

Abstract
Lung cancer remains to be one of the most common and serious types of cancer worldwide. While treatment is available, the survival rate of this cancer is still critically low due to late stage diagnosis and high frequency of drug resistance, thus highlighting the pressing need for a greater understanding of the molecular mechanisms involved in lung carcinogenesis. Studies in the past years have evidenced that microRNAs (miRNAs) are critical players in the regulation of various biological functions, including apoptosis, which is a process frequently evaded in cancer progression. Recently, miRNAs were demonstrated to possess proapoptotic or antiapoptotic abilities through the targeting of oncogenes or tumor suppressor genes. This review examines the involvement of miRNAs in the apoptotic process of lung cancer and will also touch on the promising evidence supporting the role of miRNAs in regulating sensitivity to anticancer treatment.
1. Introduction
Lung cancer remains a major health problem worldwide. In 2012 lung cancer was the most commonly diagnosed cancer worldwide making up 13.0% of the total incidence of cancer. It was also the most common cause of death from cancer worldwide, accounting for nearly one in five cancer deaths (19.4% of the total) [1]. Lung cancer is clinically divided into two main groups based upon the size and appearance of malignant cells: small cell lung cancer (SCLC) (16.8%) and non-small cell lung cancer (NSCLC) (80.4%) [2]. The most effective option for treatment of lung cancer is surgical resection, when feasible [3]. However, majority of patients are diagnosed at an advanced or metastatic stage of disease in which case chemotherapy and/or concurrent administration of chemotherapy and radiation is the most beneficial form of treatment [4]. Nevertheless, even with treatment, the 5-year survival rate in patients is only 16.6% [5], with poor survival rates mainly being attributed to late stage diagnosis and high frequency of drug resistance. Obtaining a better understanding regarding the molecular mechanisms involved in lung carcinogenesis is of utmost importance in the aim to identify the diagnostic and prognostic markers for early detection and targeted treatment of lung cancer.
Apoptosis plays an important role during development and in the maintenance of multicellular organisms through the removal of damaged, aged, or autoimmune cells [6]. The apoptotic process can be divided into the extrinsic and intrinsic pathway. Each pathway will ultimately result in the activation of cell death proteases, which in turn initiates a cascade of proteolysis involving effector caspases that carries out the completion of the apoptotic process [7]. In contrast to normal cells, cancer cells have the ability to evade apoptosis to promote cell survival under the conditions of environmental stress. There are a number of mechanisms by which cancer cells are able to suppress apoptosis. For example, the tumor suppressor gene p53 is a widely mutated gene in human tumorigenesis [8]. p53 mutation will inhibit the activation of DNA repair proteins leading to a decrease in the initiation of apoptosis [7], allowing for cells to divide and grow uncontrollably, forming malignant tumors. Furthermore, cancer cells are able to disrupt the balance between pro- (BCL-2, BCL-XL) and antiapoptotic factors (BAX, BIM, and PUMA) [9]. Increased expression of proapoptotic Bcl-2 protein contributes not only to the development of cancer but also to resistance against a wide variety of anticancer agents, such as cisplatin (DDP) and paclitaxel [10–12].
MicroRNAs (miRNAs) are a subset of noncoding RNAs of about 20 to 25 nucleotides long which posttranscriptionally regulate gene expression via inhibition of mRNA translation, by binding to specific target sites in their 3′-untranslated region (3′UTR), or inducing degradation of target mRNA through cleavage [13]. An individual miRNA is able to modulate the expression of multiple genes; correspondingly, a single target can be modulated by many miRNAs [14]. MiRNAs were reported to be involved in a vast range of biological processes, including apoptosis (see Figure 1) [15–22]. As miRNAs play a key role in an assortment of biological processes, an altered miRNA expression is likely to contribute to human diseases including cancer [23]. Previous studies have shown that compared to normal tissues, malignant tumors and tumor cell lines were found to have widespread deregulated miRNA expression [24–28]. MiRNAs are critical apoptosis regulators in tumorigenesis and cancer cells are able to manipulate miRNAs to regulate cell survival in oncogenesis. Many studies carried out in the past several years are aimed at elucidating the specific miRNAs associated with apoptosis in cancer and their related target genes. In this review we will examine the recent progress of research on miRNA-mediated regulation of apoptosis in lung cancer and its future therapeutic applications.
Figure 1.
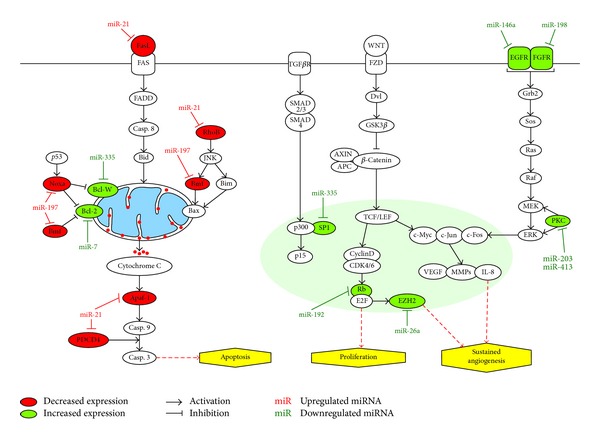
Scheme depicting up- and downregulated miRNAs and the roles they play in various biological pathways including apoptosis, proliferation, and angiogenesis.
2. Antiapoptotic miRNAs
Evasion of apoptosis is a significant hallmark of tumor progression, and one mechanism by which miRNAs influence development of cancer is through the regulation of the apoptotic process as shown in various studies [29–32]. miRNA expression can be either upregulated or downregulated and evidence has shown that dysregulated miRNAs can behave as oncogenes or tumor suppressor genes in lung cancers [18, 28, 33]. Amplification of miRNAs can lead to the downregulation of tumor suppressors or other genes that are involved in apoptosis [34].
miR-197. For example, the expression of miR-197 is increased in cancer tissues in comparison to normal specimens. Fiori et al. (2014) demonstrated that knockdown of miR-197 in NIH-H460 and A549 cells promoted induction of apoptosis, evident by the observation of caspases 3–7 activation and increased apoptotic population by Annexin staining. Furthermore, the direct interaction of miR-197 with the 3′UTR of BMF and NOXA was demonstrated by the luciferase reporter assay [35]. When activated by intra- or extracellular stimuli, proapoptotic Bmf binds to and neutralizes antiapoptotic Bcl-2 family members on the mitochondrial membrane, thus allowing proapoptotic proteins Bak and Bax to dimerize and promote the release of cytochrome c, ultimately leading to cell death [36]. Noxa is a BH-3 only proapoptotic protein transcriptionally activated by p53. Collectively, miR-197 is able to act upon different levels of the p53 pathway to counteract the induction of apoptosis, thus allowing cells to proliferate uncontrollably [35].
miR-21. miR-21 is found to be frequently upregulated in a number of cancers; however its potential role in tumorigenesis in vivo is not fully explored. Using transgenic mice with loss-of-function and gain-of-function miR-21 alleles, Hatley and colleagues elucidated the role of miR-21 in NSCLC pathogenesis in vivo [37]. It was determined that miR-21 regulates tumor proliferation and survival, which are two integral components of NSCLC pathogenesis, by targeting negative regulators of the RAS pathway as well as by targeting proapoptotic genes [37]. In regards to the apoptotic pathway, overexpression of miR-21 in vivo leads to decreased protein levels of Apaf-1, an important component of the intrinsic mitochondrial apoptotic pathway, as well as decreased expression of FasL, a key initiator of the extrinsic apoptotic pathway. Furthermore, RHOB, with a tumor suppressor role, is a target of miR-21 and its dysregulation leads to an increase in cell growth and inhibition of apoptosis [38]. Together these results suggest that relieving miR-21 downregulation of these proapoptotic and tumor suppressor genes could provide a means to enhance the effect of current chemotherapy.
miR-212. Acetylcholinesterase (AChE), a component of the cholinergic system, has the ability to influence apoptotic sensitivity both in vitro and in vivo [39–41]. In NSCLC tissues AChE levels are low and are associated with tumor aggressiveness, increase risk of postoperative recurrence, and low survival rate [42]. Lu et al. (2013) determined that AChE expression in NSCLC was posttranscriptionally modulated by miR-212 binding to its 3′UTR. Interestingly, alterations in neither AChE nor miR-212 expression significantly affected cell survival; however it was observed that during DDP-induced apoptosis miR-212 levels were reduced with a concurrent increase in AChE protein levels. This suggests that miR-212 plays a role in DDP resistance by directly inhibiting AChE and preventing apoptosis. Therefore, interference against miR-212 may potentially be a means to improve the pharmacotoxicological profile of DDP in NSCLC [43].
miR-17-5p and miR-20a. The miR-17-92 cluster, which is composed of seven miRNAs and resides in intron 3 of the C13orf25 gene at 13q31.3, is frequently overexpressed in lung cancers [44]. Matsubara et al. (2007) demonstrated that inhibition of two components of the miR-17-92 cluster, miR-17-5p, and miR-20a, with antisense oligonucleotides can induce apoptosis selectively in lung cancer cells that overexpress miR-17-92 [45]. Previously, miR-17-5p and miR-20a have been shown to directly target E2F1 [46]; thus inhibition of these miRNAs may cause the induction of apoptosis in part through the induction of E2F1 and subsequent cell cycle progression into S phase [47]. However additional studies would have to be carried out to determine the actual targets for the miR-17-92 cluster to gain a better understanding of the development of this cancer.
3. Proapoptotic miRNAs
MiRNAs that are downregulated are considered tumor suppressor genes. Tumor suppressor miRNAs usually prevent tumor development by negatively regulating oncogenes and/or genes that control cell differentiation or apoptosis [48]. MiRNAs that act as tumor suppressors can be downregulated as a result of deletions, epigenetic silencing, or loss of expression of transcription factors (see Table 2) [49].
Table 2.
Downregulated apoptosis-associated miRNAs in lung cancer.
| MicroRNA | Target genes | Function | Cell lines | In vivo models | Citation |
|---|---|---|---|---|---|
| miR-7 | BCL-2 | Suppress cell proliferation and induce cell apoptosis Inhibit cancer cell migration in vitro Reduce tumorigenicity in vivo |
A549, NCI-H1299, NCI-H1355, NCI-H460, MRC-5, HEK-293T |
Nude mice | [51] |
|
| |||||
| miR-198 | FGFR1 | Inhibit lung cancer cells proliferation Enhance cell apoptosis Inhibits growth of tumor graft in nude mouse |
A549, NCI-H460 |
Athymic BALB/c nude mice. | [69] |
|
| |||||
| miR-451 | RAB14 | Inhibit in vitro proliferation and enhance apoptosis Decrease phosphorylation of AKT and increased BAX or Bad protein level Associated with in vivo proliferation capacity |
A549, SPC-A1, NCI-H520 |
Athymic BALB/c nude mice. | [75] |
|
| |||||
| miR-192 | RB1 | Inhibit cell proliferation and promotes cell apoptosis Arrest cell in G1 phase Inhibit tumorigenesis in vivo |
A549, NCI-H460, 95D |
Athymic BALB/c nude mice. | [76] |
|
| |||||
| miR-335 | BCL-W, SP1 | Suppress proliferation and invasion ability of cells Induce apoptosis Suppress metastasis and invasiveness of cells |
A549, NCI-H1299 | N/T | [52] |
|
| |||||
| miR-608 | N/D | Increase cell death in Bcl-xL silenced cells | A549, SK-LU-1 | N/T | [55] |
|
| |||||
| miR-203 | PKCα | Decrease cell proliferation Promote cell apoptosis, but this effect only partially relies on its downregulation of PKCα |
A549 | N/T | [61] |
|
| |||||
| miR-413 | PKCε | Inhibit cell proliferation and enhance apoptosis | A549, Calu-1 | N/T | [63] |
|
| |||||
| miR-146a | EGFR | Inhibit cell growth and induces cell apoptosis Suppress motility Enhance cell proliferation inhibitory effect of TKIs and cetuximab |
NCI-H358, NCI-H1650, NCI-H1975, NCI-H292, HCC827 |
N/T | [70] |
|
| |||||
| miR-26a | EZH2 | Inhibit cell proliferation in vitroBlock G1/S phase transition and induced apoptosis Decreased metastasis capacity and invasion |
SPC-A1, A549, SK-MES-1 |
N/T | [71] |
N/D: not determined; N/T: not tested.
