed by relatives' ages).
Variants of Uncertain Significance
A total of 428 VUS were identified in 39 genes among 175 participants. Per participant, the average number of VUS across all genes was 2.1 (standard deviation, 1.5; Fig 1A). Per gene, the median number of VUS detected across all 198 participants was eight, ranging from zero (PTEN, SMAD4, SPINK1) to 36 (APC; Fig 1B). VUS protein effects were as follows: 49 intronic (11.4%), 269 missense (62.9%), 86 silent (20.1%), eight noncoding (1.9%), two in-frame codon loss (0.5%), one in-frame codon gain (0.2%), and 13 unknown (3%). Most of the VUS were novel (n = 380; 88.8%); the PolyPhen program predicted that 151 were benign (35.3%), 65 were probably damaging (15.2%), and 50 were possibly damaging (11.7%).76,77
Fig 1.
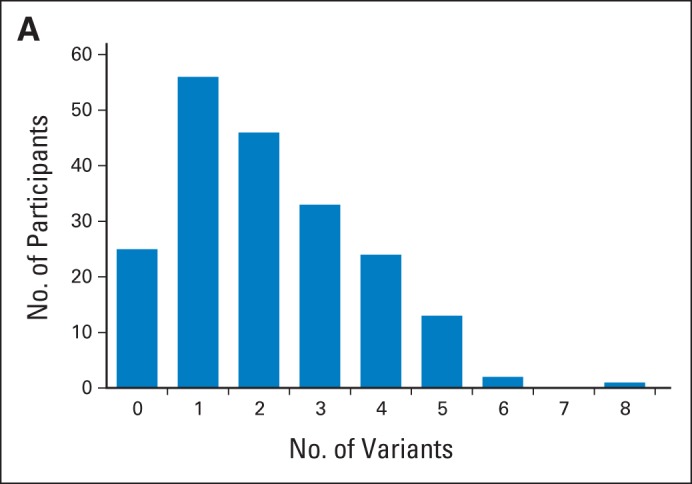
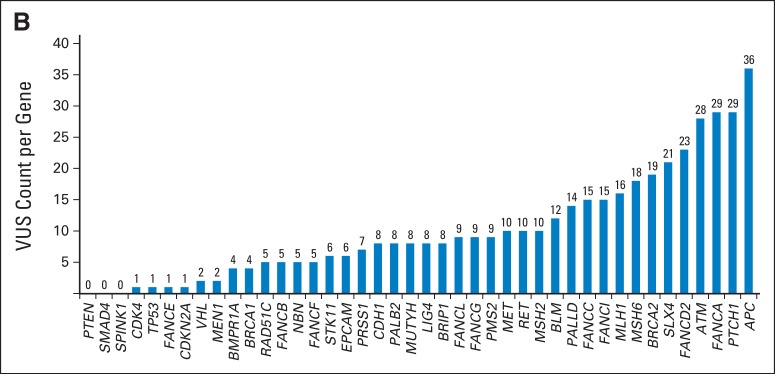
(A) Frequency of variants of uncertain significance, per participant, across 42 sequenced genes.
(B) Variants of uncertain significance (VUS) count, per gene, across 198 participants.
Clinical Interpretation of Pathogenic Variants
Of 16 pathogenic variants in genes other than BRCA1/2, we determined that 15 met our criteria for being potentially actionable (Table 3).69,78,79 One missense variant in CDKN2A (NM_000077.4:c.146T>C) was classified as likely pathogenic based on functional evidence,80–83 but with conflicting reports69; it was therefore considered not actionable.
Participant Notification and Clinical Follow-Up
Given the clinical significance of the pathogenic variants deemed potentially actionable, permission was obtained from the Stanford University IRB to contact participants again and offer them the results of their research testing. A genetic counselor (K.E.K.) attempted to call by telephone the 14 women who carried the 15 potentially actionable variants. Of these 14, three were lost to follow-up. Of the 11 women we were able to reach by telephone, 10 accepted a counseling appointment, and one was deceased but her children accepted an appointment. During counseling appointments, participants reviewed and signed a new IRB-approved informed consent document, confirming their willingness to receive study results. The appointments were led by a genetic counselor (K.E.K.) and an oncologist (A.W.K. or J.M.F.); they included an explanation of the results' estimated contribution to cancer risk, CLIA-approved confirmatory testing, and a discussion of risk-adapted screening or prevention options. Six participants (who carried the ATM, BLM, CDH1, NBN, and SLX4 variants) were advised to consider annual breast MRIs because of an estimated doubling of breast cancer risk,17,18,23,24,34–36,45,46,59–61,84,85 and six participants (who carried the CDH1, MLH1, and MUTYH variants) were advised to consider frequent colonoscopy and/or endoscopic gastroduodenoscopy (once every 1 to 2 years) due to estimated increases in gastrointestinal cancer risk.1–4,86 One participant (LS294; Table 3) who had triple-negative breast cancer at age 35 years was found to carry a frameshift MLH1 mutation consistent with Lynch syndrome. Her breast tumor was analyzed by immunohistochemistry and demonstrated absent MLH1 and PMS2 expression. She had previously undergone hysterectomy for endometrial carcinoma at age 46 years. After sequencing revealed she had Lynch syndrome, she underwent risk-reducing salpingo-oophorectomy and early colonoscopy, the latter identifying a tubular adenoma that was excised (Fig 2).
