

Abstract
Exposure to cigarette smoke during development is linked to neurodevelopmental delays and cognitive impairment including impulsivity, attention deficit disorder, and lower IQ. However, brain region specific biomolecular alterations induced by developmental cigarette smoke exposure (CSE) remain largely unexplored. In the current molecular phenotyping study, a mouse model of ‘active’ developmental CSE (serum cotinine>50 ng/mL) spanning pre-implantation through third trimester-equivalent brain development (gestational day (GD) 1 through postnatal day (PD) 21) was utilized. Hippocampus tissue collected at the time of cessation of exposure was processed for gel-based proteomic and non-targeted metabolomic profiling with Partial Least Squares-Discriminant Analysis (PLS-DA) for selection of features of interest. Ingenuity Pathway Analysis was utilized to identify candidate molecular and metabolic pathways impacted within the hippocampus. CSE impacted glycolysis, oxidative phosphorylation, fatty acid metabolism, and neurodevelopment pathways within the developing hippocampus.
Keywords: Development, cigarette smoke, tobacco, hippocampus, proteome, metabolome
1. Introduction
Cigarette smoke contains approximately 8000 toxic chemicals including nicotine, carbon monoxide, heavy metals, hydrogen cyanide, and polycyclic aromatic hydrocarbons (Borgerding and Klus 2005; Rodgman and Perfetti 2013). In the US, approximately 44 million adults smoke cigarettes including one fifth of all pregnant women (2012). Maternal cigarette smoking during pregnancy is associated with a variety of adverse pregnancy outcomes, of which low birth weight (LBW) is the most well documented (Abel 1980; Cooke 1998; Mitchell et al. 2002). Prenatal growth restriction and resultant LBW have long-lasting effects on infant and childhood growth and cognitive development (Botero and Lifshitz 1999; Das and Sysyn 2004; Gluckman et al. 2005). Epidemiological and case control studies suggest that children exposed to cigarette smoke during development exhibit aberrant behavioral and cognitive development including hyperactivity and impulsivity (Fried and Makin 1987; Roy et al. 1998; Slotkin et al. 2002), impaired learning and memory (Batstra et al. 2003; Sexton et al. 1990), perception deficits (Fried and Watkinson 2000; McCartney et al. 1994), and lower IQ with impaired intellectual development (Butler and Goldstei. H 1973; Butler and Goldstein 1973; Olds et al. 1994a, c, b).
The developmental toxicity of cigarette smoke, or its principal addictive components such as nicotine, has been widely investigated in animal models, including effects on neuro/cognition and behavior. While studies investigating the effects of developmental nicotine exposure are numerous, those examining the effects of actual inhalation exposures of cigarette smoke in animal models – as in the present study – are generally lacking. The preponderance of neurocognitive/behavioral data in mammalian animal models indicates that, when administered during fetal ontogenesis, nicotine is a potent developmental neurotoxicant with a diversity of effects on the central nervous system, including alterations in neurotransmitter signaling and resultant perturbations in the neuroanatomical and cytoarchitectural development of the brain (Ernst et al. 2001; Levin and Slotkin 1998; Levin et al. 1996; Slikker et al. 2005; Slotkin 1992). Prenatal exposure to nicotine in varied rodent models is known to: perturb early brain morphogenesis through excessive neuroepithelial cell apoptosis (Roy et al. 1998; Zhao and Reece 2005); elicit shortfalls in neuronal cell numbers through decreased cell proliferation or enhanced apoptosis (Jang et al. 2002) (Navarro et al. 1989; Slotkin 1999; Slotkin et al. 1986; Slotkin et al. 1997; Slotkin et al. 1987); alter cell size, packing density or cortical thickness (Gospe et al. 1996; Roy et al. 2002); promote abnormal gliosis at the expense of neurogenesis (Roy et al. 2002); and interfere with the development of neural circuitry (Levin and Slotkin 1998; Levin et al. 1996; Slawecki and Ehlers 2002; Slawecki et al. 2000) (Slotkin 1992; Slotkin 1999) – all in the developing brain.
The nicotine-induced “structural” alterations in the brain, noted above, correlate with neurobehavioral/cognitive “functional” deficits in exposed animals which mimic deficits seen in children whose mothers smoked during pregnancy (Cornelius and Day 2000; DiFranza et al. 2004; Ernst et al. 2001) including: increased locomotor activity (Fung 1988; Koehl et al. 2000; Nagahara and Handa 1999; Shacka et al. 1997), hyperactivity (Ajarem and Ahmad 1998; Newman et al. 1999; Sobrian et al. 2003; Vaglenova et al. 2004), impulsivity (Sobrian et al. 2003), and anxiety (Vaglenova et al. 2004). In addition, long-term impairments in attention, learning, and memory in smoke exposed animals have been observed through the aid of various paradigms such as, avoidance acquisition (Genedani et al. 1983; Peters and Ngan 1982; Vaglenova et al. 2004), radial arm maze tasks (Levin et al. 1993; Sorenson et al. 1991), and operant learning behaviors (Martin and Becker 1971). In the animal model employed in the current study, developmental CSE altered offspring neurobehavioral maturation and outcomes (Amos-Kroohs et al. 2013). Specifically, prenatal and early postnatal inhalation exposure of mice to cigarette smoke induced subnormal anxiety in various novel environments, impaired spatial learning and reference memory while sparing other behaviors (i.e. route-based learning, fear conditioning, and forced swim immobility) – supporting mounting evidence that developmental cigarette smoke exposure has long-term adverse effects on the brain, including hippocampus-mediated memory function (Amos-Kroohs et al. 2013).
Across species, the hippocampus plays an important role in orchestrating learning and memory processing (Nishitani 2003; Nishitani et al. 1998; Olton and Feustle 1981; Olton and Papas 1979; Romijn et al. 1991a), including declarative memory, spatial cognition, memory consolidation, multimodal sensory integration, habituation and novelty detection, as well as temporal information processing and sequencing (d’Hellencourt and Harry 2005; Eichenbaum 1999, 2004a, b; Fortin et al. 2004; Hammond et al. 2004; Levenson and Sweatt 2005; Shapiro and Eichenbaum 1999; Sweatt 2004; Whishaw and Jarrard 1996; Yamaguchi et al. 2004). In view of the association between pre- and peri-natal exposure to cigarette smoke/nicotine (in humans and rodents) and deficits in attention, perception, learning and memory, studies detailed in the present report investigate the effects of murine inhalation exposure to cigarette smoke during development on the molecular ontogenesis of the hippocampus. The main objective of the current study was to determine the impact of developmental CSE on the hippocampus biomolecular phenotype at the time of cessation of exposure. Utilizing a mouse model simulating “active” maternal cigarette smoking, (Esposito et al. 2008) which is characterized by attendant low birth weight in offspring and altered neurobehavioral phenotypes at maturity (Amos-Kroohs et al. 2013), hippocampus proteome and metabolome profiles were examined. The offspring were exposed to CSE (6 hrs/day, 7 days/week) from gestational day (GD) 1 through postnatal day (PD) 21 using commercial Marlboro red cigarettes, the most common brand of cigarettes smoked by young women. In parallel studies from identical offspring, developmental cigarette smoke exposure altered liver and kidney proteome profiles of the low birth weight offspring (Canales et al. 2012; Jagadapillai et al. 2012). The current study forms the basis of future studies on the persistence of these alterations past the cessation of exposure at the time of development of aberrant neurobehavioral phenotypes.
2. Materials and Methods
2.1. Animals
Adult C57BL/6J mice were purchased from Jackson Labs (Bar Harbor, ME). Animals were housed in ventilated racks and maintained in a controlled temperature/humidity environment with a 12 hour light/dark cycle and free access to Purina Lab Diet 5015 and water in the University of Louisville Research Resources Center, an AAALAC accredited vivarium. All exposure procedures were approved by the Institutional Animal Use and Care Committees of the University of Louisville and conformed to the NIH Guide for the Care and Use of Laboratory Animals. Timed pregnancies were established by overnight mating of a single mature male with two nulliparous females. The presence of a vaginal plug on the following morning was considered evidence of mating and the time considered gestational day (GD) 0.