3.1. B-Cell Lymphocyte 2 (BCL-2) Family Related miRNAs
Members of the evolutionarily conserved BCL-2 family are thought to be the central regulators of apoptosis. The expression level of BCL-2 differs for different cell types; however high levels and aberrant patterns of BCL-2 expression were reported in a wide variety of human cancers, including lung cancer [50]. Elevation of Bcl-2 protein expression contributes not only to the development of cancer but also to resistance against a wide variety of anticancer agents [10–12].
miR-7. Xiong et al. showed that miR-7 was downregulated in NSCLC cells and BCL-2 was identified as a direct target [51]. Transfection of miR-7 in A549 cells led to a significant reduction in endogenous BCL-2 mRNA and protein levels and correspondingly led to increase in the activities of caspase-3 and caspase-7 in cells with apoptotic nuclei [51]. These results thus provide evidence that BCL-2 may be involved in miR-7 mediated apoptosis induction in A549 cells.
miR-335. BCL-W, another antiapoptotic member of the BCL-2 family, was found to be a direct target of miR-335 [52]. miR-335 was downregulated in A549 and NCI-H1299 cells, and upregulation of this miRNA via transfection of miR-335 mimics led to a suppression of cell invasiveness and promotion of apoptosis. Furthermore Dyanan and Tjian (1983) discovered that miR-335 directly targeted SP1 gene, a member of the family of Sp/Kruppel-like transfection factors [53], which can enhance the activity of promoters of numerous genes involved in cell proliferation, apoptosis, differentiation, cell cycle, progression, and oncogenesis thus regulating these genes' expression [54].
miR-608. Studies in our lab identified a BCL-XL-induced miRNA, miR-608, involved in the regulation of cell death in A549 and SK-LU-1 cells [55]. BCL-XL, a major prototype of the antiapoptotic BCL-2 gene was found to be overexpressed in NSCLCs [56]. Silencing of BCL-XL in A549 and SK-LU-1 led to the significant dysregulation of a number of miRNAs, as determined through miRNA microarray, with miR-608 being the most upregulated miRNA. Upregulation of miR-608 in A549 and SK-LU-1 via miR-608 mimics led to an increase in apoptotic population, as determined by Annexin-V FITC apoptotic assay, in comparison to NP-69 cells (normal human nasopharyngeal epithelial cell line) (see Table 1) [55]. Bioinformatics analysis determined that miR-608 may be associated with various signaling pathways, primarily the phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT), wingless-type MMTV integration site family (WNT), transforming growth factor (TGF-β), mitogen activated protein kinase (MAPK), and the intrinsic pathway. However the true targets of miR-608 and its direct effects on the apoptotic process is yet to be determined.
Table 1.
Upregulated apoptosis-associated miRNAs in lung cancer.
| MicroRNA | Target genes | Function | Cell lines | In vivo models | Citation |
|---|---|---|---|---|---|
| miR-197 | BMF, NOXA | Repress p53-dependent apoptotic cascade miR inhibition decreases cell viability miR inhibition impairs cell growth and anchorage-independent colony formation |
A549, Calu-1, NIH-H460, NCI-H1299 | Nude mice | [35] |
|
| |||||
| miR-21 | SPRY1, SPRY2, BTG2, PDCD4, APAF1, FasL, RHOB | Enhance tumor proliferation and survival Inhibit apoptosis miR deletion suppresses Ras-driven transformation |
N/T |
K-rasLA2 mice CAG-miR-21 transgenic mice CAG-miR-21; K-rasLA2 compound mutant mice |
[37] |
|
| |||||
| miR-212 | AChE | Prevent apoptosis Maintain cell proliferation capacity Modulate CDDP-induced NSCLC cell apoptosis |
HEK-293T, NCI-H520, NCI-H460, SK-MES-1, BEAS-2B |
Nude mice | [43] |
|
| |||||
| miR-17-5p, miR-20a | E2F1 | miR inhibition reduces of cell growth. miR inhibition induces apoptosis and increases proportions of sub-G1 populations. |
Calu-6, A549, ACC-LC-172 |
N/T | [45] |
N/D: not determined; N/T: not tested.
3.2. Protein Kinase C (PKC) Family Related miRNAs
PKC is a serine/threonine kinase that is involved in various signal transduction pathways including those related to cellular proliferation, differentiation, and apoptosis [57–59]. PKC plays a role in lung cancer and levels of PKC proteins were found to be increased in various cell lines (A549, NCI-H1355, NCI-H1703, NCI-H157, and NCI-H1155) in comparison to primary normal human bronchial epithelial cells (NHBE) [60].
miR-203. To determine the role that miR-203 can play in the influence of cellular function, putative target prediction was carried out and PKC-α was determined to be a target [61]. Luciferase reporter assay further revealed miRNA-203 direct binding of the 3′UTR of PKC-α mRNA transcript. miR-203 negatively regulated proliferation and migration through the repression of PKC-α, and miR-203 was also able to modulate cell apoptosis. However, siRNA silencing of PKC-α resulted in a less significant apoptotic phenotype in comparison to that observed by miR-203 overexpression, thus suggesting that miR-203 may modulate multiple apoptotic genes that work together to regulate cell apoptosis [61]. Further studies must therefore be carried out to determine the additional apoptosis related targets of miR-203.
miR-143. miR-143 expression was reported to be downregulated in cancer tissues and inhibition of miR-143 promotes cell proliferation but hinders cell apoptosis. To determine the role that miR-143 plays in the apoptotic process, Akita (2002) investigated the possible targets of miR-143 and found that PKC-ε, a crucial enzyme in various cellular signaling pathways [62], was a putative target. Using the luciferase reporter assay it was determined that miR-143 specifically targets PKC-ε, and overexpression of miR-143 increases the cell apoptosis in A549 cells [63]. PKC-ε was suggested to play a role in regulating the antiapoptotic signaling pathway through the upregulation of Bcl-2 with a concurrent suppression of proapoptotic Bid [64–66]. Furthermore, PKC-ε is able to activate Akt to apply its prosurvival effects [67, 68]. Therefore, the targeting of PKC-ε could potentially be a valuable therapeutic strategy for lung cancer.
3.3. Other miRNAs
miR-198. miR-198 is downregulated in NSCLC cell lines and overexpression of this miRNA inhibits cell viability and enhances apoptosis in A549 cells. Overexpression of miR-198 induces the expression of poly(ADP-ribose) polymerase (PARP) and of cleaved caspase-3. miR-198 was also able to inhibit growth of tumor grafts in nude mouse. FGFR1, a lung cancer oncogene, which is a membrane-bound receptor tyrosine kinase that regulates proliferation via the MAPK and PI3K pathway, much like EGFR, was found to be a direct target of miR-198 [69].
miR-146a. Expression of miR-146a is low in malignant tissues in comparison to corresponding adjacent normal lung tissues. Functionally, miR-146a suppresses cell growth, inhibits cell migration and increases cellular apoptosis [70]. Upregulation of miR-146a expression via miR-146a mimic transfection resulted in the downregulation of EGFR as well as phosphorylated EGFR, both at the mRNA and at protein levels. Furthermore, downstream pathways (ERK-1/2, AKT, and STAT) were also downregulated in response to miR-146a mimic transfection, albeit with a weaker effect as that seen by cells transfected with EGFR specific siRNA. miR-146a mimic also led to the decrease of phosphorylation of the NF-κB inhibitor IκBα, but not total IκBα. Levels of phospho-NFκB, total NF-κB, and the total immune-modulating kinase, IRAK-1, were also found to be decreased following miR-146a mimic transfection, suggesting that miR-146a regulates NF-κB and IRAK-1 signaling [70].
miR-26a. miR-26a expression is downregulated in lung cancer tissues relative to normal tissues. Transfection of miR-26a into A549 cells was able to decrease cell proliferation, block the G1/S phase transition of cell cycle, and induce apoptosis [71]. The chromatin regulator enzyme EZH2, which regulates survival and metastasis of cancer cells [72], was found to be a direct target of miR-26a. Downregulation of EZH2 expression, caused by overexpression of miR-26a will transactivate downstream tumor suppressor genes DAB21P and RUNX3. DAB21P is a potent growth inhibitor that induces G0/G1 phase cell cycle arrest and could lead to apoptosis [73], while RUNX3 leads to cell cycle arrest, apoptosis, and significant decrease of tumor growth and abrogation of metastasis [74].
miR-451. Poor tumor differentiation, advance pathological state, lymph node metastasis, and poor prognosis are associated with downregulation of miR-451, which occurs in lung cancer [75]. To observe the functions of miR-451, Wang et al. (2011) upregulated miR-451 expression via mimics and observed suppressed in vitro proliferation, chromatin condensation and nuclear fragmentation upon 4′,6-diamidino-2-phenylindole (DAPI) staining, and significant caspase-3 activity. These results suggested that ectopic expression of miR-451 was able to induce an increase in apoptosis in a caspase-3 dependent manner. In addition, the RAB14 gene was identified as a direct target of miR-451. Inhibition of RAB14 led to a decrease in phosphorylation of Akt, which subsequently decreased levels of Bcl-2 protein expression and increased proapoptotic Bax or Bad protein expression. As the expression levels of RAB14 protein were inversely correlated with the expression levels of miR-451 in NSCLC tissues it was concluded that downregulation of RAB14 may be the mechanism by which miR-451 carries out its tumor suppressor functions [75].
miR-192. miR-192 was found to be downregulated in A549, NCI-H460, and 95D cell lines [76]. Cell viability was greatly decreased following miR-192 upregulation, while levels of apoptosis were elevated with induced expression of PARP protein and cleaved caspase-7, thus suggesting that miR-192 induces apoptosis through the caspase pathway. Using bioinformatics analysis, RB1 gene was determined to be a putative target of miR-192 and luciferase reporter assays confirmed direct binding of miR-192 to the 3′-UTR of this gene [76]. Since RB1 plays a vital role in regulating cell apoptosis, its downregulation was shown to induce γ-H2AX foci formation, a marker of DNA damage, and to promote apoptosis in A549 cells [77].
4. miRNA and Response to Cancer Therapy
Many cancer therapies available today aim to induce tumor-selective cell death; however resistance to chemotherapeutics is a significant obstacle to the long-term treatment and survival of NSCLC patients [78]. Presently, there are various chemotherapeutics that are being utilized in the treatment of lung cancer, including FDA approved drugs (DDP, paclitaxel, docetaxel, gemcitabine, and EGFR-TKIs), natural compounds (curcumin), and small organic compounds (PRIMA-1) (see Table 3). The association of miRNAs as regulators of malignancy and apoptosis has been widely reported; thus it is reasonable to assume that miRNAs play significant roles in sensitivity/resistance to common cancer treatments (see Figure 2) [79]. Indeed, recent studies have demonstrated miRNAs as potential agents involved in the sensitivity of lung cancer cells to cytotoxic therapy.
Table 3.
Drug-associated miRNAs in lung cancer.
| Drug | MicroRNA | Target genes | Function | Cell Lines | In vivo models | Citation |
|---|---|---|---|---|---|---|
| Docetaxel | miR-100 | Plk1 | Chemosensitize lung adenocarcinoma cells to docetaxel Suppress cell proliferation, enhance apoptosis and cell cycle arrest in the G2/M phase of cell cycle Down-regulate in vivo cell proliferating ability |
SPC-A1, A549, NCI-H1299, SPC-A1/DTX |
Nude mice | [102] |
| miR-650 | ING4 | Confer docetaxel chemoresistance both in vitro and in vivo | SPC-A1, NCI-H1299 | Athymic BALB/c nude mice | [105] | |
|
| ||||||
| Cisplatin | miR-451 | N/D | Enhance DDP chemosensitivity Inhibit growth and enhance apoptosis |
A549 | BALB/c nude mice | [84] |
| miR-31 | ABCB9 | Induced DDP resistance Antiapoptotic effect |
SPC-A1, LTEP-A2, NCI-H460, NCI-H1299 |
N/T | [86] | |
|
| ||||||
| Paclitaxel | miR-133a/b, miR-361-3p | N/D | Oncogenic miR inhibition reduces cell survival |
NCI-H1155, NCI-H1993, NCI-H358 |
N/T | [91] |
| miR-101 | EZH2 | Decrease proliferation and invasive ability of cells Sensitize cells to paclitaxel |
NCI-H226, A549, NCI-H358, 801D |
N/T | [98] | |
|
| ||||||
| Gemcitabine | miR-133b | MCL-1, BCL-W |
Increase apoptosis Gemcitabine sensitivity |
A549, NCI-H23, NCI-H2172, NCI-H226, NCI-H522, NCI-H2009, NCI-H1703 |
N/T | [111] |
|
| ||||||
| EGFR-TKI | miR-30b/c miR-221/222 | APAF-1, BIM |
Gefitinib-induced PARP cleavage Decrease cell viability Restore gefitinib sensitivity |
Calu-1, A549, PC-9, HCC827 | N/T | [123] |
| miR-214 | PTEN | Oncogenic Knockdown sensitizes cells to gefitinib |
HCC827, HCC827/GR | N/T | [126] | |
| miR-133b | EGFR | Inhibit cell's growth and invasion abilities Enhance EGFR-TKI sensitivity |
PC-9, A549, NCI-H1650, NCI-H1975 | N/T | [131] | |
|
| ||||||
| Curcumin | miRNA-186* | Caspase-10 | Inhibits cell apoptosis Down-regulation of miR-186 by curcumin induces apoptosis |
A549, A549/DDP | N/T | [154, 157] |
|
| ||||||
| TRAIL | miR-221, miR-222 | N/D | Impair TRAIL-dependent apoptosis Induce TRAIL-resistance |
Calu-1, A549, NCI-H460 |
N/T | [168] |
| miR-34a, miR-34c | PDGFR-α, PDGFR-β | Augment TRAIL response Reduce migratory and invasive capacity of cells |
NCI-H460, A549, NCI-H1299, Calu-6 NCI-H1703, |
N/T | [169] | |
| miR-212 | PED | Increase sensitivity to TRAIL Tumor suppressor |
Calu-1, NCI-H460 | N/T | [175] | |
|
| ||||||
| PRIMA-1 | miR-34a | N/D | Induce apoptosis in the lung cancer cells containing mutant p53 | A549, NCI-H211, NCI-H1155, NCI-H1299 |
N/T | [148] |
|
| ||||||
| Multidrug Resistance | miR-200bc/429 | BCL2, XIAP | Enhanced sensitivity to various anticancer drugs including VCR, CDDP, VP-16, and ADR | A549, A549/CDDP | N/T | [158] |
| miR-181b | BCL2 | Increased sensitivity to a number of anticancer drugs including VCR, 5-Fu, CDDP, VP-16, and ADR | A549, A549/CDDP | N/T | [161] | |
N/D: Not determined, N/T: Not tested.
