

Abstract
Chronic post-natal hyperoxia reduces the hypoxic ventilatory response by reducing the carotid body sensitivity to acute hypoxia as demonstrated by a reduced afferent nerve response, reduced calcium response of carotid body glomus cells and reduced catecholamine secretion in response to acute hypoxia. The present study examined whether hyperoxia alters the electrophysiological characteristics of glomus cells. Rats were treated with hyperoxia for 1 week starting at P1 or P7 and for 2 weeks starting at P1 followed by harvesting and dissociation of their carotid bodies for whole cell, perforated-patch recording. As compared to glomus cells from normoxia animals, hyperoxia treated cells showed a significant reduction in the magnitude of depolarization in response to hypoxia and anoxia, despite little change in the depolarizing response to 20 mM K+. Resting cell membrane potential in glomus cells from rats exposed to hyperoxia from P1 to P15 and studied at P15 was slightly depolarized compared to other treatment groups and normoxia-treated cells, but conductance normalized to cell size was not different among groups. We conclude that postnatal hyperoxia impairs carotid chemoreceptor hypoxia transduction at a step between hypoxia sensing and membrane depolarization. This occurs without a major change in baseline electrophysiological characteristics, suggesting altered signaling or alterations in the relative abundance of different leak channel isoforms.
Keywords: carotid body, chemoreceptor, potassium channel, oxygen sensing, development
1. Introduction
The acute hypoxic ventilatory response (HVR) is markedly attenuated in adult animals exposed to perinatal hyperoxia in the newborn period (Bavis et al., 2011a). This plasticity only occurs if the hyperoxia exposure is during the first month of life in rats, a ‘critical period’ of carotid body development (Carroll, 2003). In contrast, equivalent chronic hyperoxia exposure of adult animals does not produce lasting respiratory impairment (Ling et al, 1996). The issue is of some clinical importance because of the prevalent use of oxygen supplementation in the clinical setting, for instance, in the treatment of premature infants.
This attenuation of the HVR is due to carotid chemoreceptor dysfunction as evidenced by an impaired whole carotid sinus nerve response to moderate hypoxia, asphyxia and cyanide which are, at least in part, due to axonal loss (Bisgard et al., 2003; Erickson et al., 1998) and a reduced number of oxygen-sensing cells (glomus cells) within the carotid body (CB) (Dmitrieff et al., 2012). Besides axonal loss of chemoreceptor afferents, perinatal hyperoxia impairs the spiking response of the remaining chemoreceptor fibers, a reversible impairment that lasts less than 7–8 days following a return to normoxia (Bavis et al., 2011b).
The impairment of organ function in the immediate post-hyperoxic period appears to be due to an alteration in the biophysical response of the glomus cell to acute hypoxia. Although acute hypoxia generally raises intracellular calcium ([Ca2+]i) levels in glomus cells, the response is severely blunted by hyperoxia exposure. For instance, hyperoxia exposure of neonatal rats from P7 to P12 blunts the [Ca2+]i response to acute hypoxia challenge and nearly abolishes the response when hyperoxia is continued to P14 (Donnelly et al., 2009). As described for single-unit activity, this profound impairment of glomus cell [Ca2+]i responses to acute hypoxia can be fully reversed after 7–8 days of recovery in normoxia (Bavis et al., 2011b). As expected for the blunted calcium response, hypoxia-induced vesicle release from glomus cells is similarly reduced by hyperoxia exposure (Donnelly et al., 2009).
The mechanism of hyperoxia-induced blunting of the glomus cell [Ca2+]i response to acute hypoxia is unknown. It is well-established that the rise in [Ca2+]i with acute hypoxia is primarily due to depolarization and calcium influx via voltage-gated calcium channels; intracellular calcium release appears to play only a minor role (Buckler and Vaughan-Jones, 1994). Hyperoxia could potentially impair this process by several mechanisms. For instance, hyperoxia may ablate the oxygen-sensing signal or induce expression of channel that stabilize membrane potential at hyperpolarized levels. The present study was undertaken to determine whether perinatal hyperoxia exposure alters the resting electrophysiological characteristics of CB glomus cells and/or leads to blunting of cell membrane depolarization in response to acute hypoxia. Some of these experiments were presented at the meeting of the International Society for Arterial Chemoreception (ISAC) in 2011 (Kim et al., 2012).
2. Methods
2.1 Ethical approval
The use of animals in this study was approved by the Animal Care and Use Committee of the University of Arkansas for Medical Sciences.