2.2. Murine Cigarette Smoke Exposure Model
Dams were randomly assigned to either the SHAM or cigarette smoke exposure (CSE) groups. Exposure to cigarette smoke was performed using the Teague TE-10C cigarette smoke inhalation exposure system (Teague Enterprises, Davis, CA). Animals were exposed to cigarette smoke (CSE; commercial Marlboro Red cigarettes) or filtered ambient air (SHAM) for 6h/day throughout the entirety of gestation, and following parturition were exposed with their offspring until PD 21, as described in detail (Amos-Kroohs et al. 2013). Aged, diluted side- and mainstream smoke was delivered to the CSE chamber under conditions providing total suspended particulates (TSPs) in the range of 20–30 mg/m3 – an exposure that elevates dam/pup plasma cotinine levels (Koren et al. 1992; Koren et al. 1998) to those resembling levels in pregnant women who are ‘active’ smokers (i.e., plasma cotinine greater than 12 ng/ml (Jarvis et al. 2008). TSP levels represent the “dose” of cigarette smoke to which the animal is exposed, while plasma cotinine (primary metabolite of nicotine) serves as a biomarker of the internal dose. Control animals were sham exposed to ambient air in whole body inhalation chambers under identical conditions (temperature, humidity, flow rate) to CSE mice. Chamber conditions including total suspended particulates (TSP), carbon monoxide levels, humidity, and temperature were measured twice daily during the GD 1 through PD21 exposure period. On PD21, pups were euthanized by asphyxiation with carbon dioxide followed by thoracotomy and cardiac puncture. Tissues were harvested and stored at −80°C until analysis. The tissues utilized for the current study were from identical offspring as those utilized for our prior studies with a total of 9 offspring from individual litters representing each group (n=9 for CSE and Sham groups) (Canales et al. 2012; Jagadapillai et al. 2012).
2.3. Proteome Profiling
Individual offspring hippocampus tissue samples were homogenized in an ice cold methanol/chloroform matrix, washed with methanol/chloroform, and then pelleted by centrifugation. The dried pellets were homogenized in sample preparation buffer [7M urea, 2M thiourea, 40mM dithiothreitol (DTT)] and stored at −80°C for a short time until analysis. Protein concentration for each of the samples was determined using the Bradford Assay (Bradford 1976). For each individual hippocampus tissue protein extract (n=9 per group), four hundred micrograms of protein in rehydration buffer (8M urea, 2% CHAPS, 2 μl IPG buffer pH 3–10, 2.5 mg/ml DTT, 0.002% bromophenol blue) was applied to IPGphor Drystrips (Nonlinear, 3–11, 180 mm × 3 mm × 0.5 mm, GE Healthcare, Piscataway, NJ). First dimension separation by isoelectric focusing at 22,000 Volt hours (Vh) was performed with a hold at 100 Volts until further processing. The IEF strips were stored at −80°C for 1 hr followed by: (1) equilibration for 60 minutes in reducing buffer (6M urea, 75 mM Tris-HCl pH 8.8, 29.3% glycerol, 2% SDS, 0.002% bromophenol blue with 3.5 mg/ml DTT) and (2) equilibration in alkylating buffer (same buffer with 45 mg/ml iodoacetamide instead of DTT) for an additional 60 minutes. Second dimension SDS-PAGE separation (25cm × 20.5cm 15% polyacrylamide gels) was performed overnight (18 h; 100V). Protein spots were visualized by Colloidal Coomassie Blue G-250 (Candiano et al. 2004).
2.3.1 Image Analysis
Gels were scanned using an Epson Expression 10,000 XL scanner with transparency attachment. Densitometric analysis of gel images was performed with Progenesis SameSpots software (Nonlinear Dynamics; New Castle-on-Tyne, UK). Protein spots were detected automatically and manually adjusted (if required) for accuracy. For each protein spot, the intensity was measured, background was subtracted, and individual spot density was normalized by total pixel density of each gel. Spots with average normalized pixel depth ≤1000 relative abundance units and non-normalized areas with pixel depth below 100 were removed as noise. The averaged normalized spot abundance was compared between groups to determine fold differences in abundance. Two gel images were removed from analysis due to high non-specific background resulting in a total of 7 CSE and 9 Sham 2D gels utilized for image analysis.
2.3.2. Protein identification
Protein spots were excised and destained with 50% ethanol in 50 mM ammonium bicarbonate for a minimum of 5 washes. Excised gel spots were then dehydrated in acetonitrile (ACN), dried, and rehydrated with 10 ng/μl trypsin and 40mM ammonium bicarbonate. Proteins were digested at room temperature for approximately 18 h. Peptides were eluted in acidified acetonitrile and stored at −20°C until analysis. The mass to charge ratio of peptides was determined by direct inject LTQ/FT-ICR-MS/MS (or HPLC interface on occasion) with collision induced dissociation for structural feature identification. Peptide identification was performed with the Mascot (Matrix Sciences v 2.2.2) search algorithm utilizing the NCBInr (with decoy) database (updated June 1, 2012). Search parameters included: mammalian class, 2 missed cleavages, carbamidomethyl C variable modification, enzyme trypsin/P, and an allowed peptide charge of 1+, 2+, or 3+. Positive protein identification required a total MOWSE absolute probability entire protein score ≥100 composed of a minimum of two peptides with individual scores MOWSE absolute probability scores ≥50 (Neuhoff et al. 1990; Pappin et al. 1993).
2.3.3. Statistical analysis of proteome data
Partial Least Squares-Discriminant Analysis (PLS-DA) modeling of variance between groups with Variable Importance in Projection (VIP) rankings was used to identify protein spot features whose normalized abundance determined the differences between groups (Karp et al. 2005). Due to the limited number of gels per group (n=8 for SHAM group and n=8 for CSE group), the dataset was not split into a test and validation set.
2.3.4. Ingenuity Pathway Analysis (IPA)
Ingenuity Pathway Analysis was used to determine families and metabolic pathways of the proteins that were identified (Ingenuity Systems, 2011). The GI numbers of identified were entered into the IPA program with selected outputs including functional network mapping and interactions of proteins based on prior literature reports.
2.4 Global metabolome profiling
Hippocampus tissue was weighed and homogenized with 5 volumes of ice-cold HPLC-grade 1:2 chloroform/methanol with subsequent separation of organic and aqueous phases, washing of phases, and pooling of washes with original extracts. The samples, both aqueous and lipophilic phases, were dried under vacuum and stored at −80°C until analysis. At time of spectral collection, dual polarity spectral features were collected with chip-based nanoelectrospray (Advion TriVersa Nanomate) for direct infusion positive ion LTQ-FT-ICR-MS. The sample spray characteristics were stable (greater than 10 minutes) with ion current falling between 10–90 nA (2.1 kV spray voltage and 0.05 psi head pressure). A spectral range of 50–1000 m/z was recorded for 0.5 min with 100,000 resolution. Experimental replicates and technical replicates were injected in alternating group order.
2.4.1 Spectra processing and feature validation
Raw spectra were filtered to remove background noise prior to peak detection and shaping, followed by spectra deisotoping and removal of blank mass features. Isotopic clusters were identified using a m/z tolerance of 0.0010 Da, a minimum/maximum charge of 5, and the first allowed gap at position 3. The isotopic clustered peaks were then excluded from analysis.
2.4.2 Statistical analysis of metabolome data
Statistical analysis was performed using the R package caret (Kuhn 2008). The data (955 m/z features) containing all non-noise, non-adduct features from the two phases collected in both positive and negative polarity were normalized by mean centering and unit variance scaling prior to analysis. The PLS-DA classifier was augmented by including a recursive feature elimination (RFE) step in the algorithm. The PLS-DA with RFE procedure was performed on normalized CSE/SHAM hippocampus samples to select an optimal subset (number) of m/z features with respect to classification accuracy from the original 955 m/z feature using a double cross-validation scheme to optimize both the number of PLS components and number of peaks used for classification. Briefly, to find the optimal subset of peaks, data were randomly partitioned 25 times into 75% training and 25% test sets, selecting important peaks for different subsets based on the variable importance measure ranking. The variable importance measure is based on weighted sums of the absolute PLS loadings, where the weights are based on the relative percentage of explained variation associated with each component. For each of the 25 outer splits, an inner-loop validation using 25 random splits into test (25%) and training (75%) data was carried out to construct a PLS model with the optimal number of components for different subsets of peaks. For this inner-loop validation, classification accuracy was calculated for between 1 and 5 components, for each different subset of peaks. The optimized number of components was chosen to construct a classifier for each particular subset. After the 25 outer resampling iterations, 25 classification accuracies were obtained for each subset of peaks and these values were averaged for final selection of the optimal subset. The PLS-DA classifier with the optimal number of components using the optimal subset of peaks was used to construct the final classifier. As a final evaluation of the selected model, the data were randomly partitioned 100 times into 50% training and 50% test sets, and the final PLS-DA model was fit to the training data and classification accuracy evaluated on the test data.