Figure 2.
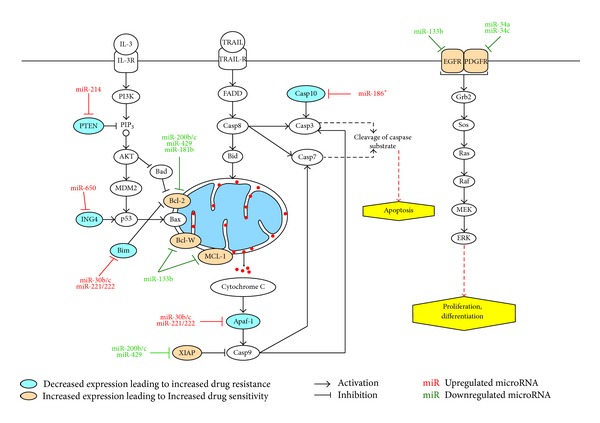
Scheme depicting the roles miRNAs play in sensitivity and resistance to common cancer treatments.
4.1. Cisplatin- (DDP-) Related miRNAs
DDP is a platinum-coordinated complex that is the most widely used chemotherapy for human NSCLC in the past two decades [80–82]. However, multiple administration of DDP results in the development of drug resistance leading to failure of treatment, as demonstrated by tumor growth or tumor relapse [78, 83]. Therefore, to overcome the treatment plateau of DDP on NSCLC, the biological mechanisms by which DDP action is enforced must be further elucidated. As miRNAs act as critical regulators in the development of drug resistance, it would be interesting to research the mechanism through which oncogenic miRNAs modulates DDP-induced apoptosis in NSCLC.
miR-451. miR-451 was downregulated in NSCLC tissues in comparison to normal lung tissues, and upregulation of miR-451 enhances DDP chemosensitivity in A549 cells by inhibiting cell growth and inducing apoptosis enhancement [84]. Bian et al. (2011) demonstrated in their study that upregulation of miR-451 enhanced caspase-3-dependent apoptosis through the inactivation of the Akt signaling pathway, which in turn decreased Bcl-2 while increasing expression of Bax protein levels. Furthermore, results of Annexin V-FITC apoptosis assay indicated that in miR-451 transfected A549 cells (A549/miR-451) a higher percentage of apoptosis was observed in comparison to mock A549 cells. Caspase-3 activity in A549/miR-451 treated with DDP was significantly increased against the control, thus suggesting that miR-451 upregulation increases chemosensitivity of A549 cells by enhancing DDP-induced apoptosis. Together these results suggest a possible strategy for treatment of human NSCLC through the combined application of DDP treatment with miR-451 upregulation [84].
miR-31. On the other hand, miR-31 is upregulated in NSCLC cell lines and was demonstrated to induce DDP resistance. To demonstrate this, Glavinas et al. (2004) transfected miR-31 mimics into DDP-sensitive SPC-A-1 cells which led to a marked increase in the resistance of SPC-A-1 cells, while transfection of miR-31 inhibitors increased sensitivity of resistant NCI-H1299 to DDP treatment. To elucidate the mechanism by which DDP resistance is induced by miR-31, bioinformatics analysis was carried out and ABCB9, a membrane transporter involved in drug uptake [85], was predicted to be a target gene. The luciferase reporter assay then confirmed direct miR-31 regulation of ABCB9 by binding to its 3′UTR [86]. Overexpression/knockdown studies indicated a significant decrease in the percentage of DDP-induced apoptotic cells when miR-31 was increased via mimics and a marked increase in DDP-induced apoptotic cells when miR-31 inhibitors were introduced, thus suggesting that miR-31 exerts an antiapoptotic effect in DDP-induced apoptosis through the inhibition of ABCB9.
4.2. Paclitaxel-Related miRNAs
Paclitaxel was the first identified member of taxanes in the list of FDA-approved anticancer drugs. This compound has been shown to have significant single-agent activity against various solid tumors [87, 88] including NSCLC [89]. However, combination of this compound with DDP or carboplatin showed superior response and improved survival rates [90].
miR-133a/b and miR-361-3p. High-throughput screening (HTS) approach was performed by Du and colleagues in 2013 to identify miRNAs that modulate lung cancer cell survival and response to paclitaxel treatment [91]. Using three NSCLC cell lines that have distinct genetic backgrounds (NCI-H1155, NCI-H1993, and NCI-H358), inhibition of two miRNAs (miR-133a/b and miR-361-3p) was found to potently decrease cell viability, although cytotoxicity of the two miRNAs vary greatly, which may be due to different endogenous expression levels of the miRNAs in each cell line. Interestingly, the inhibitors of miR-133a/b and miR-361-3p were found to reduce cell survival through different mechanisms. miR-133a/b inhibitor was able to dramatically increase apoptotic events as seen by increased percentage of cells undergoing apoptosis and increased levels of activated caspase-3. However miR-361-3p only showed a modest effect on caspase-3 activation thus suggesting that additional mechanisms are involved in the cytotoxicity of this miRNA. The effect of miRNA inhibitors on cell cycle distribution was then evaluated and results indicated that S phase arrest contributes to cytotoxicity induced by miR-133a/b and miR-361-3p inhibitors. Together these results suggest that miR-133a/b and miR-361-3p may function as oncogenes in cancer cells by regulating tumor suppressor genes.
miR-101. Increasing evidence has revealed that EZH2 has oncogenic properties, as an increased expression of EZH2 augments proliferation and invasion of cancer cells [92–94], while depletion leads to a decline in cell proliferation, increased apoptosis, and inhibition of metastatic tumor growth in vivo [95, 96]. Overexpression of EZH2 has been associated with tumor progression and cancer aggressiveness in NSCLC [97]. In a study by Zhang and colleagues (2011), it was discovered that a decreased expression of miR-101 was associated with EZH2 overexpression in NSCLC tissues [98]. Luciferase reporter assay revealed that miR-101 regulates EZH2 expression through the binding of its 3′UTR mRNA. Overexpression of miR-101 led to a decrease in EZH2 protein levels with subsequent decrease in the proliferation and invasive ability of NSCLC cells. Furthermore, overexpression of miR-101 led to a sensitization of NSCLC cells to paclitaxel.
4.3. Docetaxel-Related miRNAs
Docetaxel, a semisynthetic analog of paclitaxel, is one of the first-line chemotherapy regimens for advanced NSCLC, with genotoxic effects caused by microtubule stabilizing, apoptotic induction through microtubule bundling, and Bcl-2 blocking [99, 100].
miR-100. In a miRNA microarray profiling carried out by Rui and colleagues in 2010, miR-100 was significantly downregulated in docetaxel-resistant SPC-A1/DTX cells relative to SPC-A1 parental cells [101]. To elucidate the role that miR-100 plays in the formation of docetaxel resistance, the authors' transfected miR-100 mimics SPC-A1/DTX cells [102]. Results suggested that restoration of miR-100 expression chemosensitizes cells to docetaxel in vitro, complemented with a suppression of cell proliferation, enhancement of apoptosis, and cell cycle arrest in the G2/M phase of cell cycle. Ectopic miR-100 expression was also able to downregulate in vivo cell proliferating ability. Moreover, PLK1 gene was identified to be a direct target of miR-100. PLK1 plays a role in promotion of cell proliferation and overexpression of this gene has been observed in various human cancers [103] including NSCLC [104]. Knockdown of Plk1 protein expression by miR-100 led to a significant suppression of cell proliferation of SPC-A1/DTX, dramatic increase of early apoptosis rate, G2/M arresting population, and an increase in the response of SPC-A1/DTX cells to docetaxel both in vitro and in vivo. miR-100 was therefore concluded to function as a chemosensitizer restorer to docetaxel by targeting PLK1 and inducing the suppression of cell proliferation, enhancement of apoptosis, and mitotic arrest.
miR-650. High expression of miR-650 can be found in lung cancer tissues, and its dysregulation is correlated with advance clinical stage as a poor prognostic factor for these patients [105]. Furthermore, Huang et al. (2013) determined that the expression of miR-650 is negatively correlated with patients' response to docetaxel. Using two docetaxel-resistant cell lines (SPC-A1/DTX and H1299/DTX), the authors demonstrated that downregulation of miR-650 was able to reverse the resistance. ING4, a novel tumor suppressor gene, was then identified as the functional target of miR-650 and results from flow cytometry and Hoechst staining assays indicated that miR-650 inhibitor was able to induce an increase in caspase-3-dependent apoptosis. Cells transfected miR-650 inhibitors exhibited decreased expression of Bcl-2 protein, with an increased expression of Bax protein, led to the progression of apoptosis. The findings of this study confirmed that miR-650 was able to confer docetaxel chemoresistance through the regulation of Bcl-2/Bax expression by targeting of ING4 [105].
4.4. Gemcitabine-Related miRNA
Gemcitabine, a pyrimidine nucleoside antimetabolite, has been shown to be an effective agent most particularly when administered in combination regimes [106]. Due to its theoretical ability of interfering with the inhibition of repair of platinum-induced DNA damage, gemcitabine is the perfect partner for platinum compounds. Gemcitabine in combination with DDP represents a common first-line treatment for patients with advanced NSCLC, especially in Europe [80, 107–110].
miR-133b. miR-133b is greatly reduced in cancer tissue in comparison to adjacent normal lung tissue [111]. Prediction programs identified two common predicted targets of miR-133b, the antiapoptotic MCL-1 and BCL-W, both of which are members of the antiapoptotic BCL-2 family [112] and have previously been reported to be increased in both solid and hematological malignancies including lung cancer [113, 114]. Transfection of miR-133b using pre-miR-133b resulted in a decrease in Bcl-W and Mcl-1 protein expression with a moderate increase of apoptosis. However combination treatment of miR-133b overexpression with 24 hours treatment of gemcitabine resulted in a greater degree of cleaved PARP expression as well as apoptosis. This concludes that miR-133b is able to target prosurvival molecules and induce apoptosis in the setting of chemotherapeutic agents [111].