2.2 Animal model
Experiments were conducted on Sprague-Dawley rat pups of both sexes. Starting on postnatal day 1 (P1) or postnatal day 7 (P7) rats were placed in an environmental chamber (OxyCycler, BioSpherix) for one week or two weeks. For exposure starting at P1, pregnant rats were placed in the chamber 1–2 days prior to expected delivery and were allowed to give birth. The chamber atmosphere was maintained at 60% O2 and chamber CO2 was maintained under 0.2% by a controlled leak; both variables were continuously monitored (AnaWin2 Run-Time, ver. 2.4.17, Watlow-Anafaze). Animals remained in the chamber until studied at P8 or P15. Control animals maintained in normoxia were housed in the same room and carotid bodies were harvested at the same ages.
Three experimental groups were employed. The first utilized hyperoxia exposure from P1 to P8, which we have previously demonstrated causes a severe impairment of the afferent nerve response to hyperoxia (Donnelly et al., 2009). Comparison is made to a control group raised in normoxia from P1 to P8. The second group was exposed to hyperoxia from P1 to P15 and compared to a control group raised in normoxia from P1 to P15 to determine whether a longer treatment period produced greater effects. In the third group, rats were raised in normoxia from P1 to P7, exposed to hyperoxia from P7 to P15 and compared to control rats raised in normoxia from P1 to P15. This initial exposure to normoxia from P1 to P7 allowed for the normal postnatal maturation of chemoreceptor function to take place prior to hyperoxia exposure. These three groups are referred to as Hyper 1–8, Hyper 7–15 and Hyper 1–15 respectively.
2.3 Carotid body cell isolation and experimental groups
Carotid bodies were harvested as previously described (Wasicko et al., 2006). Rat pups were anesthetized with isoflurane, decapitated, and the heads placed in ice-cold buffered saline solution (BSS: 118 mM NaCl, 23 mM NaHCO3, 3 mM KCl, 2 mM KH2PO4, 1.2 mM CaCl2, 1 mM MgCl2, 10 mM Glucose, pH 7.2). CBs were isolated and placed in ice-cold low Ca2+/Mg2+ phosphate buffered saline solution (PBS: 137 mM NaCl, 2.8 mM KCl, 2 mM KH2PO4, 0.07 mM CaCl2, 0.05 mM MgCl2, pH 7.4). Each CB was sectioned 2~3 times and placed in a solution containing trypsin (1mg/mL) and collagenase (1mg/mL) in low Ca2+/Mg2+ PBS and incubated at 37°C for 20–25 minutes. CBs were gently triturated using a fire polished Pasteur pipette to mechanically dissociate the cells. Enzymatic digestion was continued for an additional few minutes when necessary. CB growth medium (Ham’s F-12, 10% fetal bovine serum, 23 mM glucose, 2 mM L-alanyl-glutamine (Glutamax-1), 10K units penicillin/streptomycin, and 0.08 unit/ml insulin) was added to stop enzyme activity. After brief trituration, the solution containing the dissociated CBs was centrifuged for 5 minutes at ~2000 xg using a microcentrifuge. Supernatant was removed and warm CB growth media were added to resuspend the pellet. This step was repeated to remove traces of enzymes. A small drop of suspended CB cells was placed on glass coverslips coated with poly-D-lysine, and incubated at 37°C for 40–45 minutes to allow settling and attachment of the cells. CB growth medium was further added to the cells and then incubated at 37°C for additional 2 hours. Coverslips were then transferred to the recording chamber for electrophysiological experiments. Clusters of 2–5 cells were identified and cells within this cluster that showed a granular cell surface were chosen for electrophysiological studies.
2.4 Electrophysiological studies
All voltage and current clamp recordings were performed using the perforated patch whole-cell recording technique. Experiments were conducted using an EPC9 amplifier (HEKA). Current clamp protocols were generated using Patchmaster software (HEKA). Electrodes were made from borosilicate glass capillaries (Warner Instrument Corp.). The pipette solution for all recording conditions contained (mM): 70 K2SO4, 30 KCl, 2 MgCl2, 1 EGTA and 10 HEPES; pH was adjusted to 7.2 with NaOH. The pipette tips were filled with this solution then back-filled with the same solution containing 240–360 μg/ml amphotericin B. Pipette resistance with this solution was approximately 2-3 MΩ. The bath was grounded through an Ag–AgCl pellet with a 3 M KCl agar-bridge. Only recordings in which the access resistance <100MΩ were accepted. Voltage measures were not corrected for the loss of the liquid junction potential between the pipette and bath solution (~7.4 mV) and represents a constant value in all recordings.