2.4.3 Putative Feature Identification
The neutral monoisotopic masses of the putative features of interest generated from the above statistical analyses were calculated from the selected polarity. The tentative identification of the features of interest was based on the accurate mass (<5ppm variance) obtained from a search of the Human Metabolome Database (HMDB) and Lipid MAPS with MetaboAnalyst as search engine (Xia et al. 2012). Preliminary identification was dependent on agreement of predicted and observed isotopic distribution. The results were combined and the preliminary redundant identifications were deleted. Though not positively identified by standard compound fragment comparison, the putative feature list includes several previously described hippocampus lipid species. The putative chemical class identification and the possibility of inclusion of sodium and potassium adducts for these peaks were also checked by MS/MS fragmentation pattern. In all cases of glycerolipid species, multiple acyl side chain attributions were found thus the m/z feature contained multiple related structures. For example, the neutral monoisotopic mass of 759.5799 contained PC 34:1 composed of mixture of at least 3 separate acyl side chains including 16:1, 18:0, and 18:1. Confirmation of chemical class as a phosphatidylcholine was based on neutral mass loss from the choline moiety. Un-annotated features of interest are also listed and maintain importance in differentiating the two groups. Functional annotation to metabolic pathways was performed with the freely available pathway visualization tool, MetaboAnalyst (Xia and Wishart 2011).
3. Results
3.1. Exposure Conditions and Outcomes
The inhalation exposure chamber conditions were identical to the conditions reported previously (Amos-Kroohs et al. 2013; Canales et al. 2012; Esposito et al. 2008; Jagadapillai et al. 2012). Average levels of CO and TSP in CSE chamber during GD1-PD21 were 138 ± 19.8 ppm and 25.4 ± 6.5 mg/m3, respectively, while those in SHAM chamber were lower than detection limit. At birth, the offspring exhibited low birth weight which persisted throughout the postnatal exposure period. At the discontinuation of exposure on PD21, the average weight deficit of the CSE versus the SHAM offspring was 13.2%. This decrement was reflected in organ weights suggesting that the weight deficit is a proportional decrease in mass across all organs.
3.2. Hippocampus Proteome Profile
The hippocampus 2D-SDS-PAGE gel protein spot patterns were similar between CSE and SHAM groups (Figure 1). The molecular weights of protein spots descend from ~100 kDa to ~10 kDa with the isoelectric focusing point spanning a pH of 3–10. Approximately the same number of protein spots was found on each gel without a consistent increase or decrease in spot numbers based on sample group membership. The dominant difference between gel patterns of the CSE and SHAM groups consisted of a variance in pixel depths (spot abundances) for some spots.
Figure 1. Representative 2D-SDS-PAGE gels of the urea-soluble hippocampus proteins from SHAM and CSE offspring.
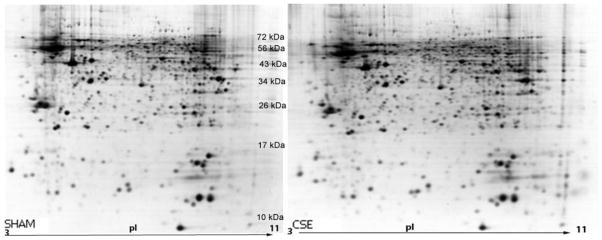
The figure depicts a side-by-side comparison of protein spot separations based on isoelectric focusing point (horizontal) and molecular weight (vertical) in the two experimental groups (SHAM-left; CSE-right). The gels are similar in number of spots without the appearance or loss of spots between groups. The contrast of these images has been uniformly altered to accentuate low abundant protein spots. Subsequent analyses of protein spot alterations between the groups were based on unaltered contrast images.
3.3. Partial Least Squares-Discriminant Analysis
A total of 1144 proteins spots remained after the noise peaks were excluded. The normalized pixel depth (abundance) of each protein spot that was found on all gels was utilized for Partial Least Squares-Discriminant Analysis (PLS-DA; PASW Statistics 18). When all non-noise protein spots were included in the analysis, the first three latent factors accounted for nearly 100% (~92% for the 1st latent factor) of variance between groups. In Figure 2, the separation between the SHAM and CSE groups based on the top three latent factors were plotted.
Figure 2. Latent factors plotted in 3D from the proteome PLS-DA model.

A graphical representation of the differences between sample groups based on the relative abundances of protein spots is shown by plotting each sample within a matrix composed of the top 3 latent factors (eigenvectors). The clear separation of the groups is visible with consistency in the hippocampus proteome profile of each biological replicate within the groups (Blue=Sham; Green=CSE). All protein spots from all 2D gels (excluding noise) were included in the calculation of VIP rankings and the graphing of the separation of groups by latent factors.
3.4. Proteins Impacted by CSE
A 2D-SDS-PAGE protein spot map is shown with color coded numbers labeling the protein spots found to be important in describing the separation of the groups (Variable Import in Projection rankings less than 1.7; decreased in blue and increased in green; Figure 3). The proteins identified as present in these spots are listed in Tables 1 and 2. The top scoring protein for each spot was reported when in great excess as evidenced by a MOWSE score greater than twice the next nearest identification. Spots with unidentified proteins are not included in the list.
Figure 3. At PD21, the urea soluble hippocampus proteome profiles are altered by developmental CSE.

The protein spots with altered abundance following developmental CSE are circled and numbered. Numbers in blue represent decreased abundance proteins and the numbers in green represent increased abundance proteins. These protein spots were ranked according to the Variable Import in Projection score (VIP ≥1.7) based on the PLS-DA model and contributed to the separation of the proteome profiles of the SHAM and CSE groups. Protein spots that were identified are listed in Tables 1–3.
Table 1.
Identification of proteins from spots with decreased abundance that contributed to the separation of hippocampus proteome profiles of offspring exposed to CSE throughout development (GD1-PD21 [PLS-DA model, VIP values (≥1.7)].
| Spot# | VIP | Protein Identification | GI Number | MOWSE Score |
|---|---|---|---|---|
| 12 | 2.09 | ATP synthase, H+ transporting, mitochondrial F1 complex, alpha subunit, isoform 1 | 6680748 | 577 |
| 14 | 2.07 | aldolase C, fructose-bisphosphate | 60687506 | 865 |
| 16 | 1.98 | proteasome (prosome, macropain) subunit, beta type 5 | 6755204 | 235 |
| 18 | 1.94 | M2-type pyruvate kinase | 1405933 | 369 |
| 25 | 1.87 | vacuolar H+ATPase B2 | 17105370 | 243 |
| 28 | 1.86 | aldolase C, fructose-bisphosphate | 60687506 | 342 |
| 33 | 1.82 | ATP synthase, H+ transporting, mitochondrial F1 complex, alpha subunit, isoform 1 | 6680748 | 620 |
| 35 | 1.80 | dihydropyrimidinase-related protein | 6714522 | 207 |
| 37 | 1.80 | ATP synthase, H+ transporting, mitochondrial F1 complex, alpha subunit, isoform 1 | 6680748 | 980 |
| 38 | 1.79 | heat shock protein (hsp60) precursor | 56383 | 699 |
| 40 | 1.78 | Protein disulfide-isomerase A3 | 1352384 | 260 |
| 41 | 1.77 | aldhehyde dehydrogenase family 5, subfamily A1 | 27369748 | 368 |
Table 2.