Fig 2.

Pathogenic variant interpretation, participant notification, and clinical follow-up.
DISCUSSION
We evaluated the clinical performance of a multiple-gene sequencing panel among 198 women meeting evidence-based practice guidelines for BRCA1/2 testing. We detected 16 pathogenic variants (11.4%; 95% CI, 7.0% to 17.7%) in other genes among women who tested negative for BRCA1/2 mutations, with 15 variants (10.6%; 95% CI, 6.5% to 16.9%) warranting discussion of more intensive screening or prevention. This is a significant yield of potentially actionable results, comparable to the 5% to 10% probability threshold endorsed by guidelines and payers for BRCA1/2 and Lynch syndrome testing.1–3 Up to 10 years after research sample donation, most participants (78.6%) could be reached for results notification, and all accepted confirmatory CLIA-approved testing and counseling. Although further research is required to guide practice, these findings provide an early signal for the clinical relevance of multiple-gene sequencing in cancer-risk assessment.
Multiple-gene sequencing panels have emerged over the last few years, with clinical availability as of 2012.8,87 However, the United States Supreme Court decision against gene patenting in June 2013 permitted the inclusion of BRCA1/2 in panels offered by several companies,87 and a concurrent drop in pricing has provided incentive for the uptake of these new products because a six-gene panel now costs no more than a two-gene test. Owing to absent data on clinical performance, expert opinion statements urge caution in using multiple-gene panels outside a clinical trial.2,8 Our current study was nested within a Clinical Cancer Genetics practice adherent to evidence-based testing guidelines,2 and its findings should generalize broadly across clinical settings. Furthermore, its results address real-world challenges of multiple-gene sequencing. Our definition of pathogenic variants as potentially actionable generally follows recommendations of the American College of Medical Genetics (for reporting of incidental findings, although some genes we reported fall outside of these recommendations)58 and other guideline organizations (for breast MRI screening with a two-fold relative risk increase, which we estimated using published literature on mutation penetrance).59–61 Notably, there are no data as yet on the risk-benefit ratio of breast MRI screening among patients with pathogenic variants in genes of moderate penetrance (eg, ATM, BLM). Given the remaining uncertainty in penetrance estimates for such variants, we cannot precisely estimate their contribution to a woman's age-specific cancer risks and, therefore, the optimal breast screening protocol and age of initiation remain unknown. A subjective component is unavoidable in interpreting unfamiliar variants11; other clinicians might make different judgements about patient notification and management. Nonetheless, our experience of results disclosure and risk-adapted intervention may inform future applications of multiple-gene sequencing.
Some of the pathogenic variants we identified would be detected by adherence to current practice guidelines. For example, the participant (LS294) who was found to carry an MLH1 mutation would now receive Lynch syndrome testing because she had endometrial carcinoma at age 46 years.1 When she was clinically assessed several years ago, however, Lynch syndrome testing was not routine for early endometrial cancer, and it is still underutilized.88,89 Moreover, this patient's early triple-negative breast cancer would place BRCA1/2 testing first in most clinicians' differential diagnoses,2 such that sequential single-syndrome testing would be slower to provide the correct answer than a multiplex approach. In contrast, other pathogenic variants lack such clear guidelines and present significant challenges. One example is the frameshift SLX4 mutations identified in participants LS347 and LS430. Recently identified as causing Fanconi's Anemia, SLX4 mutations are predicted to convey a two-fold increase in breast cancer risk that is similar to other pathway components such as PALB2. Though some publications support this risk association, SLX4 mutations appear rare among breast cancer families.45,46,84 Participants LS347 and LS430 each had a striking family history that included at least three breast cancers diagnosed before age 50 years; we therefore recommended breast MRI screenings, consistent with guidelines for women with increased risk.59–61 As another example, germline CDH1 mutations convey substantial risks of gastric and breast cancer in hereditary gastric cancer families90; the inadequacy of endoscopic screening leaves prophylactic gastrectomy as the only effective intervention.6 However, it is unknown whether patients who were found to carry CDH1 mutations incidentally, without family gastric cancer history, should undergo such life-changing surgery. Because participant LS443's CDH1 variant has not been reported among gastric cancer patients, we recommended close surveillance rather than gastrectomy. These ambiguous cases illustrate the complexities of multiple-gene sequencing in clinical practice and the potential hazards of unwarranted intervention. As a conservative approach to identified pathogenic variants of uncertain penetrance, we discussed increasing the frequency and/or intensity of cancer screening (Fig 2), but we judged the evidence insufficient to support recommendation of irreversible, invasive interventions such as prophylactic surgery. Another crucial unanswered question is whether testing negative for an identified familial mutation in genes of moderate penetrance (eg, PALB2, CHEK2) implies that a patient's risk of developing mutation-associated cancers is no greater than that of the general population; we require this missing information to enable risk-appropriate screening. Population-based studies of mutation penetrance and clinical trials of risk-reducing interventions are urgently needed to guide such difficult clinical decisions.