4.5. Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors- (EGFR-TKIs-) Related miRNAs
EGFR is a plasma membrane glycoprotein that belongs to a family of four different tyrosine kinase receptors (EGFR (ErbB1), HER2/neu (ErbB2), HER3 (ErbB3), and HER4 (ErbB4)) [115]. Dimerization of EGFR may result in cancer cell proliferation, inhibition of apoptosis, invasion, metastasis, and tumor induced neovascularization [116]. Mutations and subsequent overexpression of EGFR can be found in all histologic subtypes of NSCLC [117]. Deletion in exon 19, which removes the conserved sequence LREA, and a single point mutation in exon 21, which leads to the substitution of arginine for leucine at position 858 (L858R), are the most clinically relevant and extensively studied drug-sensitive mutations [118]. Studies have shown that these mutations preferentially bind to first generation EGFR-TKIs, gefitinib and erlotinib [119, 120]. First generation EGFR-TKIs function by selectively targeting the receptor via a competitive, reversible binding at the tyrosine kinase domain, thus leading to the inhibition of ATP binding and subsequent signal transduction and downstream functions [121]. However, acquired resistance to EGFR-TKIs in the metastatic setting is unavoidable. While the average progression-free survival (PFS) is between 10 and 16 months, treatment duration can last as short as 1 month [122]. Drug resistance therefore still remains a problem and new therapies and strategies must be developed to overcome such resistance.
miR-30b/c and miR-221/222. EGF and MET receptors control gefitinib-induced apoptosis and NSCLC tumorigenesis through the downregulation of specific oncogenic miRNAs, miR-30b/c, and miR-221/222 [123]. Using bioinformatics analysis and luciferase assays, APAF-1 and BIM (previously found to play a role in TKI sensitivity [124, 125]) were determined to be direct targets of miR-221/222 and -30b/c. To investigate the roles these miRNAs play in gefitinib-induced apoptosis, wild-type EGFR expressing NSCLC cells (Calu-1 and A549) and cells with EGFR exon-19 deletions (PC9 and HCC827) were utilized. Upon gefitinib treatment, significant downregulation of miR-30b/c and miR-221/222 with an increased BIM and APAF-1 protein levels were observed only in PC9 and HCC827 sensitive cells. To further determine the contribution of miR-30b/c and miR-221/222-mediated APAF-1 and BIM downregulation to cellular TKI response, Garofalo et al. (2012) overexpressed APAF-1 and BIM in A549 resistant cells, which consequently led to gefitinib-induced PARP cleavage. Furthermore, as miR-30b/c and miR-221/222 are regulated by MET, a strong downregulation was observed of these miRNAs when Calu-1- and A549-MET overexpressing cells were treated with MET inhibitors SU11274. Furthermore an increase in caspase-3/7 activity and decreased cell viability was observed in SU11274-treated Calu-1 cells following exposure to varying gefitinib concentrations. Together, these results suggest that MET inhibition restores gefitinib sensitivity in TKI-resistant Calu-1 through downregulation of miR-30b/c and miR-221/222 [123].
miR-214. miR-214 is significantly upregulated in gefitinib resistant lung adenocarcinoma cell line, HCC827/GR, in comparison to parental HCC827 lung adenocarcinoma cells. HCC827/GR was obtained by exposing HCC827 cells to increasing concentrations of gefitinib over six months [126]. Using dual-luciferase reporter assay, Wang et al. (2012) confirmed PTEN as a direct functional target of miR-214. PTEN encodes a 403 amino acid dual-specificity lipid and protein phosphatase which functions as a tumor suppressor in many tumors [127, 128]. Knockdown of miR-214 expression resulted in the upregulation of PTEN protein and inactivation of AKT, which is largely linked to antiapoptotic function [129, 130]. Furthermore, knockdown of miR-214 resensitized HCC827/GR to gefitinib, as demonstrated through MTS assay. miR-214 was thus concluded to potentially serve as a therapeutic target to reverse the acquired resistance of gefitinib in lung adenocarcinoma cells.
miR-133b. Expression of miR-133b is significantly downregulated in NSCLC tissues in comparison to nonneoplastic lung tissues [131], and the 3′UTR of EGFR was found to be a direct target of this miRNA thus inhibiting its expression. Treatment of EGFR-addicted lung cancer cells, PC-9 and A549 with miR-133b mimic inhibited phosphorylation of EGFR, AKT, and extracellular signal-related kinase (ERK)1/2, thus inhibiting their growth and invasion abilities. However in non-EGFR-addicted NSCLC cells NCI-H1650 and NCI-H1975, no significant changes in the expression of phosphorylated EGFR, AKT, and ERK1/2 were found. Furthermore, miR-133b was able to restore or enhance EGFR-TKI sensitivity in NSCLC cells, especially in EGFR-addicted cells. These findings reveal that transfection of miR-133b in EGFR-addicted NSCLC has the therapeutic potential for overcoming EGFR-TKI resistance [131].
4.6. PRIMA-1-Related miRNA
The tumor suppressor p53 gene regulates cell growth through the activation of the transcription of numerous genes specifically those involved in cell cycle regulation, apoptosis, and genomic stability [132–134] andhas also been implicated in the response to anticancer therapies [133]. p53 has been reported to be frequently mutated in humans cancers with mutations occurring in greater than 50% of lung tumors [135, 136]. Restoration of wild-type p53 function has led to regression of cancers in mice [137, 138], and thus efforts to treat cancers through the reactivation of p53 with a low-molecular-weight compound such as PRIMA-1 (p53-dependent reactivation and induction of massive apoptosis) [139, 140] are widely supported.
miR-34a. In a study conducted by Duan et al. (2010), the role of miR-34 family members in regulating PRIMA-1 induced apoptosis was investigated. The authors discovered that PRIMA-1 was able to upregulate miR-34a in p53 mutant cells. Previous studies have shown evidence that the miR-34 family plays a role in the regulation of cell proliferation and apoptosis [19, 141–147]. The results of this study suggest that PRIMA-1 is able to restore wild-type function to mutant p53, which will upregulate miR-34a to induce apoptosis in lung cancer cells [148].
4.7. Curcumin Related miRNA
Curcumin is a compound extracted from the rhizomes of Curcuma longa L. and studies carried out exhibited its diverse pharmacological effects which include anti-inflammatory, antioxidant, and antitumor activities [149]. Previous studies have also shown that curcumin can induce apoptosis in many types of cancer cells [150, 151], through the inhibition of NF-κB, survivin/BIRC5, and BCL-2 [152, 153]. However few studies have been carried out to report the importance of miRNA expression modulation in mediating the biological effects of curcumin.
miRNA-186*. In a study conducted by Zhang et al.(2010), curcumin was shown to have the ability to inhibit cell proliferation and induce apoptosis in A549 cells. The authors performed a cluster analysis on the expression profiles on curcumin-treated and dimethyl sulfoxide (DMSO) control-treated samples and found that miR-186* was shown to be significantly downregulated in response to curcumin treatment, thus suggesting that miR-186* may play an oncogenic role in human lung cancer cells. Inhibition of miR-186* was shown to greatly decrease cell proliferation in A549 cells and increase the induction of apoptosis. Furthermore, caspase-10 was revealed to be a direct target of miR-186 [154]. This study thus provided the first evidence that miR-186* is essential for the anticancer effects of curcumin in A549 cells and that caspase-10 may be an important target of miR-186* in preventing apoptosis.
However, even though curcumin has exhibited antitumor activity, there has been concern regarding the effects of curcumin on multidrug resistant cells [155, 156]. To analyze such effects, A549/DDP, the DDP-resistant derivative of parental A549 cells generated by coculturing parental A549 cells with 6 nm DDP to maintain the drug resistance phenotype, was utilized. In a study conducted by Zhang et al. (2010), a comprehensive miRNA profiling of untreated multidrug-resistant cell line (A549/DDP) was performed and compared against results obtained for A549/DDP cells treated with curcumin. Results showed that miR-186* was downregulated more than 2.5-fold compared to levels in control cells. The antiapoptotic effects of miR-186* in A549/DDP cells were investigated and it was found that transfection of miR-186* mimics led to an inhibition of apoptosis in comparison to that in the control, thus suggesting that miR-186* plays an oncogenic role in this cell line. To confirm the role miR-186* plays in curcumin-induced A549/DDP apoptosis, flow cytometry was used to detect the rate of apoptosis in A549/DDP cells treated with curcumin, control cells, or curcumin combined with miR-186* mimic cells. Results indicated that apoptosis in the combination group was significantly decreased in comparison to cells treated with curcumin [157]. These findings reveal that curcumin is able to induce apoptosis in the multidrug resistant cell line by downregulating miR-186*.
4.8. Multidrug Resistance
miR-200bc/429. In 2012, Zhu et al. reported that the miR-200bc/429 cluster was downregulated in multidrug-resistant A549/DDP cells, in comparison to parental A549 cell [158]. Recent studies have suggested that aberrant DNA methylation of the promoter region of the miR-200bc/429 cluster may be a critical mechanism leading to dysregulated expression level of the miR-200 family [159, 160]. While the roles of the two sequence clusters of miR-200 family on the epithelial-to-mesenchymal transition of tumor cells are well studied, the role that this miRNA family plays on apoptosis has been minimally studied. Zhu et al. demonstrated using MTT that transfection of miR-200bc/429 cluster mimics into A549/DDP greatly enhanced sensitivity of this cell line to various anticancer drugs including vincristine (VCR), etoposide (VP-16), adriamycin (ADR), and DDP. It was found that miR-200bc/429 cluster was able to modulate multidrug resistance (MDR) in lung cancer cell lines, at least in part by inhibiting the antiapoptotic Bcl-2 and XIAP protein expression, thus affecting the mitochondrial release of cytochrome c. Therapeutic methods that target the miR-200bc/429 clusters thus provides a promising method to enhance treatment effect of NSCLC.
miR-181b. In another study by Zhu et al. (2010), miR-181b was also found to be downregulated in multidrug-resistant A549/DDP cells, in comparison to parental A549 cell line [161]. To determine whether miR-181b has a direct role in MDR development, MTT assay was performed revealing that all A549/DDP cells transfected with miR-181b mimic exhibited a significant increase in sensitivity to a number of anticancer drugs including 5-fluorouracil (5-Fu), VCR, DDP, VP-16, and ADR. Bioinformatics analysis predicted antiapoptotic BCL-2 as a potential target of miR-181, with two conserved target sites in the 3′UT region. Transfection of miR-181a in A549/DDP cells led to a significant decrease in Bcl-2 protein levels, as demonstrated by Western blot. Furthermore, A549/DDP miR-181b transfected cells also led to an increase in apoptosis as detected by flow cytometry. Together these results demonstrate miR-181b's ability to modulate the development of MDR in lung cancer cell lines, at least in part, by modulation of apoptosis through the targeting of the antiapoptotic BCL-2.
4.9. TRAIL-Related miRNAs
The Apo2L/tumor necrosis factor- (TNF-) α-related apoptosis inducing ligand (TRAIL) is a member of the TNF family that is known to induce apoptosis in various cancers [162]. Treatment of transformed cells with TRAIL has been shown to successfully induce apoptosis both in vitro and in vivo [162, 163]; however a wide range of human cancer cells are resistant to TRAIL-induced apoptosis [164].
miR-221 and -222. To identify the mechanisms by which miRNAs may play a role in TRAIL resistance, Garofalo et al. (2008) carried out a genome wide profiling of miRNAs in three different lung cancer cell lines (A459, Calu-1, and NCI-H460) and found that miR-221 and miR-222 were markedly upregulated in TRAIL-resistant cells. In TRAIL sensitive cells NCI-H460, TRAIL was able to induce the activation of the caspase cascade, evaluated by the appearance of cleaved fragments. However, transfection of NCI-H460 cells with pre-miRs-221 and -222 caused a significant reduction of TRAIL-mediated cell death machinery activation. Further experiments deduced that miR-221 and-222 directly targeted p27Kip1, and inhibition of p27Kip1 via pre-miR-221 and -222 transfection led to an increase in cell resistance to TRAIL as assessed by Annexin V staining, and PARP and caspase-8 activation. Taken together, the authors' results demonstrate that increased levels of miR-221 and -222 may modulate sensitivity of NSCLC cells to TRAIL with important implications in the design of new therapeutic agents.
miR-34a and miR-34c. In another study, miR-34a and miR-34c expression were found to be significantly downregulated in NSCLC cells and lung tumors in comparison to normal lung tissues. Performing a bioinformatics search, Garofalo et al. (2013) determined that PDGFR-α and PDGFR-β were targets of these miRNAs; both of which have been reported to be overexpressed and associated with poor outcome in lung cancer [165]. Through targeting PDGFR-α and PDGFR-β, miR-34a/c were able to decrease invasiveness as well as increase TRAIL-induced apoptosis. TRAIL resistance is common in lung tumors and it has been reported that PDGFR-α and PDGFR-β regulate the PI3K/Akt and ERK1/2 pathways [166, 167], which play a role in TRAIL-induced apoptosis [168]. Phosphorylation levels of ERKs were found to be decreased following ectopic expression of miR-34a/c; additionally caspase-3/7 assay revealed an increase in TRAIL sensitivity. This study demonstrates that inhibition of PDGFR-α and PDGFR-β by miR-34a/c is able to antagonize tumorigenicity and increase sensitivity to TRAIL-induced cell death [169].
miR-212. PED/PEA-15 is a death effector domain (DED) family member, which has been implicated in the processes of cell growth and metabolism [170–172]. Furthermore, PED/PEA-15 has a broad range of antiapoptotic ability, being able to inhibit both the intrinsic and extrinsic apoptotic pathways [171, 173]. Zanca and colleagues (2008) reported that PED/PEA-15 overexpression plays a role in TRAIL resistance in NSCLC [174]; however the mechanism that regulates its expression is not well known. In further studies, Incoronato et al. (2010) reported that NSCLC-affected lung tissue has an increased expression of PED/PEA-15 with a concurrent downregulation of miR-212 and decreased response to TRAIL treatment [175]. miR-212 negatively regulates PED/PEA-15 by directly binding to its 3′UTR. miR-212 downregulation has previously been reported to be involved in lung cancer response to chemotherapy, in particular to docetaxel [101]. In this study, transfection of NSCLC Calu-1 cells with pre-miR-212 led to a decrease in PED/PEA-15 expression with increased caspase-8 activation following treatment with TRAIL, indicating increased sensitivity of Calu-1 cells to TRAIL-mediated cell death. Therefore, the expression of miR-212 could be used to predict therapeutic response to TRAIL in lung cancer.