Identification of proteins from spots with increased abundance that contributed to the separation of hippocampus proteome profiles of offspring exposed to CSE throughout development (GD1-PD21 [PLS-DA model, VIP values (≥1.7)].
| Spot# | VIP | Protein Identification | GI Number | MOWSE Score |
|---|---|---|---|---|
| 52 | 2.58 | ATP synthase, H+ transporting, mitochondrial F1 complex, O subunit | 20070412 | 249 |
| 53 | 2.58 | aldhehyde dehydrogenase family 5, subfamily A1 | 27369748 | 415 |
| 55 | 2.31 | myotrophin | 6679961 | 158 |
| 57 | 2.24 | ATP synthase beta subunit | 1374715 | 222 |
| 58 | 2.22 | ATP synthase, H+ transporting, mitochondrial F1 complex, alpha subunit, isoform 1 | 6680748 | 909 |
| 59 | 2.14 | isovaleryl coenzyme A dehydrogenase | 9789985 | 216 |
| 61 | 2.09 | mitochondrial aldehyde dehydrogenase 2 | 6753036 | 486 |
| 64 | 2.03 | heterogeneous nuclear ribonucleoprotein A1 | 8393547 | 235 |
| 65 | 2.01 | septin 2 a | 6754816 | 219 |
| 66 | 2.00 | ATP synthase, H+ transporting, mitochondrial F1 complex, O subunit | 20070412 | 200 |
| 69 | 1.93 | poly(rC) binding protein 1 | 6754994 | 190 |
| 72 | 1.91 | purine rich element binding protein A | 6679573 | 345 |
| 75 | 1.89 | myelin basic protein | 199051 | 154 |
| 76 | 1.85 | aconitase 2, mitochondrial | 18079339 | 520 |
| 79 | 1.83 | annexin A5 | 6753060 | 334 |
| 82 | 1.80 | cyclophilin CyP-S1 | 53035 | 318 |
| 83 | 1.80 | cofilin 1, non-muscle | 6680924 | 123 |
| 84 | 1.79 | CDCrel-1AI | 19909845 | 346 |
| 87 | 1.78 | sirtuin 2 (silent mating type information regulation 2, homolog) 2 (S. cerevisiae), isoform CRA_b | 148692166 | 236 |
| 90 | 1.77 | myelin basic protein | 199051 | 114 |
| 91 | 1.76 | 78 kDa glucose-regulated protein | 2506545 | 211 |
| 92 | 1.75 | Atp5b protein | 23272966 | 768 |
| 93 | 1.75 | bisphosphoglycerate mutase 1 | 114326546 | 351 |
| 94 | 1.74 | acetyl-Coenzyme A acetyltransferase 1 precursor | 21450129 | 137 |
| 95 | 1.74 | 6-phosphogluconolactonase | 13384778 | 386 |
| 96 | 1.74 | Protein NipSnap homolog 2 | 17380131 | 150 |
| 98 | 1.71 | alpha-tubulin isotype M-alpha-2 | 202210 | 237 |
3.5. Ingenuity Pathway Analysis-Pathways with Altered Protein Abundance
The proteins identified in the top VIP1 rank (VIP1 >1.7) were input to the Ingenuity Pathway Analysis (IPA) algorithm to identify potential hippocampus metabolic networks impacted by developmental CSE. The identified proteins of the top VIP1 rank (VIP1 >1.7) are shown in the shaded shapes within the networks. Figure 4 depicts the impact of proteins with increased and decreased abundance (identified spots in Tables 1 and 2). In the CSE group, the H+-transporting two-sector ATPase, Tubulin, ERK1/2 (extracellular signal-regulated kinases), and NFKB (complex) represent central nodes of the network.
Figure 4. The cellular compromise, nucleic acid metabolism, and small molecule biochemistry pathways are impacted in the hippocampus by developmental CSE.

Proteins identified as contributing to the separation of the groups (CSE and Sham; PD21) are shadowed and connected to the network by arrows denoting directionality of impact. Several of these proteins also are designated members of the Cancer, hematological disease, and reproductive system disease network that was also impacted by developmental CSE. In the associated figure, solid lines indicated a direct interaction while dotted lines indicate an indirect interaction. Geometric shapes identify classes of proteins: phosphatases (triangle), kinases (inverted triangle), enzymes (vertical diamond), transcription regulators (horizontal ellipse), transporters (trapezoid), and other important molecules (circles).
The following protein were identified as feeding into/out of the ERK1/2 and NFKB (complex) nodes: Cyclophilin CyP-S1 (Peptidylprolyl isomerase B, PPIB) (Spot 82), mitochondrial aldehyde dehydrogenase 2 (ALDH2) (Spot 61), Annexin A5 (ANXA5, Spot 79), Hspd1 protein (Hsp60, Spot 38), myotrophin (MTPN, Spot 55), and Cofilin 1 (CFL1, Spot 83).
The following protein were identified as feeding into/out of the H+-transporting two-sector ATPase node: Vacuolar H+ATPase B2 (ATP6V1B2, Spot 25), Mitochondrial F1 complex, alpha subunit 1 (ATP5A1, Spots 12, 33, and 37), ATP synthase beta subunit (ATP5B, Spots 57 and 92), and ATP synthase, H+ transporting, mitochondrial F1 complex, O subunit (ATP5O, Spots 52 and 66).
The following proteins were identified as feeding into/out of the Tubulin node: Sirtuin 2 (SIRT2, Spot 87), Dihydropyrimidinase-like 5 (DPYSL5, Spot 35), Myelin basic protein (MBP, Spots 75 and 90), and α-tubulin isotype M-alpha-2 (TUBA1B, Spot 98). The following protein were identified as feeding into/out-of the minor node: Heterogeneous nuclear ribonucleoprotein A1 (HNRNPA1, Spot 64), Poly(rC)-binding protein 1 (PCBP1, Spot 69), and PURA (Spot 72).
3.6 Hippocampus Metabolome Features of Interest Impacted by Developmental CSE
As shown in Figure 5A, a single PLS component described greater than 90% of the separation between the groups when all m/z features of both polarities and each extract phase were combined and then analyzed for import in describing the separation of the metabolome profiles of the hippocampus in the two groups. This PLS-DA model includes 35 features of interest that were selected based on fit with the training set and that yielded sensitivity, specificity, and accuracy of the model greater than 90% (Figure 5B) Boxplots describing the differences in raw intensity values for the 35 m/z features of interest found to be significantly altered in the hippocampus by developmental CSE are shown if Figure 6. The features of interest are separated into origin phase and ion polarity.
Figure 5.

Figure 5A: Latent factors plotted in 3D from the metabolome PLS-DA model. A graphical representation of the differences between sample groups based on the relative abundances of m/z features is shown by plotting each sample within a single latent factor. All m/z features (excluding noise and isotopically linked m/z features) were included in the calculation of VIP rankings and the graphing of the separation of groups by latent factors
Figure 5B: A PLS-DA model using 1 component with 35 m/z features was fit to the training data obtained from randomly partitioning the data 100 times into training and test sets. The table below shows the average performance of the fitted models over the 100 test sets.
Figure 6.

Spectral intensities of metabolite features of interest that differ between groups in the hippocampus of offspring exposed to CSE throughout development (GD1-PD21). Features of interest are subdivided into four categories based on polarity and directionality of impact.
Tentative identification of 11 of the features of interest based on accurate mass, isotopic abundance agreement with predicted values, and limited structural validation are listed in Table 3. Nine of the eleven features are lipid species. The 11 features were imported into MetaboAnalyst for pathway elucidation. Impacted pathways (based on uncorrected p-value scores) included arachidonic acid metabolism, glycerophospholipid metabolism, and sphingolipid metabolism (Figure 7).
Table 3.