The standard bicarbonate-buffered perfusion solution contained (mM): 117 NaCl, 4.5 KCl, 23 NaHCO3, 1 MgCl2, 2.5 CaCl2, and 12 glucose equilibrated with either 21% O2/5% CO2 /balance N2 or 0% O2/5% CO2 / balance N2 at 35°C: pH was adjusted to 7.4 with NaOH. The same perfusion solution was used for measurement of responses to high extracellular K+ except for equimolar substitution of 20 mM KCl for NaCl. Osmolarity was 290–300 mOsm for all solutions. The temperature of the perfusion solutions was kept at 35.0±0.5°C and the rate of perfusion was 1.8–2.2 ml/min. The partial pressure of O2 was monitored in the perfusion line using a flow-through probe (OM-4 Oxygen Meter, Microelectrode, Inc.) and in the chamber using needle oxygen electrode (Diamond General Development Corp, Ann Arbor, MI); electrodes were calibrated using 0% O2 solution (gassed with 95% N2/5% CO2/ balance N2 for 60 min followed by addition of 0.5 mM dithionite) and 21% O2 solution (gassed with air/5% CO2 for 60 min at 35°C). Solutions were delivered to the recording chamber through gas-impermeable stainless steel tubing. By using a small volume (<100 μl) recording chamber, the bath exchange could be achieved in <0.5 min.
The passive membrane characteristics of membrane capacitance (Cm), membrane resistance (Rm), and access resistance (Ra) were estimated from the response to a step hyperpolarization of 20 mV from a holding potential of −80 mV. The current response was fitted to a single-order exponential. Ra was estimated from the magnitude of the current step at the start of the hyperpolarization and Rm was estimated from the steady-state current response. Cm was estimated by dividing the time constant of the exponential by Ra. The normalized conductance was calculated by 1/ (Cm × Rm).
The membrane depolarization was recorded in current clamp mode (I=0). Perforated patch recordings were formed on CB glomus cells during perfusion with a bicarbonate-buffered solution previously gassed with air/5% CO2 gas mixture for ~60 min. After measurement of baseline electrophysiologic parameters, high K+ extracellular solution was applied for 1–2 min. The effects of hypoxia and anoxia were studied by switching between normoxic, hypoxic (0% O2) and anoxic (0% O2 with glucose oxidase + catalase or dithionite) perfusion solution for 2–3 min. Although the hypoxic solution was bubbled with 0% oxygen, some oxygen was added to the perfusate by diffusion in the open chamber, such that chamber oxygen tension was approximately 5–6 torr. Anoxic solutions were achieved by adding an oxygen scavenger, glucose oxidase (30 U/ml) and catalase (160 U/ml) (GOC) or 0.5 mM sodium dithionite (dithio: Na2S2O4) into hypoxic solution (0% O2). Experiments were excluded if they did not exhibit recovery of membrane potential after hypoxia challenge.
2.5 Data analysis
Data obtained from current clamp records were analyzed with Origin 8 (Microcal Corp). All values are presented as mean ± SEM; n is the number of independent observations, considered as separate clusters of cells. Statistical comparisons for maximal membrane depolarization were performed using one way ANOVA with Tukey-Kramer test for post-hoc multiple comparisons among all groups (>3 groups) using GraphPad Prism 4.02. The glomus cell membrane characteristics were performed by unpaired t-test (two-tailed) for P8 and Hyper 1–8, and one way ANOVA with Tukey-Kramer test for post-hoc multiple comparisons among P15, Hyper 7–15 and Hyper 1–15. A p-value <0.05 was considered statistically significant.
3. Results
3.1 Effect of hyperoxia-treatment on baseline properties
Despite the major effect of postnatal hyperoxia exposure on chemoreceptor function, hyperoxia exposure had little effect on the baseline electrophysiological parameters of glomus cells. Resting membrane potential was not different between experimental and control groups, except for a slightly less negative resting Vm in the group treated with hyperoxia from P1-P15 (Table 1). Input resistance (Rm) was significantly increased in the Hyper 1–8 group (Table 1), but this was associated with a significant reduction in membrane capacitance (Table 1). After normalization of cell conductance to cell surface area (capacitance), hyperoxia treatment had no significant effect on conductance for any treatment group (Table 1). Thus, hyperoxia treatment does not appear to affect the normalized resting conductance of CB glomus cells.
Table 1.
Electrophysiological membrane characteristics of CB glomus cells: Cell capacitance (Cm), membrane resistance (Rm), normalized conductance and resting membrane potentials (Vm) were measured and calculated from CB glomus cells of normoxia pups and glomus cells of hyperoxia treated pups by perforated patch clamp.