5. Conclusions
In terms of molecular events occurring in tumors, evasion of apoptosis is an important hallmark of tumor progression. Recent evidence has exhibited deregulated miRNAs to play a role in the apoptotic process. In lung cancer, upregulated miRNAs have been shown to serve as oncogenes, targeting tumor suppressor, and/or proapoptotic genes, while downregulated miRNAs can function as tumor suppressors, targeting oncogenic and/or antiapoptotic genes. Additionally, studies have also indicated that miRNAs play a significant role in altering sensitivity and resistance to cytotoxic treatment. Targeting of specific miRNAs could therefore potentially be used as valuable therapeutics for lung cancer. Together, these studies have illustrated the importance for further studies and validation of miRNAs and their targets. Furthermore, there is a serious shortage in research being carried out in miRNA-regulated apoptosis in SCLC. As SCLC accounts for 16.8% of lung cancer incidence and is a highly aggressive form of lung cancer it would be of great interest to determine the functions of miRNAs in regulation of apoptosis in this lung cancer subtype.
Acknowledgments
This work was supported by the High Impact Research Grant (HIR) (H-21001-F000036), the University of Malaya Postgraduate Research Fund (PPP) (PV058-2011B), and the Centre for Research in Biotechnology for Agriculture (CEBAR) RU Operation Grant (RU005C-2014). All funders had no role in decision to publish or preparation of the paper.
Abbreviations
- γ-H2AX:
Gamma, H2A histone family, member X
- 3′UTR:
3′-Untranslated region
- 5-Fu:
5-Fluorouracil
- ABCB9:
ATP-binding cassette, subfamily B (MDR/TAP), member 9
- AChE:
Acetylcholinesterase
- ADR:
Adriamycin
- Akt:
Protein kinase B
- Apaf-1:
Apoptotic peptidase activating factor 1
- BAK:
BCL2-antagonist/killer 1
- BAX:
BCL2-associated X protein
- BCL-2:
B-cell CLL/Lymphoma
- BCL-W:
BCL2-like 2
- BCL-XL:
BCL2-like 1
- BIM:
BCL2-like 11 (apoptosis facilitator)
- BIRC5:
Baculoviral IAP repeat containing 5
- BMF:
Bcl-2-modifying factor
- BTG2:
BTG family, member 2
- DAB2IP:
Disabled homolog 2-interacting protein
- DDP:
Cisplatin
- DMSO:
Dimethyl sulfoxide
- E2F1:
E2F transcription factor 1
- EGFR:
Epidermal growth factor receptor
- ERK1:
Mitogen-activated protein kinase 3
- ERK2:
Mitogen-activated protein kinase 1
- EZH2:
Histone-lysine N-methyltransferase
- FasL:
Fas ligand
- FDA:
Food and drug administration
- FGFR1:
Fibroblast growth factor receptor 1
- FITC:
Fluorescein isothiocyanate
- HTS:
High-throughput screening
- IκBα:
Nuclear factor of kappa light polypeptide gene enhancer in B cells inhibitor alpha
- ING4:
Inhibitor of growth family, member 4
- IRAK-1:
Interleukin-1 receptor-associated kinase 1
- MAPK:
Mitogen activated protein kinase
- MCL1:
Molecule myeloid leukemia 1
- MDR:
Multidrug resistance
- miRNA:
MicroRNA
- NF-κB:
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NSCLC:
Non-small cell lung cancer
- NOXA:
Phorbol-12-myristate-13-acetate-induced protein 1
- p27Kip1:
Cyclin-dependent kinase inhibitor 1B
- p53:
Tumor protein p53
- PDCD4:
Programmed cell death 4 (neoplastic transformation inhibitor)
- PDGFR-α:
Platelet-derived growth factor receptor-alpha polypeptide
- PDGFR-β:
Platelet-derived growth factor receptor-beta polypeptide
- PEA-15:
Astrocytic phosphoprotein PEA-15
- PED:
Preimplantation embryonic development
- PI3K/AKT:
Phosphatidylinositol 3-kinase/protein kinase B
- PKC:
Protein kinase C
- PKC-α:
Protein kinase C-alpha
- PKC-ε:
Protein kinase C-epsilon
- PLK1:
Polo-like kinase 1
- PRIMA-1:
p53-dependent reactivation and induction of massive apoptosis
- PFS:
progression-free survival
- PUMA:
BCL2 binding component 3
- RAB14:
RAB14, member RAS oncogene family
- RB1:
Retinoblastoma 1
- RHOB:
Ras homolog family member B
- RUNX3:
Runt-related transcription factor 3
- SCLC:
Small cell lung cancer
- SP1:
Specificity Protein 1
- SPRY1:
Sprouty homolog 1, antagonist of FGF signaling (Drosophila)
- SPRY2:
Sprouty homolog 2 (Drosophila)
- STAT:
Signal transducer and activator of transcription
- TRAIL:
Apo2L/tumor necrosis factor- (TNF-) α-related apoptosis inducing ligand
- TGF-β:
Transforming growth factor-beta
- VCR:
Vincristine
- VP-16:
Etoposide
- WNT:
Wingless-type MMTV integration site family
- XIAP:
X-linked inhibitor of apoptosis.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.GLOBOCAN 2012: Estimated Cancer Incidence, Mortality and Prevalence Worldwide in 2012. International Agency for Search on Cancer. World Health Organization; 2014. [Google Scholar]
- 2.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2008. CA: Cancer Journal for Clinicians. 2008;58(2):71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 3.Molina JR, Yang P, Cassivi SD, Schild SE, Adjei AA. Non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clinic Proceedings. 2008;83(5):584–594. doi: 10.4065/83.5.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pfister DG, Johnson DH, Azzoli CG, et al. American Society of Clinical Oncology treatment of unresectable non-small-cell lung cancer guideline: update 2003. Journal of Clinical Oncology. 2004;22(2):330–353. doi: 10.1200/JCO.2004.09.053. [DOI] [PubMed] [Google Scholar]
- 5.SEER Cancer Statistics Review 1975–2010. Surveillance. Epidemiology, and End Results Program. National Cancer Institute; 2013. [Google Scholar]
- 6.Sorenson CM. Bcl-2 family members and disease. Biochimica et Biophysica Acta. 2004;1644(2-3):169–177. doi: 10.1016/j.bbamcr.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 7.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 8.Wang XW, Harris CC. p53 tumor-suppressor gene: clues to molecular carcinogenesis. Journal of Cellular Physiology. 1997;173:247–255. doi: 10.1002/(SICI)1097-4652(199711)173:2<247::AID-JCP30>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 9.Daniel JC, Smythe WR. The role of Bcl-2 family members in non-small cell lung cancer. Seminars in Thoracic and Cardiovascular Surgery. 2004;16(1):19–27. doi: 10.1053/j.semtcvs.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 10.Fisher TC, Milner AE, Gregory CD, et al. bcl-2 Modulation of apoptosis induced by anticancer drugs: resistance to thymidylate stress is independent of classical resistance pathways. Cancer Research. 1993;53(14):3321–3326. [PubMed] [Google Scholar]
- 11.Miyashita T, Reed JC. Bcl-2 oncoprotein blocks chemotherapy-induced apoptosis in a human leukemia cell line. Blood. 1993;81(1):151–157. [PubMed] [Google Scholar]
- 12.Tang C, Willingham MC, Reed JC, et al. High levels of p26BCL-2 oncoprotein retard taxol-induced apoptosis in human pre-B leukemia cells. Leukemia. 1994;8(11):1960–1969. [PubMed] [Google Scholar]
- 13.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 14.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120(1):15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 15.Calin GA, Dumitru CD, Shimizu M, et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(24):15524–15529. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cimmino A, Calin GA, Fabbri M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(39):13944–13949. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han J, Goldstein LA, Gastman BR, Rabinowich H. Interrelated roles for Mcl-1 and BIM in regulation of TRAIL-mediated mitochondrial apoptosis. The Journal of Biological Chemistry. 2006;281(15):10153–10163. doi: 10.1074/jbc.M510349200. [DOI] [PubMed] [Google Scholar]
- 18.Volinia S, Calin GA, Liu C-G, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(7):2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He L, He X, Lim LP, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447(7148):1130–1134. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mott JL, Kobayashi S, Bronk SF, Gores GJ. mir-29 regulates Mcl-1 protein expression and apoptosis. Oncogene. 2007;26(42):6133–6140. doi: 10.1038/sj.onc.1210436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Woods K, Thomson JM, Hammond SM. Direct regulation of an oncogenic micro-RNA cluster by E2F transcription factors. The Journal of Biological Chemistry. 2007;282(4):2130–2134. doi: 10.1074/jbc.C600252200. [DOI] [PubMed] [Google Scholar]
- 22.Tsang WP, Kwok TT. Let-7a microRNA suppresses therapeutics-induced cancer cell death by targeting caspase-3. Apoptosis. 2008;13(10):1215–1222. doi: 10.1007/s10495-008-0256-z. [DOI] [PubMed] [Google Scholar]
- 23.Wiemer EAC. The role of microRNAs in cancer: no small matter. European Journal of Cancer. 2007;43(10):1529–1544. doi: 10.1016/j.ejca.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 24.Calin GA, Liu C-G, Sevignani C, et al. MicroRNA profiling reveals distinct signatures in B cell chronic lymphocytic leukemias. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(32):11755–11760. doi: 10.1073/pnas.0404432101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iorio MV, Ferracin M, Liu C-G, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Research. 2005;65(16):7065–7070. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- 26.Lyng MB, Lænkholm A-V, Søkilde R, Gravgaard KH, Litman T, Ditzel HJ. Global microRNA expression profiling of high-risk ER+ breast cancers from patients receiving adjuvant Tamoxifen mono-therapy: a DBCG study. PLoS ONE. 2012;7(5) doi: 10.1371/journal.pone.0036170.e36170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ciafrè SA, Galardi S, Mangiola A, et al. Extensive modulation of a set of microRNAs in primary glioblastoma. Biochemical and Biophysical Research Communications. 2005;334(4):1351–1358. doi: 10.1016/j.bbrc.2005.07.030. [DOI] [PubMed] [Google Scholar]
- 28.Yanaihara N, Caplen N, Bowman E, et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell. 2006;9(3):189–198. doi: 10.1016/j.ccr.2006.01.025. [DOI] [PubMed] [Google Scholar]
- 29.Jovanovic M, Hengartner MO. miRNAs and apoptosis: RNAs to die for. Oncogene. 2006;25(46):6176–6187. doi: 10.1038/sj.onc.1209912. [DOI] [PubMed] [Google Scholar]
- 30.Lima RT, Busacca S, Almeida GM, Gaudino G, Fennell DA, Vasconcelos MH. MicroRNA regulation of core apoptosis pathways in cancer. European Journal of Cancer. 2011;47(2):163–174. doi: 10.1016/j.ejca.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 31.Li C, Hashimi SM, Good DA, et al. Apoptosis and microRNA aberrations in cancer. Clinical and Experimental Pharmacology and Physiology. 2012;39(8):739–746. doi: 10.1111/j.1440-1681.2012.05700.x. [DOI] [PubMed] [Google Scholar]
- 32.Wang Y, Lee CGL. MicroRNA and cancer—focus on apoptosis. Journal of Cellular and Molecular Medicine. 2009;13(1):12–23. doi: 10.1111/j.1582-4934.2008.00510.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Son JW, Kim YJ, Cho HM, et al. MicroRNA expression profiles in Korean non-small cell lung cancer. Tuberculosis and Respiratory Diseases. 2009;67(5):413–421. [Google Scholar]
- 34.Miska EA. How microRNAs control cell division, differentiation and death. Current Opinion in Genetics and Development. 2005;15(5):563–568. doi: 10.1016/j.gde.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 35.Fiori ME, Barbini C, Haas TL, et al. Antitumor effect of miR-197 targeting in p53 wild-type lung cancer. Cell Death & Differentiation. 2014;21(5):774–782. doi: 10.1038/cdd.2014.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Puthalakath H, Villunger A, O’Reilly LA, et al. Bmf: a proapoptotic BH3-only protein regulated by interaction with the myosin V actin motor complex, activated by anoikis. Science. 2001;293(5536):1829–1832. doi: 10.1126/science.1062257. [DOI] [PubMed] [Google Scholar]