Putative identification of metabolite features contributing to the separation of groups in the hippocampus metabolome profiles of offspring exposed to CSE throughout development (GD1-PD21).
| Neutral Mass | Mass dev (ppm) | Structure | ID | Metlin ID | Functional Group Validated (Y/N) |
|---|---|---|---|---|---|
| Positive Polarity | |||||
| 203.1152 | 0.33 | C9H17NO4 | L-acetylcarnitine | 956 | N |
| 257.2351 | 1.06 | C15H31NO2 | Sphingosine | 64626 | Y |
| 267.0962 | 0.34 | C10H13N5O4 | Adenosine | 86 | N |
| 730.6006 | 3.26 | C41H83N2O6P | SM (d18:0/18:1) | 41584 | Y |
| 733.5641 | 3.57 | C40H80NO8P | Dipalmitoylphosphatidylcholine | 39306 | Y |
| 759.5799 | 3.58 | C42H82NO8P | PC 34:1 | 39118 | Y |
| 761.5951 | 3.05 | C42H84NO8P | PC 34:0 | 39142 | Y |
| 835.5385 | 3.36 | C46H78NO10P | PS 40:6 | 40825 | Y |
| 829.5656 | 4.92 | C48H80NO8P | PC 40:8 | 39688 | Y |
| Negative Polarity | |||||
| 274.0458 | 0.78 | C7H15O9P | Heptulose-2-phosphate | 66168 | N |
| 304.2406 | 0.90 | C20H32O2 | Arachidonic Acid | 24087 | N |
Figure 7. Metabolite features with putative identifications were analyzed for potential impact on meabolic pathways.

Arachidonic acid, glycerophospholipid, and sphingolipid metabolism pathways were impacted in the hippocampus of offspring exposed to CSE throughout development (GD1-PD21).
4. Discussion
The current study was designed to assess the impact of developmental CSE on the biomolecular phenotype of the hippocampus in an animal model previously found to exhibit hippocampus dependent neurobehavioral alterations. In humans, the structural development of hippocampus is completed prenatally with the functional maturation continuing into infancy (Arnold and Trojanowski 1996a, b). However in the mouse, both structural and functional development continues postnatally with structural maturation not complete until between 2 and 3 weeks of age (Arnold and Trojanowski 1996a; Bayer 1980a, b; Grove and Tole 1999; Tole et al. 1997; Tole and Grove 2001; Woodhams et al. 1989; Woodhams and Webb 1989). The postnatal development period, critical for the mouse to establish neural differentiation and gene expression patterns, corresponds to the third trimester of human fetal brain growth (Dobbing 1981; Dobbing and Sands 1981; Morgane et al. 2002; Romijn et al. 1991).
Cigarette smoke is a strong oxidizing agent that induces a state of systemic oxidative stress. Previous studies have documented that smokers possess increased markers of oxidative stress and reduced antioxidant levels in serum (Ozguner et al. 2005). Though the hippocampus is not heavily vascularized, chronic CSE increases markers of oxidative stress in the various brain structures (Ho et al. 2012). As reported in our previous studies concerning the impact of developmental CSE on tissue proteomes, the expression and activity of antioxidant enzymes was impacted in liver and kidney tissue of the identical offspring utilized in the current study indicating a state of tissue oxidative stress (Canales et al. 2012; Jagadapillai et al. 2012). In the hippocampus of these offspring, alterations in antioxidant protein expression were not noted, however a trend toward decreases in total glutathione (GSH + 2GSSG; p=0.09), reduced glutathione (GSH, p=0.09), and glutathione-S-transferase activity (15% reduction, p=0.07), and HSP60 precursor protein abundance were found, along with increased abundances of stress response/chaperone proteins including cyclophilin and HSP78. These findings are interpreted as further evidence of an ongoing systemic oxidative stress. CSE-induced oxidative stress leads to DNA, lipid, and protein oxidation with the eventual outcome being compromised cellular function (Bernhard and Wang 2007).
Cell morphology and migration during morphogenesis, as well as tissue repair and regeneration, is dependent on precisely timed cytoskeletal reorganization. In the current study, increased abundances of several cytoskeletal proteins were found, including myelin basic protein, annexin A5, cofilin, and α-tubulin Mα2 coupled to an increased abundance of Sirt2, a microtubule deacetylase. Together, these findings suggest aberrant timing of cytoskeletal reorganization in the hippocampus that could reflect atypical cell and nerve sheath patterning, or accelerated growth to compensate for delays in cellular patterning associated with the low birth weight phenotype induced by developmental CSE. Coupled with findings of altered arachidonic acid, glycerolipid and sphingolipid metabolism, these data suggest that hippocampus cell morphology and membrane fluidity are altered by developmental CSE.
In the hippocampus of the CSE group, several proteins involved in the glycolysis pathway were altered. Aldolase C and pyruvate kinase were decreased in abundance, while phosphoglycerate mutase 1 was increased in abundance. Pyruvate kinase serves as a regulatory enzyme in gluconeogenesis though it and phosphoglycerate mutase are part of the glycolysis cycle. The liver is the primary site of gluconeogenesis with transport of glucose through the serum and across the blood brain barrier. Coupled with prior findings of suppressed gluconeogenic activity in the liver of these offspring (Canales et al. 2012), and an ~13% decrease in fed blood glucose levels at PD21, these findings indicate that developmental CSE depresses tissue glucose availability and reflect a mild hypoglycemia.
The oxidative phosphorylation pathway in the hippocampus was also affected by developmental CSE. ATP synthase (H+ transporting, mitochondrial F1 complex, alpha subunit 1) and Vacuolar H+ATPase B2 were decreased in abundance. In contrast, ATP synthase, (H+ transporting, mitochondrial F1 complex, O subunit) and aconitase 2 were increased in abundance. Aconitase 2, which converts citrate to isocitrate in the tricarboxylic acid cycle, generates NADH for the oxidative phosphorylation pathway. Oxidative phosphorylation, rather than glycolysis, is involved in powering the presynaptic and postsynaptic mechanisms controlling brain information processing (Hall et al. 2012). Coupled with the impact on glucose availability, these findings indicate that developmental CSE impacts hippocampus mitochondrial dependent energy supply pathways.
The abundance of two proteins [Acetyl-Coenzyme A acetyltransferase 1 (ACAT; Spot 94) and Isovaleryl coenzyme A dehydrogenase (leucine catabolism; Spot 59)] contributing to branched chain amino acid catabolism were increased in the hippocampus of the CSE offspring. ACAT, a thiolase, plays a key role in multiple biochemical pathways including the fatty acid β-oxidation pathway. Together with the suppression of cellular energy supply pathways, it seems probable that hippocampus mitochondrial function is impaired by developmental CSE.
In summary, the current study examined the impact of developmental CSE spanning GD1 through PD21 on the global proteome and metabolome profiles of hippocampus tissue at the time of cessation of insult. The current study examined the impact of aged, dilute cigarette smoke as a complex mixture in reference to filtered air and as such does not differentiate impact based on nicotine, combustion byproducts such as carbon monoxide, or particulate matter. Developmental CSE induced systemic oxidative stress and impaired hippocampus cell structure proteins and energy supply availability. In conjunction with our prior studies of the impact of developmental CSE on the kidney and liver proteome profiles, we propose that suppression of gluconeogenesis within the CSE offspring liver, with attempted compensatory glucose production by the kidney, is insufficient to support the normal level energetic processes within the hippocampus. Coupled with the findings of developmental CSE (GD1-PD21) as causative of systemic oxidative stress, we propose that a lack of antioxidant balance and mild hypoglycemia impair the functional development of the hippocampus. The question of whether these alterations in the hippocampus biomolecular phenotype persist past the cessation of exposure, and the import of ongoing hypoglycemia in the development of cognitive and behavioral abnormalities in these offspring, is the subject of future studies.
Supplementary Material
Highlights.