A major concern is the discovery of variants of uncertain significance, which do not contribute to risk assessment and may prompt anxiety and overtreatment. With widespread BRCA1/2 testing, the prevalence of VUS has declined to an acceptably low rate of 2% to 5%.91 Predictably, we found that sequencing 42 genes identified VUS in many participants (88%), averaging 2.1 VUS per participant. Our demonstration of this anticipated finding (that sequencing more genes yields more VUS, most of them novel) speaks to nascent efforts toward clinical whole-genome analysis. Moreover, it raises the question of a break-even point: what is the best set and number of genes to maximize information and minimize noise? Consistent with our IRB-approved protocol, we elected not to recontact about VUS, because these results lack practical implications. Outside a study, however, such a decision might conflict with patient autonomy, and the current standard of clinical VUS reporting will likely pertain.2 Therefore, it is crucial to develop methods for reclassifying VUS quickly and to communicate evolving VUS interpretations according to patients' preferences and understanding. This priority underscores the critical importance of sharing open databases of VUS identified through various genetic sequencing efforts, public and private.
Our study has limitations. The 42 genes that we selected reflect published literature but an optimal multiple-gene panel for routine diagnostic use remains to be defined. We conducted this study in an academic center, within a specialized Clinical Cancer Genetics service that is not universally available. Although participants did not differ significantly from the source population on most measured criteria, it is possible that they had subtle features of personal or family history that suggested greater inherited risk, which might have increased their willingness to donate a research blood sample. Because they were clinically accrued, participants do not represent the entire United States population. Another limitation is the absence of patient-reported preferences and outcomes, which will be a critical consideration in translating next-generation sequencing into practice. Our current results may inform the design of studies that address these important unanswered questions.
To our knowledge, this is the first clinical evaluation of next-generation sequencing among patients referred for breast and ovarian cancer risk assessment. Undoubtedly, multiple-gene sequencing raises many questions about results interpretation and patient counseling. Our study demonstrates an early signal for the clinical relevance of multiple-gene sequencing and provides a strong rationale for future research to define its most effective use.
Glossary Terms
- BRCA1:
a tumor suppressor gene known to play a role in repairing DNA breaks. Mutations in this gene are associated with increased risks of developing breast or ovarian cancer.
- BRCA2:
a tumor suppressor gene whose protein product is involved in repairing chromosomal damage. Although structurally different from BRCA1, BRCA2 has cellular functions similar to BRCA1. BRCA2 binds to RAD51 to fix DNA breaks caused by irradiation and other environmental agents. Also known as the breast cancer 2 early onset gene.
- CDH1
cadherin 1 gene. Mutations in this gene are correlated with gastric, breast, colorectal, thyroid, and ovarian cancers.
- Lynch syndrome:
hereditary nonpolyposis colorectal cancer (HNPCC). A cancer syndrome characterized by Henry T. Lynch in 1966, this genetic condition has a high risk of colon cancer as well as other cancers including endometrial, ovary, stomach, small intestine, hepatobiliary tract, upper urinary tract, brain, and skin.
- MLH1 (MutL homolog 1):
a DNA mismatch repair enzyme. MLH1 is responsible for overall fidelity of DNA replication.
Footnotes
See accompanying editorial on page 1987
Processed as a Rapid Communication manuscript.
Supported by the Breast Cancer Research Foundation, a Stanford University Cancer Institute Developmental Research Award, the Jan Weimer Junior Faculty Chair in Breast Oncology at Stanford University, National Institutes of Health Clinical and Translational Science Award No. UL1 RR025744, and InVitae.
Presented in part at the American Society of Clinical Oncology Breast Cancer Symposium, San Francisco, CA, September 7-9, 2013, and at the American Society of Human Genetics Annual Meeting, Boston, MA, October 22-26, 2013.
Terms in blue are defined in the glossary, found at the end of this article and online at www.jco.org.
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Although all authors completed the disclosure declaration, the following author(s) and/or an author's immediate family member(s) indicated a financial or other interest that is relevant to the subject matter under consideration in this article. Certain relationships marked with a “U” are those for which no compensation was received; those relationships marked with a “C” were compensated. For a detailed description of the disclosure categories, or for more information about ASCO's conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors.