- 37.Hatley ME, Patrick DM, Garcia MR, et al. Modulation of K-Ras-dependent lung tumorigenesis by MicroRNA-21. Cancer Cell. 2010;18(3):282–293. doi: 10.1016/j.ccr.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim D-M, Chung K-S, Choi S-J, et al. RhoB induces apoptosis via direct interaction with TNFAIP1 in HeLa cells. International Journal of Cancer. 2009;125(11):2520–2527. doi: 10.1002/ijc.24617. [DOI] [PubMed] [Google Scholar]
- 39.Ye W, Gong X, Xie J, et al. AChE deficiency or inhibition decreases apoptosis and p53 expression and protects renal function after ischemia/reperfusion. Apoptosis. 2010;15(4):474–487. doi: 10.1007/s10495-009-0438-3. [DOI] [PubMed] [Google Scholar]
- 40.Zhang X, Lu L, Liu S, Ye W, Wu J, Zhang X. Acetylcholinesterase deficiency decreases apoptosis in dopaminergic neurons in the neurotoxin model of Parkinson’s disease. International Journal of Biochemistry & Cell Biology. 2013;45(2):265–272. doi: 10.1016/j.biocel.2012.11.015. [DOI] [PubMed] [Google Scholar]
- 41.Zhang XJ, Yang L, Zhao Q, et al. Induction of acetylcholinesterase expression during apoptosis in various cell types. Cell Death & Differentiation. 2002;9(8):790–800. doi: 10.1038/sj.cdd.4401034. [DOI] [PubMed] [Google Scholar]
- 42.Zhao Y, Wang X, Wang T, et al. Acetylcholinesterase, a key prognostic predictor for hepatocellular carcinoma, suppresses cell growth and induces chemosensitization. Hepatology. 2011;53(2):493–503. doi: 10.1002/hep.24079. [DOI] [PubMed] [Google Scholar]
- 43.Lu L, Zhang X, Zhang B, Wu J, Zhang X. Synaptic acetylcholinesterase targeted by microRNA-212 functions as a tumor suppressor in non-small cell lung cancer. The International Journal of Biochemistry & Cell Biology. 2013;45:2530–2540. doi: 10.1016/j.biocel.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 44.Hayashita Y, Osada H, Tatematsu Y, et al. A polycistronic MicroRNA cluster, miR-17-92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Research. 2005;65(21):9628–9632. doi: 10.1158/0008-5472.CAN-05-2352. [DOI] [PubMed] [Google Scholar]
- 45.Matsubara H, Takeuchi T, Nishikawa E, et al. Apoptosis induction by antisense oligonucleotides against miR-17-5p and miR-20a in lung cancers overexpressing miR-17-92. Oncogene. 2007;26(41):6099–6105. doi: 10.1038/sj.onc.1210425. [DOI] [PubMed] [Google Scholar]
- 46.O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435(7043):839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- 47.MacLeod K. pRb and E2f-1 in mouse development and tumorigenesis. Current Opinion in Genetics and Development. 1999;9(1):31–39. doi: 10.1016/s0959-437x(99)80005-7. [DOI] [PubMed] [Google Scholar]
- 48.Zhang B, Pan X, Cobb GP, Anderson TA. microRNAs as oncogenes and tumor suppressors. Developmental Biology. 2007;302(1):1–12. doi: 10.1016/j.ydbio.2006.08.028. [DOI] [PubMed] [Google Scholar]
- 49.Ruan K, Fang X, Ouyang G. MicroRNAs: novel regulators in the hallmarks of human cancer. Cancer Letters. 2009;285(2):116–126. doi: 10.1016/j.canlet.2009.04.031. [DOI] [PubMed] [Google Scholar]
- 50.Hockenbery DM, Zutter M, Hickey W, Nahm M, Korsmeyer SJ. BCL2 protein is topographically restricted in tissues characterized by apoptotic cell death. Proceedings of the National Academy of Sciences of the United States of America. 1991;88(16):6961–6965. doi: 10.1073/pnas.88.16.6961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xiong S, Zheng Y, Jiang P, Liu R, Liu X, Chu Y. MicroRNA-7 inhibits the growth of human non-small cell lung cancer A549 cells through targeting BCL-2. International Journal of Biological Sciences. 2011;7(6):805–814. doi: 10.7150/ijbs.7.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang H, Li M, Zhang R, et al. Effect of miR-335 upregulation on the apoptosis and invasion of lung cancer cell A549 and H1299. Tumor Biology. 2013;34:3101–3109. doi: 10.1007/s13277-013-0878-9. [DOI] [PubMed] [Google Scholar]
- 53.Dynan WS, Tjian R. The promoter-specific transcription factor Sp 1 binds to upstream sequences in the SV40 early promoter. Cell. 1983;35(1):79–87. doi: 10.1016/0092-8674(83)90210-6. [DOI] [PubMed] [Google Scholar]
- 54.Suske G, Bruford E, Philipsen S. Mammalian SP/KLF transcription factors: bring in the family. Genomics. 2005;85(5):551–556. doi: 10.1016/j.ygeno.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 55.Othman N, In LLA, Harikrishna JA, Hasima N. Bcl-xL silencing induces alterations in hsa-miR-608 expression and subsequent cell death in A549 and SK-LU1 human lung adenocarcinoma cells. PLoS ONE. 2013;8(12) doi: 10.1371/journal.pone.0081735.e81735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Soini Y, Kinnula V, Kaarteenaho-Wiik R, Kurttila E, Linnainmaa K, Pääkkö P. Apoptosis and expression of apoptosis regulating proteins bcl-2, mcl-1, bcl-X, and bax in malignant mesothelioma. Clinical Cancer Research. 1999;5(11):3508–3515. [PubMed] [Google Scholar]
- 57.Nishizuka Y. Protein kinase C and lipid signaling for sustained cellular responses. FASEB Journal. 1995;9(7):484–496. [PubMed] [Google Scholar]
- 58.Liu WS, Heckman CA. The sevenfold way of PKC regulation. Cellular Signalling. 1998;10(8):529–542. doi: 10.1016/s0898-6568(98)00012-6. [DOI] [PubMed] [Google Scholar]
- 59.Livneh E, Fishman DD. Linking protein kinase c to cell-cycle control. European Journal of Biochemistry. 1997;248(1):1–9. doi: 10.1111/j.1432-1033.1997.t01-4-00001.x. [DOI] [PubMed] [Google Scholar]
- 60.Clark AS, West KA, Blumberg PM, Dennis PA. Altered protein kinase C (PKC) isoforms in non-small cell lung cancer cells: PKCδ promotes cellular survival and chemotherapeutic resistance. Cancer Research. 2003;63(4):780–786. [PubMed] [Google Scholar]
- 61.Wang C, Wang X, Lian H, et al. miR-203 inhibits cell proliferation and migration of lung cancer cells by targeting PKCα . PLoS ONE. 2013;8(9) doi: 10.1371/journal.pone.0073985.e73985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Akita Y. Protein kinase C-ε (PKC-ε): its unique structure and function. Journal of Biochemistry. 2002;132(6):847–852. doi: 10.1093/oxfordjournals.jbchem.a003296. [DOI] [PubMed] [Google Scholar]
- 63.Zhang N, Su Y, Xu L. Targeting PKCe by miR-143 regulates cell apoptosis in lung cancer. FEBS Letters. 2013;587:3661–3667. doi: 10.1016/j.febslet.2013.09.018. [DOI] [PubMed] [Google Scholar]
- 64.McJilton MA, Van Sikes C, Wescott GG, et al. Protein kinase Cε interacts with Bax and promotes survival of human prostate cancer cells. Oncogene. 2003;22(39):7958–7968. doi: 10.1038/sj.onc.1206795. [DOI] [PubMed] [Google Scholar]
- 65.Ding L, Wang H, Lang W, Xiao L. Protein kinase C-ε promotes survival of lung cancer cells by suppressing apoptosis through dysregulation of the mitochondrial caspase pathway. The Journal of Biological Chemistry. 2002;277(38):35305–35313. doi: 10.1074/jbc.M201460200. [DOI] [PubMed] [Google Scholar]
- 66.Sivaprasad U, Shankar E, Basu A. Downregulation of Bid is associated with PKCε-mediated TRAIL resistance. Cell Death & Differentiation. 2007;14(4):851–860. doi: 10.1038/sj.cdd.4402077. [DOI] [PubMed] [Google Scholar]
- 67.Okhrimenko H, Lu W, Xiang C, Hamburger N, Kazimirsky G, Brodie C. Protein kinase C-ε regulates the apoptosis and survival of glioma cells. Cancer Research. 2005;65(16):7301–7309. doi: 10.1158/0008-5472.CAN-05-1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu D, Thakore CU, Wescott GG, McCubrey JA, Terrian DM. Integrin signaling links protein kinase Cε to the protein kinase B/Akt survival pathway in recurrent prostate cancer cells. Oncogene. 2004;23(53):8659–8672. doi: 10.1038/sj.onc.1207900. [DOI] [PubMed] [Google Scholar]
- 69.Yang J, Zhao H, Xin Y, Fan L. MicroRNA-198 inhibits proliferation and induces apoptosis of lung cancer cells via targeting FGFR1. Journal of Cellular Biochemistry. 2013;115(5):987–995. doi: 10.1002/jcb.24742. [DOI] [PubMed] [Google Scholar]
- 70.Chen G, Umelo IA, Lv S, et al. miR-146a inhibits cell growth, cell migration and induces apoptosis in non-small cell lung cancer cells. PLoS ONE. 2013;8(3) doi: 10.1371/journal.pone.0060317.e60317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dang X, Ma A, Yang L, et al. MicroRNA-26a regulates tumorigenic properties of EZH2 in human lung carcinoma cells. Cancer Genetics. 2012;205(3):113–123. doi: 10.1016/j.cancergen.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 72.Cao Q, Yu J, Dhanasekaran SM, et al. Repression of E-cadherin by the polycomb group protein EZH2 in cancer. Oncogene. 2008;27(58):7274–7284. doi: 10.1038/onc.2008.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xie D, Gore C, Zhou J, et al. DAB2IP coordinates both PI3K-Akt and ASK1 pathways for cell survival and apoptosis. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(47):19878–19883. doi: 10.1073/pnas.0908458106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wei D, Gong W, Oh SC, et al. Loss of RUNX3 expression significantly affects the clinical outcome of gastric cancer patients and its restoration causes drastic suppression of tumor growth and metastasis. Cancer Research. 2005;65(11):4809–4816. doi: 10.1158/0008-5472.CAN-04-3741. [DOI] [PubMed] [Google Scholar]
- 75.Wang R, Wang Z-X, Yang J-S, Pan X, De W, Chen L-B. MicroRNA-451 functions as a tumor suppressor in human non-small cell lung cancer by targeting ras-related protein 14 (RAB14) Oncogene. 2011;30(23):2644–2658. doi: 10.1038/onc.2010.642. [DOI] [PubMed] [Google Scholar]
- 76.Feng S, Cong S, Zhang X, et al. MicroRNA-192 targeting retinoblastoma 1 inhibits cell proliferation and induces cell apoptosis in lung cancer cells. Nucleic Acids Research. 2011;39(15):6669–6678. doi: 10.1093/nar/gkr232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ebi H, Sato T, Sugito N, et al. Counterbalance between RB inactivation and miR-17-92 overexpression in reactive oxygen species and DNA damage induction in lung cancers. Oncogene. 2009;28(38):3371–3379. doi: 10.1038/onc.2009.201. [DOI] [PubMed] [Google Scholar]
- 78.Chang A. Chemotherapy, chemoresistance and the changing treatment landscape for NSCLC. Lung Cancer. 2011;71(1):3–10. doi: 10.1016/j.lungcan.2010.08.022. [DOI] [PubMed] [Google Scholar]
- 79.Lynam-Lennon N, Maher SG, Reynolds JV. The roles of microRNA in cancer and apoptosis. Biological Reviews. 2009;84(1):55–71. doi: 10.1111/j.1469-185X.2008.00061.x. [DOI] [PubMed] [Google Scholar]
- 80.Schiller JH, Harrington D, Belani CP, et al. Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. The New England Journal of Medicine. 2002;346(2):92–98. doi: 10.1056/NEJMoa011954. [DOI] [PubMed] [Google Scholar]
- 81.Wang G, Reed E, Li QQ. Molecular basis of cellular response to cisplatin chemotherapy in non-small cell lung cancer. Oncology Reports. 2004;12(5):955–965. [PubMed] [Google Scholar]
- 82.Eaton KD, Martins RG. Maintenance chemotherapy in non-small cell lung cancer. JNCCN Journal of the National Comprehensive Cancer Network. 2010;8(7):815–821. doi: 10.6004/jnccn.2010.0058. [DOI] [PubMed] [Google Scholar]
- 83.Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nature Reviews Drug Discovery. 2006;5(3):219–234. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- 84.Bian H-B, Pan X, Yang J-S, Wang Z-X, De W. Upregulation of microRNA-451 increases cisplatin sensitivity of non-small cell lung cancer cell line (A549) Journal of Experimental and Clinical Cancer Research. 2011;30(1, article 20) doi: 10.1186/1756-9966-30-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Glavinas H, Krajcsi P, Cserepes J, Sarkadi B. The role of ABC transporters in drug resistance, metabolism and toxicity. Current Drug Delivery. 2004;1(1):27–42. doi: 10.2174/1567201043480036. [DOI] [PubMed] [Google Scholar]