Developmental CSE (GD1-PD21)
Hippocampus proteome alterations
Hippocampus metabolome alterations
Acknowledgments
Research in the present study was supported in part by a COBRE grant (P20 GM103453-10) from the National Institute of General Medical Sciences at the National Institutes of Health, by PHS grant NIH R21DA027466, and by the University of Louisville CREAM Center from NSF EPSCoR grant EPS-0447479.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- 2012 Current cigarette smoking among adults - United States. MMWR Morb Mortal Wkly Rep. 2011;61:889–894. [PubMed] [Google Scholar]
- Abel EL. Smoking during pregnancy: a review of effects on growth and development of offspring. Hum Biol. 1980;52:593–625. [PubMed] [Google Scholar]
- Ajarem JS, Ahmad M. Prenatal nicotine exposure modifies behavior of mice through early development. Pharmacol Biochem Behav. 1998;59:313–318. doi: 10.1016/s0091-3057(97)00408-5. [DOI] [PubMed] [Google Scholar]
- Amos-Kroohs RM, Williams MT, Braun AA, Graham DL, Webb CL, Birtles TS, Greene RM, Vorhees CV, Pisano MM. Neurobehavioral phenotype of C57BL/6J mice prenatally and neonatally exposed to cigarette smoke. Neurotoxicol Teratol. 2013;35:34–45. doi: 10.1016/j.ntt.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold SE, Trojanowski JQ. Human fetal hippocampal development: I. Cytoarchitecture, myeloarchitecture, and neuronal morphologic features. J Comp Neurol. 1996a;367:274–292. doi: 10.1002/(SICI)1096-9861(19960401)367:2<274::AID-CNE9>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Arnold SE, Trojanowski JQ. Human fetal hippocampal development: II. The neuronal cytoskeleton. J Comp Neurol. 1996b;367:293–307. doi: 10.1002/(SICI)1096-9861(19960401)367:2<293::AID-CNE10>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Batstra L, Hadders-Algra M, Neeleman J. Effect of antenatal exposure to maternal smoking on behavioural problems and academic achievement in childhood: prospective evidence from a Dutch birth cohort. Early Hum Dev. 2003;75:21–33. doi: 10.1016/j.earlhumdev.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Bayer SA. Development of the hippocampal region in the rat. I. Neurogenesis examined with 3H-thymidine autoradiography. J Comp Neurol. 1980a;190:87–114. doi: 10.1002/cne.901900107. [DOI] [PubMed] [Google Scholar]
- Bayer SA. Development of the hippocampal region in the rat. II. Morphogenesis during embryonic and early postnatal life. J Comp Neurol. 1980b;190:115–134. doi: 10.1002/cne.901900108. [DOI] [PubMed] [Google Scholar]
- Bernhard D, Wang XL. Smoking, oxidative stress and cardiovascular diseases - Do anti-oxidative therapies fail? Curr Med Chem. 2007;14:1703–1712. doi: 10.2174/092986707781058959. [DOI] [PubMed] [Google Scholar]
- Borgerding M, Klus H. Analysis of complex mixtures--cigarette smoke. Exp Toxicol Pathol. 2005;57(Suppl 1):43–73. doi: 10.1016/j.etp.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Botero D, Lifshitz F. Intrauterine growth retardation and long-term effects on growth. Curr Opin Pediatr. 1999;11:340–347. doi: 10.1097/00008480-199908000-00013. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Butler NR, Goldstei H. Smoking in Pregnancy and Subsequent Child Development. Br Med J. 1973;4:573–575. doi: 10.1136/bmj.4.5892.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler NR, Goldstein H. Smoking in pregnancy and subsequent child development. Br Med J. 1973;4:573–575. doi: 10.1136/bmj.4.5892.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canales L, Chen J, Kelty E, Musah S, Webb C, Pisano MM, Neal RE. Developmental cigarette smoke exposure: liver proteome profile alterations in low birth weight pups. Toxicology. 2012;300:1–11. doi: 10.1016/j.tox.2012.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candiano G, Bruschi M, Musante L, Santucci L, Ghiggeri GM, Carnemolla B, Orecchia P, Zardi L, Righetti PG. Blue silver: a very sensitive colloidal Coomassie G-250 staining for proteome analysis. Electrophoresis. 2004;25:1327–1333. doi: 10.1002/elps.200305844. [DOI] [PubMed] [Google Scholar]
- Cooke RW. Smoking, intra-uterine growth retardation and sudden infant death syndrome. Int J Epidemiol. 1998;27:238–241. doi: 10.1093/ije/27.2.238. [DOI] [PubMed] [Google Scholar]
- Cornelius MD, Day NL. The effects of tobacco use during and after pregnancy on exposed children. Alcohol Res Health. 2000;24:242–249. [PMC free article] [PubMed] [Google Scholar]
- d’Hellencourt CL, Harry GJ. Molecular profiles of mRNA levels in laser capture microdissected murine hippocampal regions differentially responsive to TMT-induced cell death. Journal of Neurochemistry. 2005;93:206–220. doi: 10.1111/j.1471-4159.2004.03017.x. [DOI] [PubMed] [Google Scholar]
- Das UG, Sysyn GD. Abnormal fetal growth: intrauterine growth retardation, small for gestational age, large for gestational age. Pediatr Clin North Am. 2004;51:639–654. viii. doi: 10.1016/j.pcl.2004.01.004. [DOI] [PubMed] [Google Scholar]
- DiFranza JR, Aligne CA, Weitzman M. Prenatal and postnatal environmental tobacco smoke exposure and children’s health. Pediatrics. 2004;113:1007–1015. [PubMed] [Google Scholar]
- Dobbing J. The Development of the Brain. Developmental Medicine and Child Neurology. 1981;23:101–101. [Google Scholar]
- Dobbing J, Sands J. Vulnerability of Developing Brain Not Explained by Cell Number Cell-Size Hypothesis. Early Human Development. 1981;5:227–231. doi: 10.1016/0378-3782(81)90030-x. [DOI] [PubMed] [Google Scholar]
- Eichenbaum H. The hippocampus and mechanisms of declarative memory. Behavioural Brain Research. 1999;103:123–133. doi: 10.1016/s0166-4328(99)00044-3. [DOI] [PubMed] [Google Scholar]
- Eichenbaum H. Hippocampus: Cognitive processes and neural representations that underlie declarative memory. Neuron. 2004a;44:109–120. doi: 10.1016/j.neuron.2004.08.028. [DOI] [PubMed] [Google Scholar]
- Eichenbaum H. An Information Processing Framework for Memory Representation by the Hippocampus. Cognitive Neurosciences Iii. (3) 2004b:679–690. [Google Scholar]
- Ernst M, Moolchan ET, Robinson ML. Behavioral and neural consequences of prenatal exposure to nicotine. J Am Acad Child Adolesc Psychiatry. 2001;40:630–641. doi: 10.1097/00004583-200106000-00007. [DOI] [PubMed] [Google Scholar]
- Esposito ER, Horn KH, Greene RM, Pisano MM. An animal model of cigarette smoke-induced in utero growth retardation. Toxicology. 2008;246:193–202. doi: 10.1016/j.tox.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin NJ, Wright SP, Eichenbaum H. Recollection-like memory retrieval in rats is dependent on the hippocampus. Nature. 2004;431:188–191. doi: 10.1038/nature02853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fried PA, Makin JE. Neonatal behavioural correlates of prenatal exposure to marihuana, cigarettes and alcohol in a low risk population. Neurotoxicol Teratol. 1987;9:1–7. doi: 10.1016/0892-0362(87)90062-6. [DOI] [PubMed] [Google Scholar]
- Fried PA, Watkinson B. Visuoperceptual functioning differs in 9- to 12-year olds prenatally exposed to cigarettes and marihuana. Neurotoxicol Teratol. 2000;22:11–20. doi: 10.1016/s0892-0362(99)00046-x. [DOI] [PubMed] [Google Scholar]
- Fung YK. Postnatal behavioural effects of maternal nicotine exposure in rats. J Pharm Pharmacol. 1988;40:870–872. doi: 10.1111/j.2042-7158.1988.tb06290.x. [DOI] [PubMed] [Google Scholar]