Employment or Leadership Position: Emily E. Hare, InVitae (C); Yuya Kobayashi, InVitae (C); Stephen E. Lincoln, InVitae (C); Michele Cargill, InVitae (C) Consultant or Advisory Role: Uri Ladabaum, Exact Sciences (C); James M. Ford, InVitae (U) Stock Ownership: Emily E. Hare, InVitae; Yuya Kobayashi, InVitae; Stephen E. Lincoln, InVitae, Illumina; Michele Cargill, InVitae Honoraria: None Research Funding: Allison W. Kurian, InVitae; James M. Ford, InVitae Expert Testimony: None Patents, Royalties, and Licenses: None Other Remuneration: None
AUTHOR CONTRIBUTIONS
Conception and design: Allison W. Kurian, Uri Ladabaum, Michele Cargill, James M. Ford
Financial support: Allison W. Kurian, James M. Ford
Administrative support: Meredith A. Mills, Lisa McPherson
Provision of study materials or patients: Allison W. Kurian, Lisa McPherson, James M. Ford
Collection and assembly of data: Allison W. Kurian, Emily E. Hare, Meredith A. Mills, Lisa McPherson, Valerie McGuire, Yuya Kobayashi, Stephen E. Lincoln, Michele Cargill, James M. Ford
Data analysis and interpretation: Allison W. Kurian, Emily E. Hare, Kerry E. Kingham, Alice S. Whittemore, Uri Ladabaum, Yuya Kobayashi, Stephen E. Lincoln, Michele Cargill, James M. Ford
Manuscript writing: All authors
Final approval of manuscript: All authors
REFERENCES
- 1.Burt RW, Cannon JA, David DS, et al. Colorectal Cancer Screening, National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology, 2013. http://www.nccn.org.
- 2.Daly M. Genetic/Familial High-Risk Assessment: Breast and Ovarian, National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology, 2013. http://www.nccn.org. [DOI] [PubMed]
- 3.Daly MB, Axilbund JE, Buys S, et al. Genetic/familial high-risk assessment: Breast and ovarian. J Natl Compr Canc Netw. 2010;8:562–594. doi: 10.6004/jnccn.2010.0043. [DOI] [PubMed] [Google Scholar]
- 4.Ladabaum U, Wang G, Terdiman J, et al. Strategies to identify the Lynch syndrome among patients with colorectal cancer: A cost-effectiveness analysis. Ann Intern Med. 2011;155:69–79. doi: 10.7326/0003-4819-155-2-201107190-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Domchek SM, Friebel TM, Singer CF, et al. Association of risk-reducing surgery in BRCA1 or BRCA2 mutation carriers with cancer risk and mortality. JAMA. 2010;304:967–975. doi: 10.1001/jama.2010.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Norton JA, Ham CM, Van Dam J, et al. CDH1 truncating mutations in the E-cadherin gene: An indication for total gastrectomy to treat hereditary diffuse gastric cancer. Ann Surg. 2007;245:873–879. doi: 10.1097/01.sla.0000254370.29893.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rebbeck TR, Kauff ND, Domchek SM. Meta-analysis of risk reduction estimates associated with risk-reducing salpingo-oophorectomy in BRCA1 or BRCA2 mutation carriers. J Natl Cancer Inst. 2009;26:1331–1337. doi: 10.1093/jnci/djn442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Domchek SM, Bradbury A, Garber JE, et al. Multiplex genetic testing for cancer susceptibility: Out on the high wire without a net? J Clin Oncol. 2013;31:1267–1270. doi: 10.1200/JCO.2012.46.9403. [DOI] [PubMed] [Google Scholar]
- 9.Walsh T, Lee MK, Casadei S, et al. Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing. Proc Natl Acad Sci U S A. 2010;107:12629–12633. doi: 10.1073/pnas.1007983107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chute CG, Kohane IS. Genomic medicine, health information technology, and patient care. JAMA. 2013;309:1467–1468. doi: 10.1001/jama.2013.1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McGuire AL, McCullough LB, Evans JP. The indispensable role of professional judgment in genomic medicine. JAMA. 2013;309:1465–1466. doi: 10.1001/jama.2013.1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tuckson RV, Newcomer L, De Sa JM. Accessing genomic medicine: affordability, diffusion, and disparities. JAMA. 2013;309:1469–1470. doi: 10.1001/jama.2013.1468. [DOI] [PubMed] [Google Scholar]
- 13.Lowe HJ, Ferris TA, Hernandez PM, et al. STRIDE: An integrated standards-based translational research informatics platform. AMIA Annu Symp Proc. 2009;2009:391–395. [PMC free article] [PubMed] [Google Scholar]
- 14.Weber SC, Seto T, Olson C, et al. Oncoshare: Lessons learned from building an integrated multi-institutional database for comparative effectiveness research. AMIA Annu Symp Proc. 2012;2012:970–978. [PMC free article] [PubMed] [Google Scholar]