- 86.Dong Z, Zhong Z, Yang L, Wang S, Gong Z. MicroRNA-31 inhibits cisplatin-induced apoptosis in non-small cell lung cancer cells by regulating the drug transporter ABCB9. Cancer Letters. 2014;343:249–257. doi: 10.1016/j.canlet.2013.09.034. [DOI] [PubMed] [Google Scholar]
- 87.Mcguire WP, Hoskins WJ, Brady MF, et al. Cyclophosphamide and cisplatin compared with paclitaxel and cisplatin in patients with stage III and stage IV ovarian cancer. The New England Journal of Medicine. 1996;334(1):1–6. doi: 10.1056/NEJM199601043340101. [DOI] [PubMed] [Google Scholar]
- 88.Henderson IC, Berry DA, Demetri GD, et al. Improved outcomes from adding sequential paclitaxel but not from escalating doxorubicin dose in an adjuvant chemotherapy regimen for patients with node-positive primary breast cancer. Journal of Clinical Oncology. 2003;21(6):976–983. doi: 10.1200/JCO.2003.02.063. [DOI] [PubMed] [Google Scholar]
- 89.Bonomi P, Kim K, Fairclough D, Cella D, Kugler J, Rowinsky E. Comparison of survival for stage IIIB versus stage IV non-small cell lung cancer (NSCLC) patients with etoposide-cisplatin versus taxol-cisplatin: an Eastern Cooperative Oncology Group (ECOG) trial. Proceedings of American Society of Clinical Oncology; 1997; p. p. 454a. [DOI] [PubMed] [Google Scholar]
- 90.Belani CP, Lee J, Socinski MA, et al. Randomized phase III trial comparing cisplatin/etopside versus carboplatin/paclitaxel in advanced and metastatic non-small cell lung cancer (NSCLC). Proceedings of American Society of Clinical Oncology; 1998; p. p. 455a. [DOI] [PubMed] [Google Scholar]
- 91.Du L, Borkowski R, Zhao Z, et al. A high-throughput screen identifies miRNA inhibitors regulating lung cancer cell survival and response to paclitaxel. RNA Biology. 2013;10:1700–1713. doi: 10.4161/rna.26541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Karanikolas BDW, Figueiredo ML, Wu L. Comprehensive evaluation of the role of EZH2 in the growth, invasion, and aggression of a panel of prostate cancer cell lines. Prostate. 2010;70(6):675–688. doi: 10.1002/pros.21112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sellers WR, Loda M. The EZH2 polycomb transcriptional repressor—a marker or mover of metastatic prostate cancer? Cancer Cell. 2002;2(5):349–350. doi: 10.1016/s1535-6108(02)00187-3. [DOI] [PubMed] [Google Scholar]
- 94.Kleer CG, Cao Q, Varambally S, et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(20):11606–11611. doi: 10.1073/pnas.1933744100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wagener N, Holland D, Bulkescher J, et al. The enhancer of zeste homolog 2 gene contributes to cell proliferation and apoptosis resistance in renal cell carcinoma cells. International Journal of Cancer. 2008;123(7):1545–1550. doi: 10.1002/ijc.23683. [DOI] [PubMed] [Google Scholar]
- 96.Ougolkov AV, Bilim VN, Billadeau DD. Regulation of pancreatic tumor cell proliferation and chemoresistance by the histone methyltransferase enhancer of zeste homologue 2. Clinical Cancer Research. 2008;14(21):6790–6796. doi: 10.1158/1078-0432.CCR-08-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kikuchi J, Kinoshita I, Shimizu Y, et al. Distinctive expression of the polycomb group proteins Bmi1 polycomb ring finger oncogene and enhancer of zeste homolog 2 in nonsmall cell lung cancers and their clinical and clinicopathologic significance. Cancer. 2010;116(12):3015–3024. doi: 10.1002/cncr.25128. [DOI] [PubMed] [Google Scholar]
- 98.Zhang J-G, Guo J-F, Liu D-L, Liu Q, Wang J-J. MicroRNA-101 exerts tumor-suppressive functions in non-small cell lung cancer through directly targeting enhancer of zeste homolog 2. Journal of Thoracic Oncology. 2011;6(4):671–678. doi: 10.1097/JTO.0b013e318208eb35. [DOI] [PubMed] [Google Scholar]
- 99.Yvon A-MC, Wadsworth P, Jordan MA. Taxol suppresses dynamics of individual microtubules in living human tumor cells. Molecular Biology of the Cell. 1999;10(4):947–959. doi: 10.1091/mbc.10.4.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lyseng-Williamson KA, Fenton C. Docetaxel: a review of its use in metastatic breast cancer. Drugs. 2005;65(17):2513–2531. doi: 10.2165/00003495-200565170-00007. [DOI] [PubMed] [Google Scholar]
- 101.Rui W, Bing F, Hai-Zhu S, Wei D, Long-Bang C. Identification of microRNA profiles in docetaxel-resistant human non-small cell lung carcinoma cells (SPC-A1) Journal of Cellular and Molecular Medicine. 2010;14(1-2):206–214. doi: 10.1111/j.1582-4934.2009.00964.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Feng B, Wang R, Chen L-B. MiR-100 resensitizes docetaxel-resistant human lung adenocarcinoma cells (SPC-A1) to docetaxel by targeting Plk1. Cancer Letters. 2012;317(2):184–191. doi: 10.1016/j.canlet.2011.11.024. [DOI] [PubMed] [Google Scholar]
- 103.Eckerdt F, Yuan J, Strebhardt K. Polo-like kinases and oncogenesis. Oncogene. 2005;24(2):267–276. doi: 10.1038/sj.onc.1208273. [DOI] [PubMed] [Google Scholar]
- 104.Wolf G, Elez R, Doermer A, et al. Prognostic significance of polo-like kinase (PLK) expression in non-small cell lung cancer. Oncogene. 1997;14(5):543–549. doi: 10.1038/sj.onc.1200862. [DOI] [PubMed] [Google Scholar]
- 105.Huang J-Y, Cui S-Y, Chen Y-T, et al. MicroRNA-650 was a prognostic factor in human lung adenocarcinoma and confers the docetaxel chemoresistance of lung adenocarcinoma cells via regulating Bcl-2/Bax expression. PLoS ONE. 2013;8(8) doi: 10.1371/journal.pone.0072615.e72615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Peters GJ, Van Der Wilt CL, Van Moorsel CJA, Kroep JR, Bergman AM, Ackland SP. Basis for effective combination cancer chemotherapy with antimetabolites. Pharmacology and Therapeutics. 2000;87(2-3):227–253. doi: 10.1016/s0163-7258(00)00086-3. [DOI] [PubMed] [Google Scholar]
- 107.Crinò L, Scagliotti GV, Ricci S, et al. Gemcitabine and cisplatin versus mitomycin, ifosfamide, and cisplatin in advanced non-small-cell lung cancer: a randomized phase III study of the Italian Lung Cancer Project. Journal of Clinical Oncology. 1999;17(11):3522–3530. doi: 10.1200/JCO.1999.17.11.3522. [DOI] [PubMed] [Google Scholar]
- 108.Cardenal F, Paz López-Cabrerizo M, Antón A, et al. Randomized phase III study of gemcitabine-cisplatin versus etoposide-cisplatin in the treatment of locally advanced or metastatic non-small-cell lung cancer. Journal of Clinical Oncology. 1999;17(1):12–18. doi: 10.1200/JCO.1999.17.1.12. [DOI] [PubMed] [Google Scholar]
- 109.Sandler AB, Nemunaitis J, Denham C, et al. Phase III trial of gemcitabine plus cisplatin versus cisplatin alone in patients with locally advanced or metastatic non-small-cell lung cancer. Journal of Clinical Oncology. 2000;18(1):122–130. doi: 10.1200/JCO.2000.18.1.122. [DOI] [PubMed] [Google Scholar]
- 110.Scagliotti GV, De Marinis F, Rinaldi M, et al. Phase III randomized trial comparing three platinum-based doublets in advanced non-small-cell lung cancer. Journal of Clinical Oncology. 2002;20(21):4285–4291. doi: 10.1200/JCO.2002.02.068. [DOI] [PubMed] [Google Scholar]
- 111.Crawford M, Batte K, Yu L, et al. MicroRNA 133B targets pro-survival molecules MCL-1 and BCL2L2 in lung cancer. Biochemical and Biophysical Research Communications. 2009;388(3):483–489. doi: 10.1016/j.bbrc.2009.07.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Thomadaki H, Scorilas A. BCL2 family of apoptosis-related genes: functions and clinical implications in cancer. Critical Reviews in Clinical Laboratory Sciences. 2006;43(1):1–67. doi: 10.1080/10408360500295626. [DOI] [PubMed] [Google Scholar]
- 113.Song L, Coppola D, Livingston S, Cress D, Haura EB. Mcl-1 regulates survival and sensitivity to diverse apoptotic stimuli in human non-small cell lung cancer cells. Cancer Biology and Therapy. 2005;4(3):267–276. doi: 10.4161/cbt.4.3.1496. [DOI] [PubMed] [Google Scholar]
- 114.Kawasaki T, Yokoi S, Tsuda H, et al. BCL2L2 is a probable target for novel 14q11.2 amplification detected in a non-small cell lung cancer cell line. Cancer Science. 2007;98(7):1070–1077. doi: 10.1111/j.1349-7006.2007.00491.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mendelsohn J, Baselga J. Status of epidermal growth factor receptor antagonists in the biology and treatment of cancer. Journal of Clinical Oncology. 2003;21(14):2787–2799. doi: 10.1200/JCO.2003.01.504. [DOI] [PubMed] [Google Scholar]
- 116.Ciardiello F, Tortora G. Drug therapy: EGFR antagonists in cancer treatment. The New England Journal of Medicine. 2008;358(11):1096–1174. doi: 10.1056/NEJMra0707704. [DOI] [PubMed] [Google Scholar]
- 117.Yeh P, Chen H, Andrews J, Naser R, Pao W, Horn L. DNA-mutation Inventory to Refine and Enhance Cancer Treatment (DIRECT): a catalog of clinically relevant cancer mutations to enable genome-directed anticancer therapy. Clinical Cancer Research. 2013;19(7):1894–1901. doi: 10.1158/1078-0432.CCR-12-1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ohashi K, Maruvka YE, Michor F, Pao W. Epidermal growth factor receptor tyrosine kinase inhibitor-resistant disease. Journal of Clinical Oncology. 2013;31(8):1070–1080. doi: 10.1200/JCO.2012.43.3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yun C-H, Boggon TJ, Li Y, et al. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell. 2007;11(3):217–227. doi: 10.1016/j.ccr.2006.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yun C-H, Mengwasser KE, Toms AV, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(6):2070–2075. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Costanzo R, Montanino A, Di Maio M, et al. Advanced non small cell lung cancer with epidermal growth factor receptor mutations: current evidence and future perspectives. Expert Review of Anticancer Therapy. 2013;13(10):1207–1218. doi: 10.1586/14737140.2013.845092. [DOI] [PubMed] [Google Scholar]
- 122.Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. The New England Journal of Medicine. 2010;362(25):2380–2388. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- 123.Garofalo M, Romano G, Di Leva G, et al. EGFR and MET receptor tyrosine kinase-altered microRNA expression induces tumorigenesis and gefitinib resistance in lung cancers. Nature Medicine. 2012;18(1):74–82. doi: 10.1038/nm.2577. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 124.Cragg MS, Kuroda J, Puthalakath H, Huang DCS, Strasser A. Gefitinib-induced killing of NSCLC cell lines expressing mutant EGFR requires BIM and can be enhanced by BH3 mimetics. PLoS Medicine. 2007;4(10):1681–1690. doi: 10.1371/journal.pmed.0040316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Deng W-G, Kwon J, Ekmekcioglu S, Poindexter NJ, Grimm EA. IL-24 gene transfer sensitizes melanoma cells to erlotinib through modulation of the Apaf-1 and Akt signaling pathways. Melanoma Research. 2011;21(1):44–56. doi: 10.1097/CMR.0b013e3283382155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wang Y-S, Wang Y-H, Xia H-P, Zhou S-W, Schmid-Bindert G, Zhou C-C. MicroRNA-214 regulates the acquired resistance to gefitinib via the PTEN/AKT pathway in EGFR-mutant cell lines. Asian Pacific Journal of Cancer Prevention. 2012;13(1):255–260. doi: 10.7314/apjcp.2012.13.1.255. [DOI] [PubMed] [Google Scholar]
- 127.Grant S, Qiao L, Dent P. Roles of ERBB family receptor tyrosine kinases, and downstream signaling pathways, in the control of cell growth and survival. Front Biosci. 2002;7:d376–389. doi: 10.2741/grant. [DOI] [PubMed] [Google Scholar]