- Genedani S, Bernardi M, Bertolini A. Sex-linked differences in avoidance learning in the offspring of rats treated with nicotine during pregnancy. Psychopharmacology (Berl) 1983;80:93–95. doi: 10.1007/BF00427504. [DOI] [PubMed] [Google Scholar]
- Gluckman PD, Hanson MA, Morton SM, Pinal CS. Life-long echoes--a critical analysis of the developmental origins of adult disease model. Biol Neonate. 2005;87:127–139. doi: 10.1159/000082311. [DOI] [PubMed] [Google Scholar]
- Gospe SM, Jr, Zhou SS, Pinkerton KE. Effects of environmental tobacco smoke exposure in utero and/or postnatally on brain development. Pediatr Res. 1996;39:494–498. doi: 10.1203/00006450-199603000-00018. [DOI] [PubMed] [Google Scholar]
- Grove EA, Tole S. Patterning events and specification signals in the developing hippocampus. Cereb Cortex. 1999;9:551–561. doi: 10.1093/cercor/9.6.551. [DOI] [PubMed] [Google Scholar]
- Hall CN, Klein-Flugge MC, Howarth C, Attwell D. Oxidative Phosphorylation, Not Glycolysis, Powers Presynaptic and Postsynaptic Mechanisms Underlying Brain Information Processing. Journal of Neuroscience. 2012;32:8940–8951. doi: 10.1523/JNEUROSCI.0026-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond RS, Tull LE, Stackman RW. On the delay-dependent involvement of the hippocampus in object recognition memory. Neurobiol Learn Mem. 2004;82:26–34. doi: 10.1016/j.nlm.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Ho JWM, Ho PWL, Liu HF, So DHF, Chan KH, Tse ZHM, Kung MHW, Ramsden DB, Ho SL. UCP4 is a target effector of the NF-kappa B c-Rel prosurvival pathway against oxidative stress. Free Radical Bio Med. 2012;53:383–394. doi: 10.1016/j.freeradbiomed.2012.05.002. [DOI] [PubMed] [Google Scholar]
- Jagadapillai R, Chen J, Canales L, Birtles T, Pisano MM, Neal RE. Developmental cigarette smoke exposure: kidney proteome profile alterations in low birth weight pups. Toxicology. 2012;299:80–89. doi: 10.1016/j.tox.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang MH, Shin MC, Jung SB, Lee TH, Bahn GH, Kwon YK, Kim EH, Kim CJ. Alcohol and nicotine reduce cell proliferation and enhance apoptosis in dentate gyrus. Neuroreport. 2002;13:1509–1513. doi: 10.1097/00001756-200208270-00004. [DOI] [PubMed] [Google Scholar]
- Jarvis MJ, Fidler J, Mindell J, Feyerabend C, West R. Assessing smoking status in children, adolescents and adults: cotinine cut-points revisited. Addiction. 2008;103:1553–1561. doi: 10.1111/j.1360-0443.2008.02297.x. [DOI] [PubMed] [Google Scholar]
- Karp NA, Griffin JL, Lilley KS. Application of partial least squares discriminant analysis to two-dimensional difference gel studies in expression proteomics. Proteomics. 2005;5:81–90. doi: 10.1002/pmic.200400881. [DOI] [PubMed] [Google Scholar]
- Koehl M, Bjijou Y, Le Moal M, Cador M. Nicotine-induced locomotor activity is increased by preexposure of rats to prenatal stress. Brain Res. 2000;882:196–200. doi: 10.1016/s0006-8993(00)02803-1. [DOI] [PubMed] [Google Scholar]
- Koren G, Klein J, Forman R, Graham K, Phan MK. Biological markers of intrauterine exposure to cocaine and cigarette smoking. Dev Pharmacol Ther. 1992;18:228–236. [PubMed] [Google Scholar]
- Koren G, Pastuszak A, Ito S. Drugs in pregnancy. N Engl J Med. 1998;338:1128–1137. doi: 10.1056/NEJM199804163381607. [DOI] [PubMed] [Google Scholar]
- Kuhn M. Building Predictive Models in R Using the caret Package. J Stat Softw. 2008;28:1–26. [Google Scholar]
- Levenson JM, Sweatt JD. Epigenetic mechanisms in memory formation. Nat Rev Neurosci. 2005;6:108–118. doi: 10.1038/nrn1604. [DOI] [PubMed] [Google Scholar]
- Levin E, Slotkin T. Developmental neurotoxicity of nicotine. In: Slikker W, Chang LW, editors. Handbook of Developmental Neurotoxicology. Academic Press; San Diego: 1998. pp. 587–615. [Google Scholar]
- Levin ED, Briggs SJ, Christopher NC, Rose JE. Prenatal nicotine exposure and cognitive performance in rats. Neurotoxicol Teratol. 1993;15:251–260. doi: 10.1016/0892-0362(93)90006-a. [DOI] [PubMed] [Google Scholar]
- Levin ED, Wilkerson A, Jones JP, Christopher NC, Briggs SJ. Prenatal nicotine effects on memory in rats: pharmacological and behavioral challenges. Brain Res Dev Brain Res. 1996;97:207–215. doi: 10.1016/s0165-3806(96)00144-7. [DOI] [PubMed] [Google Scholar]
- Martin JC, Becker RF. The effects of maternal nicotine absorption or hypoxic episodes upon appetitive behavior of rat offspring. Dev Psychobiol. 1971;4:133–147. doi: 10.1002/dev.420040205. [DOI] [PubMed] [Google Scholar]
- McCartney JS, Fried PA, Watkinson B. Central auditory processing in school-age children prenatally exposed to cigarette smoke. Neurotoxicol Teratol. 1994;16:269–276. doi: 10.1016/0892-0362(94)90048-5. [DOI] [PubMed] [Google Scholar]
- Mitchell EA, Thompson JM, Robinson E, Wild CJ, Becroft DM, Clark PM, Glavish N, Pattison NS, Pryor JE. Smoking, nicotine and tar and risk of small for gestational age babies. Acta Paediatr. 2002;91:323–328. doi: 10.1080/08035250252834003. [DOI] [PubMed] [Google Scholar]
- Morgane PJ, Mokler DJ, Galler JR. Effects of prenatal protein malnutrition on the hippocampal formation. Neurosci Biobehav R. 2002;26:471–483. doi: 10.1016/s0149-7634(02)00012-x. [DOI] [PubMed] [Google Scholar]
- Nagahara AH, Handa RJ. Loss of nicotine-induced effects on locomotor activity in fetal alcohol-exposed rats. Neurotoxicol Teratol. 1999;21:647–652. doi: 10.1016/s0892-0362(99)00040-9. [DOI] [PubMed] [Google Scholar]
- Navarro HA, Seidler FJ, Schwartz RD, Baker FE, Dobbins SS, Slotkin TA. Prenatal exposure to nicotine impairs nervous system development at a dose which does not affect viability or growth. Brain Res Bull. 1989;23:187–192. doi: 10.1016/0361-9230(89)90146-9. [DOI] [PubMed] [Google Scholar]
- Neuhoff V, Stamm R, Pardowitz I, Arold N, Ehrhardt W, Taube D. Essential problems in quantification of proteins following colloidal staining with coomassie brilliant blue dyes in polyacrylamide gels, and their solution. Electrophoresis. 1990;11:101–117. doi: 10.1002/elps.1150110202. [DOI] [PubMed] [Google Scholar]
- Newman MB, Shytle RD, Sanberg PR. Locomotor behavioral effects of prenatal and postnatal nicotine exposure in rat offspring. Behav Pharmacol. 1999;10:699–706. doi: 10.1097/00008877-199911000-00017. [DOI] [PubMed] [Google Scholar]
- Olds DL, Henderson CR, Jr, Tatelbaum R. Intellectual impairment in children of women who smoke cigarettes during pregnancy. Pediatrics. 1994a;93:221–227. [PubMed] [Google Scholar]
- Olds DL, Henderson CR, Jr, Tatelbaum R. Prevention of intellectual impairment in children of women who smoke cigarettes during pregnancy. Pediatrics. 1994b;93:228–233. [PubMed] [Google Scholar]
- Olds DL, Henderson CR, Tatelbaum R. Intellectual Impairment in Children of Women Who Smoke Cigarettes during Pregnancy. Pediatrics. 1994c;93:221–227. [PubMed] [Google Scholar]
- Ozguner F, Koyu A, Cesur G. Active smoking causes oxidative stress and decreases blood melatonin levels. Toxicology and Industrial Health. 2005;21:21–26. doi: 10.1191/0748233705th211oa. [DOI] [PubMed] [Google Scholar]
- Pappin DJ, Hojrup P, Bleasby AJ. Rapid identification of proteins by peptide-mass fingerprinting. Curr Biol. 1993;3:327–332. doi: 10.1016/0960-9822(93)90195-t. [DOI] [PubMed] [Google Scholar]