- 15.Kurian AW, Mitani A, Desai M, et al. Breast cancer treatment across health care systems: Linking electronic medical records and state registry data to enable outcomes research. Cancer. 2014;120:103–111. doi: 10.1002/cncr.28395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Redston M, Nathanson KL, Yuan ZQ, et al. The APCI1307K allele and breast cancer risk. Nat Genet. 1998;20:13–14. doi: 10.1038/1666. [DOI] [PubMed] [Google Scholar]
- 17.Renwick A, Thompson D, Seal S, et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet. 2006;38:873–875. doi: 10.1038/ng1837. [DOI] [PubMed] [Google Scholar]
- 18.Broberg K, Huynh E, Schlawicke Engstrom K, et al. Association between polymorphisms in RMI1, TOP3A, and BLM and risk of cancer, a case-control study. BMC Cancer. 2009;9:140. doi: 10.1186/1471-2407-9-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saetrom P, Biesinger J, Li SM, et al. A risk variant in an miR-125b binding site in BMPR1B is associated with breast cancer pathogenesis. Cancer Res. 2009;69:7459–7465. doi: 10.1158/0008-5472.CAN-09-1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen S, Parmigiani G. Meta-analysis of BRCA1 and BRCA2 penetrance. J Clin Oncol. 2007;25:1329–1333. doi: 10.1200/JCO.2006.09.1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mavaddat N, Peock S, Frost D, et al. Cancer risks for BRCA1 and BRCA2 mutation carriers: Results from prospective analysis of EMBRACE. J Natl Cancer Inst. 2013;105:812–822. doi: 10.1093/jnci/djt095. [DOI] [PubMed] [Google Scholar]
- 22.Seal S, Thompson D, Renwick A, et al. Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat Genet. 2006;38:1239–1241. doi: 10.1038/ng1902. [DOI] [PubMed] [Google Scholar]
- 23.Kaurah P, MacMillan A, Boyd N, et al. Founder and recurrent CDH1 mutations in families with hereditary diffuse gastric cancer. JAMA. 2007;297:2360–2372. doi: 10.1001/jama.297.21.2360. [DOI] [PubMed] [Google Scholar]
- 24.Pharoah PD, Guilford P, Caldas C. Incidence of gastric cancer and breast cancer in CDH1 (E-cadherin) mutation carriers from hereditary diffuse gastric cancer families. Gastroenterology. 2001;121:1348–1353. doi: 10.1053/gast.2001.29611. [DOI] [PubMed] [Google Scholar]
- 25.Dean JL, McClendon AK, Hickey TE, et al. Therapeutic response to CDK4/6 inhibition in breast cancer defined by ex vivo analyses of human tumors. Cell Cycle. 2012;11:2756–2761. doi: 10.4161/cc.21195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Debniak T, Górski B, Huzarski T, et al. A common variant of CDKN2A (p16) predisposes to breast cancer. J Med Genet. 2005;42:763–765. doi: 10.1136/jmg.2005.031476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang L, Zhang C, Li Y, et al. A non-synonymous polymorphism Thr115Met in the EpCAM gene is associated with an increased risk of breast cancer in Chinese population. Breast Cancer Res Treat. 2011;126:487–495. doi: 10.1007/s10549-010-1094-6. [DOI] [PubMed] [Google Scholar]
- 28.Barroso E, Pita G, Arias JI, et al. The Fanconi anemia family of genes and its correlation with breast cancer susceptibility and breast cancer features. Breast Cancer Res Treat. 2009;118:655–660. doi: 10.1007/s10549-009-0439-5. [DOI] [PubMed] [Google Scholar]
- 29.Kuschel B, Auranen A, McBride S, et al. Variants in DNA double-strand break repair genes and breast cancer susceptibility. Hum Mol Genet. 2002;11:1399–1407. doi: 10.1093/hmg/11.12.1399. [DOI] [PubMed] [Google Scholar]
- 30.Lemmens I, Van de Ven WJ, Kas K, et al. Identification of the multiple endocrine neoplasia type 1 (MEN1) gene: The European Consortium on MEN1. Hum Mol Genet. 1997;6:1177–1183. doi: 10.1093/hmg/6.7.1177. [DOI] [PubMed] [Google Scholar]
- 31.Neklason DW, Done MW, Sargent NR, et al. Activating mutation in MET oncogene in familial colorectal cancer. BMC Cancer. 2011;11:424. doi: 10.1186/1471-2407-11-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Win AK, Lindor NM, Young JP, et al. Risks of primary extracolonic cancers following colorectal cancer in lynch syndrome. J Natl Cancer Inst. 2012;104:1363–1372. doi: 10.1093/jnci/djs351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rennert G, Lejbkowicz F, Cohen I, et al. MutYH mutation carriers have increased breast cancer risk. Cancer. 2012;118:1989–1993. doi: 10.1002/cncr.26506. [DOI] [PubMed] [Google Scholar]