- 128.Chang F, Lee JT, Navolanic PM, et al. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: a target for cancer chemotherapy. Leukemia. 2003;17(3):590–603. doi: 10.1038/sj.leu.2402824. [DOI] [PubMed] [Google Scholar]
- 129.Tamura M, Gu J, Matsumoto K, Aota S-I, Parsons R, Yamada KM. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science. 1998;280(5369):1614–1617. doi: 10.1126/science.280.5369.1614. [DOI] [PubMed] [Google Scholar]
- 130.Sulis ML, Parsons R. PTEN: from pathology to biology. Trends in Cell Biology. 2003;13(9):478–483. doi: 10.1016/s0962-8924(03)00175-2. [DOI] [PubMed] [Google Scholar]
- 131.Liu L, Shao X, Gao W, et al. MicroRNA-133b inhibits the growth of non-small-cell lung cancer by targeting the epidermal growth factor receptor. FEBS Journal. 2012;279(20):3800–3812. doi: 10.1111/j.1742-4658.2012.08741.x. [DOI] [PubMed] [Google Scholar]
- 132.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88(3):323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 133.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408(6810):307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 134.El-Hizawi S, Lagowski JP, Kulesz-Martin M, Albor A. Induction of gene amplification as a gain-of-function phenotype of mutant p53 proteins. Cancer Research. 2002;62(11):3264–3270. [PubMed] [Google Scholar]
- 135.Olivier M, Eeles R, Hollstein M, Khan MA, Harris CC, Hainaut P. The IARC TP53 database: new online mutation analysis and recommendations to users. Human Mutation. 2002;19(6):607–614. doi: 10.1002/humu.10081. [DOI] [PubMed] [Google Scholar]
- 136.Mitsudomi T, Steinberg SM, Nau MM, et al. p53 gene mutations in non-small-cell lung cancer cell lines and their correlation with the presence of ras mutations and clinical features. Oncogene. 1992;7(1):171–180. [PubMed] [Google Scholar]
- 137.Ventura A, Kirsch DG, McLaughlin ME, et al. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445(7128):661–665. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- 138.Xue W, Zender L, Miething C, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445(7128):656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Foster BA, Coffey HA, Morin MJ, Rastinejad F. Pharmacological rescue of mutant p53 conformation and function. Science. 1999;286(5449):2507–2510. doi: 10.1126/science.286.5449.2507. [DOI] [PubMed] [Google Scholar]
- 140.Bykov VJN, Issaeva N, Shilov A, et al. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nature Medicine. 2002;8(3):282–288. doi: 10.1038/nm0302-282. [DOI] [PubMed] [Google Scholar]
- 141.Tarasov V, Jung P, Verdoodt B, et al. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G 1-arrest. Cell Cycle. 2007;6(13):1586–1593. doi: 10.4161/cc.6.13.4436. [DOI] [PubMed] [Google Scholar]
- 142.Corney DC, Flesken-Nikitin A, Godwin AK, Wang W, Nikitin AY. MicroRNA-34b and MicroRNA-34c are targets of p53 and cooperate in control of cell proliferation and adhesion-independent growth. Cancer Research. 2007;67(18):8433–8438. doi: 10.1158/0008-5472.CAN-07-1585. [DOI] [PubMed] [Google Scholar]
- 143.Bommer GT, Gerin I, Feng Y, et al. p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Current Biology. 2007;17(15):1298–1307. doi: 10.1016/j.cub.2007.06.068. [DOI] [PubMed] [Google Scholar]
- 144.Raver-Shapira N, Marciano E, Meiri E, et al. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Molecular Cell. 2007;26(5):731–743. doi: 10.1016/j.molcel.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 145.Chang T-C, Wentzel EA, Kent OA, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Molecular Cell. 2007;26(5):745–752. doi: 10.1016/j.molcel.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Welch C, Chen Y, Stallings RL. MicroRNA-34a functions as a potential tumor suppressor by inducing apoptosis in neuroblastoma cells. Oncogene. 2007;26(34):5017–5022. doi: 10.1038/sj.onc.1210293. [DOI] [PubMed] [Google Scholar]
- 147.Yamakuchi M, Ferlito M, Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(36):13421–13426. doi: 10.1073/pnas.0801613105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Duan W, Gao L, Wu X, et al. MicroRNA-34a is an important component of PRIMA-1-induced apoptotic network in human lung cancer cells. International Journal of Cancer. 2010;127(2):313–320. doi: 10.1002/ijc.25049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kuttan R, Bhanumathy P, Nirmala K, George MC. Potential anticancer activity of turmeric (Curcuma longa) Cancer Letters. 1985;29(2):197–202. doi: 10.1016/0304-3835(85)90159-4. [DOI] [PubMed] [Google Scholar]
- 150.Chiu T-L, Su C-C. Curcumin inhibits proliferation and migration by increasing the Bax to Bcl-2 ratio and decreasing NF-κBp65 expression in breast cancer MDA-MB-231 cells. International Journal of Molecular Medicine. 2009;23(4):469–475. doi: 10.3892/ijmm_00000153. [DOI] [PubMed] [Google Scholar]
- 151.Johnson SM, Gulhati P, Arrieta I, et al. Curcumin inhibits proliferation of colorectal carcinoma by modulating Akt/mTOR signaling. Anticancer Research. 2009;29(8):3185–3190. [PMC free article] [PubMed] [Google Scholar]
- 152.Chendil D, Ranga RS, Meigooni D, Sathishkumar S, Ahmed MM. Curcumin confers radiosensitizing effect in prostate cancer cell line PC-3. Oncogene. 2004;23(8):1599–1607. doi: 10.1038/sj.onc.1207284. [DOI] [PubMed] [Google Scholar]
- 153.Sandur SK, Ichikawa H, Pandey MK, et al. Role of pro-oxidants and antioxidants in the anti-inflammatory and apoptotic effects of curcumin (diferuloylmethane) Free Radical Biology and Medicine. 2007;43(4):568–580. doi: 10.1016/j.freeradbiomed.2007.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Zhang J, Du Y, Wu C, et al. Curcumin promotes apoptosis in human lung adenocarcinoma cells through miR-186* signaling pathway. Oncology Reports. 2010;24(5):1217–1223. doi: 10.3892/or_00000975. [DOI] [PubMed] [Google Scholar]
- 155.Choi BH, Kim CG, Lim Y, Shin SY, Lee YH. Curcumin down-regulates the multidrug-resistance mdr1b gene by inhibiting the PI3K/Akt/NFκB pathway. Cancer Letters. 2008;259(1):111–118. doi: 10.1016/j.canlet.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 156.Simoni D, Rizzi M, Rondanin R, et al. Antitumor effects of curcumin and structurally β-diketone modified analogs on multidrug resistant cancer cells. Bioorganic and Medicinal Chemistry Letters. 2008;18(2):845–849. doi: 10.1016/j.bmcl.2007.11.021. [DOI] [PubMed] [Google Scholar]
- 157.Zhang J, Zhang T, Ti X, et al. Curcumin promotes apoptosis in A549/DDP multidrug-resistant human lung adenocarcinoma cells through an miRNA signaling pathway. Biochemical and Biophysical Research Communications. 2010;399(1):1–6. doi: 10.1016/j.bbrc.2010.07.013. [DOI] [PubMed] [Google Scholar]
- 158.Zhu W, Xu H, Zhu D, et al. miR-200bc/429 cluster modulates multidrug resistance of human cancer cell lines by targeting BCL2 and XIAP. Cancer Chemotherapy and Pharmacology. 2012;69(3):723–731. doi: 10.1007/s00280-011-1752-3. [DOI] [PubMed] [Google Scholar]
- 159.Adam L, Zhong M, Choi W, et al. miR-200 expression regulates epithelial-to-mesenchymal transition in bladder cancer cells and reverses resistance to epidermal growth factor receptor therapy. Clinical Cancer Research. 2009;15(16):5060–5072. doi: 10.1158/1078-0432.CCR-08-2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Li A, Omura N, Hong S-M, et al. Pancreatic cancers epigenetically silence SIP1 and hypomethylate and overexpress miR-200a/200b in association with elevated circulating miR-200a and miR-200b levels. Cancer Research. 2010;70(13):5226–5237. doi: 10.1158/0008-5472.CAN-09-4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zhu W, Shan X, Wang T, Shu Y, Liu P. MiR-181b modulates multidrug resistance by targeting BCL2 in human cancer cell lines. International Journal of Cancer. 2010;127(11):2520–2529. doi: 10.1002/ijc.25260. [DOI] [PubMed] [Google Scholar]
- 162.Schaefer U, Voloshanenko O, Willen D, Walczak H. TRAIL: a multifunctional cytokine. Frontiers in Bioscience. 2007;12(10):3813–3824. doi: 10.2741/2354. [DOI] [PubMed] [Google Scholar]
- 163.Falschlehner C, Emmerich CH, Gerlach B, Walczak H. TRAIL signalling: decisions between life and death. International Journal of Biochemistry & Cell Biology. 2007;39(7-8):1462–1475. doi: 10.1016/j.biocel.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 164.Garofalo M, Quintavalle C, Di Leva G, et al. MicroRNA signatures of TRAIL resistance in human non-small cell lung cancer. Oncogene. 2008;27(27):3845–3855. doi: 10.1038/onc.2008.6. [DOI] [PubMed] [Google Scholar]
- 165.Rossi G, Cavazza A, Marchioni A, et al. Role of chemotherapy and the receptor tyrosine kinases KIT, PDGFRα, PDGFRβ, and Met in large-cell neuroendocrine carcinoma of the lung. Journal of Clinical Oncology. 2005;23(34):8774–8785. doi: 10.1200/JCO.2005.02.8233. [DOI] [PubMed] [Google Scholar]
- 166.Dierov J, Xu Q, Dierova R, Carroll M. Tel/platelet-derived growth factor receptor β activates phosphatidylinositol 3 (PI3) kinase and requires PI3 kinase to regulate the cell cycle. Blood. 2002;99(5):1758–1765. doi: 10.1182/blood.v99.5.1758. [DOI] [PubMed] [Google Scholar]
- 167.Yu J, Deuel TF, Kim H-RC. Platelet-derived growth factor (PDGF) receptor-α activates c-Jun NH2-terminal kinase-1 and antagonizes PDGF receptor-β-induced phenotypic transformation. The Journal of Biological Chemistry. 2000;275(25):19076–19082. doi: 10.1074/jbc.M910329199. [DOI] [PubMed] [Google Scholar]
- 168.Garofalo M, Di Leva G, Romano G, et al. miR-221&222 Regulate TRAIL Resistance and Enhance Tumorigenicity through PTEN and TIMP3 Downregulation. Cancer Cell. 2009;16(6):498–509. doi: 10.1016/j.ccr.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 169.Garofalo M, Jeon Y-J, Nuovo GJ, et al. MiR-34a/c-dependent PDGFR-α/β downregulation inhibits tumorigenesis and enhances TRAIL-induced apoptosis in lung cancer. PLoS ONE. 2013;8(6) doi: 10.1371/journal.pone.0067581.e67581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Condorelli G, Vigliotta G, Cafieri A, et al. PED/PEA-15: an anti-apoptotic molecule that regulates FAS/TNFR1-induced apoptosis. Oncogene. 1999;18(31):4409–4415. doi: 10.1038/sj.onc.1202831. [DOI] [PubMed] [Google Scholar]
- 171.Condorelli G, Vigliotta G, Iavarone C, et al. PED/PEA-15 gene controls glucose transport and is overexpressed in type 2 diabetes mellitus. The EMBO Journal. 1998;17(14):3858–3866. doi: 10.1093/emboj/17.14.3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Renault F, Formstecher E, Callebaut I, Junier M-P, Chneiweiss H. The multifunctional protein PEA-15 is involved in the control of apoptosis and cell cycle in astrocytes. Biochemical Pharmacology. 2003;66(8):1581–1588. doi: 10.1016/s0006-2952(03)00514-8. [DOI] [PubMed] [Google Scholar]
- 173.Kitsberg D, Formstecher E, Fauquet M, et al. Knock-out of the neural death effector domain protein PEA-15 demonstrates that its expression protects astrocytes from TNFα-induced apoptosis. Journal of Neuroscience. 1999;19(19):8244–8251. doi: 10.1523/JNEUROSCI.19-19-08244.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Zanca C, Garofalo M, Quintavalle C, et al. PED is overexpressed and mediates TRAIL resistance in human non-small cell lung cancer. Journal of Cellular and Molecular Medicine. 2008;12(6A):2416–2426. doi: 10.1111/j.1582-4934.2008.00283.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Incoronato M, Garofalo M, Urso L, et al. miR-212 increases tumor necrosis factor-related apoptosis-inducing ligand sensitivity in non-small cell lung cancer by targeting the antiapoptotic protein PED. Cancer Research. 2010;70(9):3638–3646. doi: 10.1158/0008-5472.CAN-09-3341. [DOI] [PubMed] [Google Scholar]