- Peters MA, Ngan LL. The effects of totigestational exposure to nicotine on pre- and postnatal development in the rat. Arch Int Pharmacodyn Ther. 1982;257:155–167. [PubMed] [Google Scholar]
- Rodgman A, Perfetti TA. Summary. The Chemical Components of Tobacco and Tobacco Smoke. 2. CRC Press; 2013. pp. 1471–1474. [Google Scholar]
- Romijn HJ, Hofman MA, Gramsbergen A. At What Age Is the Developing Cerebral-Cortex of the Rat Comparable to That of the Full-Term Newborn Human Baby. Early Human Development. 1991;26:61–67. doi: 10.1016/0378-3782(91)90044-4. [DOI] [PubMed] [Google Scholar]
- Roy TS, Andrews JE, Seidler FJ, Slotkin TA. Nicotine evokes cell death in embryonic rat brain during neurulation. J Pharmacol Exp Ther. 1998;287:1136–1144. [PubMed] [Google Scholar]
- Roy TS, Seidler FJ, Slotkin TA. Prenatal nicotine exposure evokes alterations of cell structure in hippocampus and somatosensory cortex. J Pharmacol Exp Ther. 2002;300:124–133. doi: 10.1124/jpet.300.1.124. [DOI] [PubMed] [Google Scholar]
- Sexton M, Fox NL, Hebel JR. Prenatal exposure to tobacco: II. Effects on cognitive functioning at age three. Int J Epidemiol. 1990;19:72–77. doi: 10.1093/ije/19.1.72. [DOI] [PubMed] [Google Scholar]
- Shacka JJ, Fennell OB, Robinson SE. Prenatal nicotine sex-dependently alters agonist-induced locomotion and stereotypy. Neurotoxicol Teratol. 1997;19:467–476. doi: 10.1016/s0892-0362(97)00063-9. [DOI] [PubMed] [Google Scholar]
- Shapiro ML, Eichenbaum H. Hippocampus as a memory map: Synaptic plasticity and memory encoding by hippocampal neurons. Hippocampus. 1999;9:365–384. doi: 10.1002/(SICI)1098-1063(1999)9:4<365::AID-HIPO4>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Slawecki CJ, Ehlers CL. Lasting effects of adolescent nicotine exposure on the electroencephalogram, event related potentials, and locomotor activity in the rat. Brain Res Dev Brain Res. 2002;138:15–25. doi: 10.1016/s0165-3806(02)00455-8. [DOI] [PubMed] [Google Scholar]
- Slawecki CJ, Thomas JD, Riley EP, Ehlers CL. Neonatal nicotine exposure alters hippocampal EEG and event-related potentials (ERPs) in rats. Pharmacol Biochem Behav. 2000;65:711–718. doi: 10.1016/s0091-3057(99)00258-0. [DOI] [PubMed] [Google Scholar]
- Slikker W, Jr, Xu ZA, Levin ED, Slotkin TA. Mode of action: disruption of brain cell replication, second messenger, and neurotransmitter systems during development leading to cognitive dysfunction--developmental neurotoxicity of nicotine. Crit Rev Toxicol. 2005;35:703–711. doi: 10.1080/10408440591007421. [DOI] [PubMed] [Google Scholar]
- Slotkin T. Prenatal exposure to nicotine: What can we learn from animal models. In: Zagon IS, Slotkin TA, editors. Maternal Substance Abuse and the Developing Nervous System. Academic Press; San Diego: 1992. pp. 97–124. [Google Scholar]
- Slotkin TA. Developmental cholinotoxicants: nicotine and chlorpyrifos. Environ Health Perspect. 1999;107(Suppl 1):71–80. doi: 10.1289/ehp.99107s171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotkin TA, Greer N, Faust J, Cho H, Seidler FJ. Effects of maternal nicotine injections on brain development in the rat: ornithine decarboxylase activity, nucleic acids and proteins in discrete brain regions. Brain Res Bull. 1986;17:41–50. doi: 10.1016/0361-9230(86)90159-0. [DOI] [PubMed] [Google Scholar]
- Slotkin TA, McCook EC, Seidler FJ. Cryptic brain cell injury caused by fetal nicotine exposure is associated with persistent elevations of c-fos protooncogene expression. Brain Res. 1997;750:180–188. doi: 10.1016/s0006-8993(96)01345-5. [DOI] [PubMed] [Google Scholar]
- Slotkin TA, Orband-Miller L, Queen KL, Whitmore WL, Seidler FJ. Effects of prenatal nicotine exposure on biochemical development of rat brain regions: maternal drug infusions via osmotic minipumps. J Pharmacol Exp Ther. 1987;240:602–611. [PubMed] [Google Scholar]
- Slotkin TA, Pinkerton KE, Auman JT, Qiao D, Seidler FJ. Perinatal exposure to environmental tobacco smoke upregulates nicotinic cholinergic receptors in monkey brain. Brain Res Dev Brain Res. 2002;133:175–179. doi: 10.1016/s0165-3806(02)00281-x. [DOI] [PubMed] [Google Scholar]
- Sobrian SK, Marr L, Ressman K. Prenatal cocaine and/or nicotine exposure produces depression and anxiety in aging rats. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:501–518. doi: 10.1016/S0278-5846(03)00042-3. [DOI] [PubMed] [Google Scholar]
- Sorenson CA, Raskin LA, Suh Y. The effects of prenatal nicotine on radial-arm maze performance in rats. Pharmacol Biochem Behav. 1991;40:991–993. doi: 10.1016/0091-3057(91)90117-k. [DOI] [PubMed] [Google Scholar]
- Sweatt JD. Hippocampal function in cognition. Psychopharmacology. 2004;174:99–110. doi: 10.1007/s00213-004-1795-9. [DOI] [PubMed] [Google Scholar]
- Tole S, Christian C, Grove EA. Early specification and autonomous development of cortical fields in the mouse hippocampus. Development. 1997;124:4959–4970. doi: 10.1242/dev.124.24.4959. [DOI] [PubMed] [Google Scholar]
- Tole S, Grove EA. Detailed field pattern is intrinsic to the embryonic mouse hippocampus early in neurogenesis. J Neurosci. 2001;21:1580–1589. doi: 10.1523/JNEUROSCI.21-05-01580.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaglenova J, Birru S, Pandiella NM, Breese CR. An assessment of the long-term developmental and behavioral teratogenicity of prenatal nicotine exposure. Behav Brain Res. 2004;150:159–170. doi: 10.1016/j.bbr.2003.07.005. [DOI] [PubMed] [Google Scholar]
- Whishaw IQ, Jarrard LE. Evidence for extrahippocampal involvement in place learning and hippocampal involvement in path integration. Hippocampus. 1996;6:513–524. doi: 10.1002/(SICI)1098-1063(1996)6:5<513::AID-HIPO4>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Woodhams PL, Calvert R, Dunnett SB. Monoclonal antibody G10 against microtubule-associated protein 1x distinguishes between growing and regenerating axons. Neuroscience. 1989;28:49–59. doi: 10.1016/0306-4522(89)90231-5. [DOI] [PubMed] [Google Scholar]
- Woodhams PL, Webb M. A developmentally regulated axonal glycoprotein (7-8D2 antigen) with a restricted distribution in mature rat brain. Neuroscience. 1989;32:417–434. doi: 10.1016/0306-4522(89)90090-0. [DOI] [PubMed] [Google Scholar]
- Xia JG, Mandal R, Sinelnikov IV, Broadhurst D, Wishart DS. MetaboAnalyst 2.0-a comprehensive server for metabolomic data analysis. Nucleic Acids Research. 2012;40:W127–W133. doi: 10.1093/nar/gks374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia JG, Wishart DS. Web-based inference of biological patterns, functions and pathways from metabolomic data using MetaboAnalyst. Nat Protoc. 2011;6:743–760. doi: 10.1038/nprot.2011.319. [DOI] [PubMed] [Google Scholar]
- Yamaguchi S, Hale LA, D’Esposito M, Knight RT. Rapid prefrontal-hippocampal habituation to novel events. Journal of Neuroscience. 2004;24:5356–5363. doi: 10.1523/JNEUROSCI.4587-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Reece EA. Nicotine-induced embryonic malformations mediated by apoptosis from increasing intracellular calcium and oxidative stress. Birth Defects Res B Dev Reprod Toxicol. 2005;74:383–391. doi: 10.1002/bdrb.20052. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.