- 34.Bogdanova N, Feshchenko S, Schürmann P, et al. Nijmegen Breakage Syndrome mutations and risk of breast cancer. Int J Cancer. 2008;122:802–806. doi: 10.1002/ijc.23168. [DOI] [PubMed] [Google Scholar]
- 35.Seemanová E, Jarolim P, Seeman P, et al. Cancer risk of heterozygotes with the NBN founder mutation. J Natl Cancer Inst. 2007;99:1875–1880. doi: 10.1093/jnci/djm251. [DOI] [PubMed] [Google Scholar]
- 36.Zhang ZH, Yang LS, Huang F, et al. Current evidence on the relationship between two polymorphisms in the NBS1 gene and breast cancer risk: A meta-analysis. Asian Pac J Cancer Prev. 2012;13:5375–5379. doi: 10.7314/apjcp.2012.13.11.5375. [DOI] [PubMed] [Google Scholar]
- 37.Rahman N, Seal S, Thompson D, et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet. 2007;39:165–167. doi: 10.1038/ng1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pogue-Geile KL, Chen R, Bronner MP, et al. Palladin mutation causes familial pancreatic cancer and suggests a new cancer mechanism. PLoS Med. 2006;3:e516. doi: 10.1371/journal.pmed.0030516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wagner K, Grzybowska E, Butkiewicz D, et al. High-throughput genotyping of a common deletion polymorphism disrupting the TRY6 gene and its association with breast cancer risk. BMC Genet. 2007;8:41. doi: 10.1186/1471-2156-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee SJ, Do IG, Lee J, et al. Gastric cancer (GC) patients with hedgehog pathway activation: PTCH1 and GLI2 as independent prognostic factors. Target Oncol. 2013 doi: 10.1007/s11523-013-0253-1. [DOI] [PubMed] [Google Scholar]
- 41.Pilarski R, Eng C. Will the real Cowden syndrome please stand up (again)? Expanding mutational and clinical spectra of the PTEN hamartoma tumour syndrome. J Med Genet. 2004;41:323–326. doi: 10.1136/jmg.2004.018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tan MH, Mester JL, Ngeow J, et al. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res. 2012;18:400–407. doi: 10.1158/1078-0432.CCR-11-2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meindl A, Hellebrand H, Wiek C, et al. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat Genet. 2010;42:410–414. doi: 10.1038/ng.569. [DOI] [PubMed] [Google Scholar]
- 44.Machens A, Lorenz K, Sekulla C, et al. Molecular epidemiology of multiple endocrine neoplasia 2: Implications for RET screening in the new millenium. Eur J Endocrinol. 2013;168:307–314. doi: 10.1530/EJE-12-0919. [DOI] [PubMed] [Google Scholar]
- 45.Landwehr R, Bogdanova NV, Antonenkova N, et al. Mutation analysis of the SLX4/FANCP gene in hereditary breast cancer. Breast Cancer Res Treat. 2011;130:1021–1028. doi: 10.1007/s10549-011-1681-1. [DOI] [PubMed] [Google Scholar]
- 46.Shah S, Kim Y, Ostrovnaya I, et al. Assessment of mutations in hereditary breast cancers. PLoS One. 2013;8:e66961. doi: 10.1371/journal.pone.0066961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tram E, Ibrahim-Zada I, Briollais L, et al. Identification of germline alterations of the mad homology 2 domain of SMAD3 and SMAD4 from the Ontario site of the breast cancer family registry (CFR) Breast Cancer Res. 2011;13:R77. doi: 10.1186/bcr2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Soon WW, Miller LD, Black MA, et al. Combined genomic and phenotype screening reveals secretory factor SPINK1 as an invasion and survival factor associated with patient prognosis in breast cancer. EMBO Mol Med. 2011;3:451–464. doi: 10.1002/emmm.201100150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hearle N, Schumacher V, Menko FH, et al. Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin Cancer Res. 2006;12:3209–3215. doi: 10.1158/1078-0432.CCR-06-0083. [DOI] [PubMed] [Google Scholar]
- 50.Lim W, Olschwang S, Keller JJ, et al. Relative frequency and morphology of cancers in STK11 mutation carriers. Gastroenterology. 2004;126:1788–1794. doi: 10.1053/j.gastro.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 51.Gonzalez KD, Noltner KA, Buzin CH, et al. Beyond Li Fraumeni Syndrome: Clinical characteristics of families with p53 germline mutations. J Clin Oncol. 2009;27:1250–1256. doi: 10.1200/JCO.2008.16.6959. [DOI] [PubMed] [Google Scholar]
- 52.Kong W, He L, Richards EJ, et al. Upregulation of miRNA-155 promotes tumour angiogenesis by targeting VHL and is associated with poor prognosis and triple-negative breast cancer. Oncogene. 2014;33:679–689. doi: 10.1038/onc.2012.636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jacobs K, Paul J, Nilsen G, et al. Accurate detection of small and large copy number events from targeted next-generation sequence data. Presented at the American Society of Human Genetics Annual Meeting; October 22-26, 2013; Boston, MA. [Google Scholar]
- 54.Richards CS, Bale S, Bellissimo DB, et al. ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet Med. 2008;10:294–300. doi: 10.1097/GIM.0b013e31816b5cae. [DOI] [PubMed] [Google Scholar]
- 55.Abecasis GR, Auton A, Brooks LD, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sherry ST, Ward MH, Kholodov M, et al. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Exome Variant Server. NHLBI GO Exome Sequencing Project (ESP) http://evs.gs.washington.edu/EVS/
- 58.Green RC, Berg JS, Grody WW, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15:565–574. doi: 10.1038/gim.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Saslow D, Boetes C, Burke W, et al. American Cancer Society guidelines for breast screening with MRI as an adjunct to mammography. CA Cancer J Clin. 2007;57:75–89. doi: 10.3322/canjclin.57.2.75. [DOI] [PubMed] [Google Scholar]
- 60.Mainiero MB, Lourenco A, Mahoney MC, et al. ACR appropriateness criteria breast cancer screening. J Am Coll Radiol. 2013;10:11–14. doi: 10.1016/j.jacr.2012.09.036. [DOI] [PubMed] [Google Scholar]
- 61.Bevers TB, Bonaccio E, Buys SS, et al. Breast Cancer Screening and Diagnosis, Version 2.2013, National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology, 2013. http://www.nccn.org.
- 62.Burt RW. Strategies for colon cancer screening with considerations of cost and access to care. J Natl Compr Canc Netw. 2010;8:2–5. doi: 10.6004/jnccn.2010.0002. [DOI] [PubMed] [Google Scholar]
- 63.Robson ME, Storm CD, Weitzel J, et al. American Society of Clinical Oncology policy statement update: Genetic and genomic testing for cancer susceptibility. J Clin Oncol. 2010;28:893–901. doi: 10.1200/JCO.2009.27.0660. [DOI] [PubMed] [Google Scholar]
- 64.Buzin CH, Gatti RA, Nguyen VQ, et al. Comprehensive scanning of the ATM gene with DOVAM-S. Hum Mutat. 2003;21:123–131. doi: 10.1002/humu.10158. [DOI] [PubMed] [Google Scholar]
- 65.Rouleau E, Lefol C, Bourdon V, et al. Quantitative PCR high-resolution melting (qPCR-HRM) curve analysis, a new approach to simultaneously screen point mutations and large rearrangements: Application to MLH1 germline mutations in Lynch syndrome. Hum Mutat. 2009;30:867–875. doi: 10.1002/humu.20947. [DOI] [PubMed] [Google Scholar]
- 66.Szmola R, Sahin-Tóth M. Uncertainties in the classification of human cationic trypsinogen (PRSS1) variants as hereditary pancreatitis-associated mutations. J Med Genet. 2010;47:348–350. doi: 10.1136/jmg.2009.072751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ali M, Kim H, Cleary S, et al. Characterization of mutant MUTYH proteins associated with familial colorectal cancer. Gastroenterology. 2008;135:499–507. doi: 10.1053/j.gastro.2008.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Eliason K, Hendrickson BC, Judkins T, et al. The potential for increased clinical sensitivity in genetic testing for polyposis colorectal cancer through the analysis of MYH mutations in North American patients. J Med Genet. 2005;42:95–96. doi: 10.1136/jmg.2004.025973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lal G, Liu L, Hogg D, et al. Patients with both pancreatic adenocarcinoma and melanoma may harbor germline CDKN2A mutations. Genes Chromosomes Cancer. 2000;27:358–361. [PubMed] [Google Scholar]
- 70.Bai H, Jones S, Guan X, et al. Functional characterization of two human MutY homolog (hMYH) missense mutations (R227W and V232F) that lie within the putative hMSH6 binding domain and are associated with hMYH polyposis. Nucleic Acids Res. 2005;33:597–604. doi: 10.1093/nar/gki209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sieber OM, Lipton L, Crabtree M, et al. Multiple colorectal adenomas, classic adenomatous polyposis, and germ-line mutations in MYH. N Engl J Med. 2003;348:791–799. doi: 10.1056/NEJMoa025283. [DOI] [PubMed] [Google Scholar]
- 72.Seemanova E, Sperling K, Neitzel H, et al. Nijmegen breakage syndrome (NBS) with neurological abnormalities and without chromosomal instability. J Med Genet. 2006;43:218–224. doi: 10.1136/jmg.2005.035287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Suriano G, Oliveira C, Ferreira P, et al. Identification of CDH1 germline missense mutations associated with functional inactivation of the E-cadherin protein in young gastric cancer probands. Hum Mol Genet. 2003;12:575–582. doi: 10.1093/hmg/ddg048. [DOI] [PubMed] [Google Scholar]
- 74.McConville CM, Stankovic T, Byrd PJ, et al. Mutations associated with variant phenotypes in ataxia-telangiectasia. Am J Hum Genet. 1996;59:320–330. [PMC free article] [PubMed] [Google Scholar]
- 75.Mitui M, Nahas SA, Du LT, et al. Functional and computational assessment of missense variants in the ataxia-telangiectasia mutated (ATM) gene: Mutations with increased cancer risk. Hum Mutat. 2009;30:12–21. doi: 10.1002/humu.20805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: Server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sunyaev S, Ramensky V, Koch I, et al. Prediction of deleterious human alleles. Hum Mol Genet. 2001;10:591–597. doi: 10.1093/hmg/10.6.591. [DOI] [PubMed] [Google Scholar]
- 78.Kim JC, Roh SA, Koo KH, et al. Genotyping possible polymorphic variants of human mismatch repair genes in healthy Korean individuals and sporadic colorectal cancer patients. Fam Cancer. 2004;3:129–137. doi: 10.1023/B:FAME.0000039919.66461.8f. [DOI] [PubMed] [Google Scholar]
- 79.Kim YM, Choe CG, Cho SK, et al. Three novel germline mutations in MLH1 and MSH2 in families with Lynch syndrome living on Jeju island, Korea. BMB Rep. 2010;43:693–697. doi: 10.5483/BMBRep.2010.43.10.693. [DOI] [PubMed] [Google Scholar]
- 80.Miller PJ, Duraisamy S, Newell JA, et al. Classifying variants of CDKN2A using computational and laboratory studies. Hum Mutat. 2011;32:900–911. doi: 10.1002/humu.21504. [DOI] [PubMed] [Google Scholar]
- 81.Ranade K, Hussussian CJ, Sikorski RS, et al. Mutations associated with familial melanoma impair p16INK4 function. Nat Genet. 1995;10:114–116. doi: 10.1038/ng0595-114. [DOI] [PubMed] [Google Scholar]
- 82.Reymond A, Brent R. p16 proteins from melanoma-prone families are deficient in binding to Cdk4. Oncogene. 1995;11:1173–1178. [PubMed] [Google Scholar]
- 83.Walker GJ, Gabrielli BG, Castellano M, et al. Functional reassessment of P16 variants using a transfection-based assay. Int J Cancer. 1999;82:305–312. doi: 10.1002/(sici)1097-0215(19990719)82:2<305::aid-ijc24>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 84.Bakker JL, van Mil SE, Crossan G, et al. Analysis of the novel fanconi anemia gene SLX4/FANCP in familial breast cancer cases. Hum Mutat. 2013;34:70–73. doi: 10.1002/humu.22206. [DOI] [PubMed] [Google Scholar]
- 85.Cybulski KE, Howlett NG. FANCP/SLX4: A Swiss army knife of DNA interstrand crosslink repair. Cell Cycle. 2011;10:1757–1763. doi: 10.4161/cc.10.11.15818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Morak M, Laner A, Bacher U, et al. MUTYH-associated polyposis: Variability of the clinical phenotype in patients with biallelic and monoallelic MUTYH mutations and report on novel mutations. Clin Genet. 2010;78:353–363. doi: 10.1111/j.1399-0004.2010.01478.x. [DOI] [PubMed] [Google Scholar]
- 87.Offit K, Bradbury A, Storm C, et al. Gene patents and personalized cancer care: Impact of the myriad case on clinical oncology. J Clin Oncol. 2013;31:2743–2748. doi: 10.1200/JCO.2013.49.7388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Backes FJ, Mitchell E, Hampel H, et al. Endometrial cancer patients and compliance with genetic counseling: Room for improvement. Gynecol Oncol. 2011;123:532–536. doi: 10.1016/j.ygyno.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 89.Kalloger SE, Allo G, Mulligan AM, et al. Use of mismatch repair immunohistochemistry and microsatellite instability testing: Exploring Canadian practices. Am J Surg Pathol. 2012;36:560–569. doi: 10.1097/PAS.0b013e31823f3b28. [DOI] [PubMed] [Google Scholar]
- 90.Chun N, Ford JM. Genetic testing by cancer site: Stomach. Cancer J. 2012;18:355–363. doi: 10.1097/PPO.0b013e31826246dc. [DOI] [PubMed] [Google Scholar]
- 91.Hall MJ, Reid JE, Burbidge LA, et al. BRCA1 and BRCA2 mutations in women of different ethnicities undergoing testing for hereditary breast-ovarian cancer. Cancer. 2009;115:2222–2233. doi: 10.1002/cncr.24200. [DOI] [PMC free article] [PubMed] [Google Scholar]