| Cm (pF) | Rm (GΩ) | Norm. Conduct. (nS/pF) |
Vm (mV) | |
|---|---|---|---|---|
| Nomoxia 8 (P8) | 3.15 ± 0.18 (22) | 1.48 ± 0.14 (15) | 0.25±0.04 (15) | −45.15 ± 1.69 (19) |
| Hyperoxia 1–8 (Hyper 1–8) | 2.15 ± 0.16 (8) ** | 2.23 ± 0.36 (5) * | 0.24±0.04 (5) | −42.20 ± 2.93 (5) |
| Normoxia 15 (P15) | 2.99 ± 0.12 (24) | 2.23 ± 0.31 (13) | 0.20±0.03 (13) | −45.70 ± 2.01 (18) |
| Hyperoxia 7–15 (Hyper 7–15) | 2.68 ± 0.09 (31) | 1.94 ± 0.21 (21) | 0.23±0.03 (21) | −41.91 ± 0.97 (17) |
| Hyperoxia 1–15 (Hyper 1–15) | 2.50 ± 0.12 (19) * | 2.04 ± 0.24 (13) | 0.22±0.03 (13) | −37.65 ± 1.47 (11) * |
p<0.05 and
p<0.01 different from same-age normoxia control.
“n” shown in parentheses.
3.2 CB glomus cell membrane depolarization responses to elevated extracellular K+ and hypoxia
Membrane depolarization responses to acute hypoxia, anoxia and 20 mM KCl were studied in Hyper 1–8, Hyper 7–15 and Hyper 1–15 groups and compared to age matched normoxia control pups at P8 and P15. Representative current clamp traces are shown in figure 1.
Figure 1.
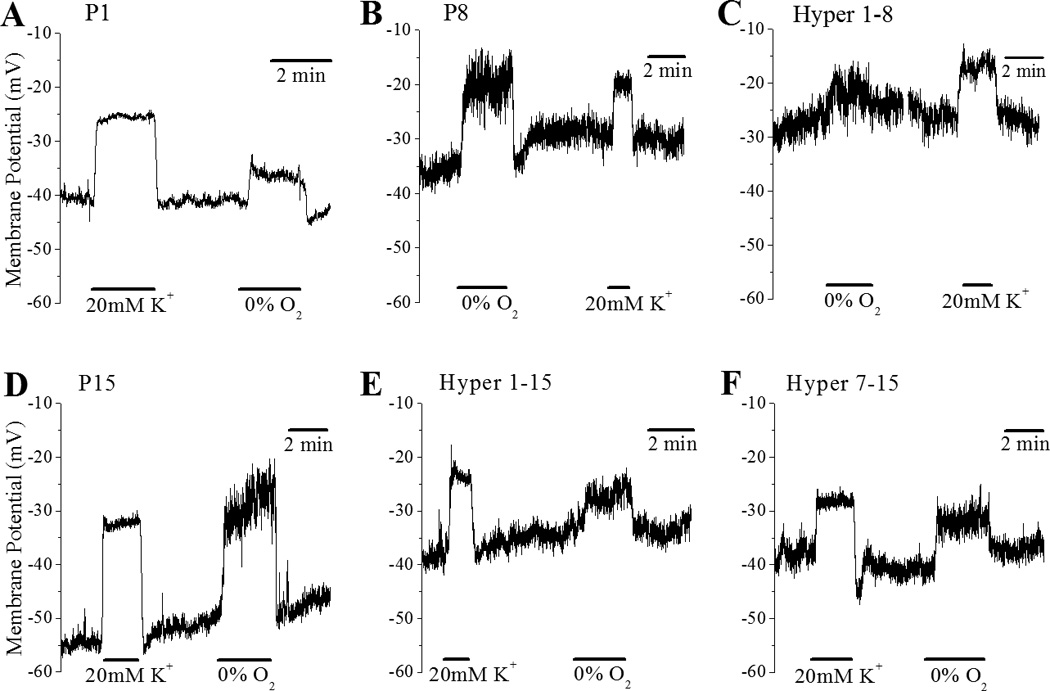
The effects of 20mM KCl and hypoxia (0% O2) on membrane potential in dissociated CB glomus cells at various ages, P1 (A), P8 (B & C) and P15 (D-F), which were reared in normoxia (A,B and D) or in various durations of hyperoxia exposure (60% O2), from P1 to P8 (C), from P1 to P15 (E) and from P7 to P15 (F).
3.2.1 Development
Consistent with previous findings from our laboratory (Wasicko et al., 2006), there were no developmental changes in the magnitude of depolarization in response to 20 mM K+ in normoxia controls at P1, P8 and P15 cell (Fig. 2A). In sharp contrast, the magnitude of hypoxia induced depolarization in response to 0% O2 (hypoxia) more than doubled by P8, from 7.11±0.84 mV at P1 to 17.01±1.36 mV at P8 and 17.56±0.93 mV at P15 (Fig. 2B). Postnatal maturation was also associated with an approximate doubling of the glomus cell depolarization response to anoxia, from 12.65±1.35 mV at P1 (n=8) to 23.96±1.85 mV (n=26) and 24.35±1.56 mV (n= 25) at P8 and P15 respectively (p<0.001).
Figure 2.
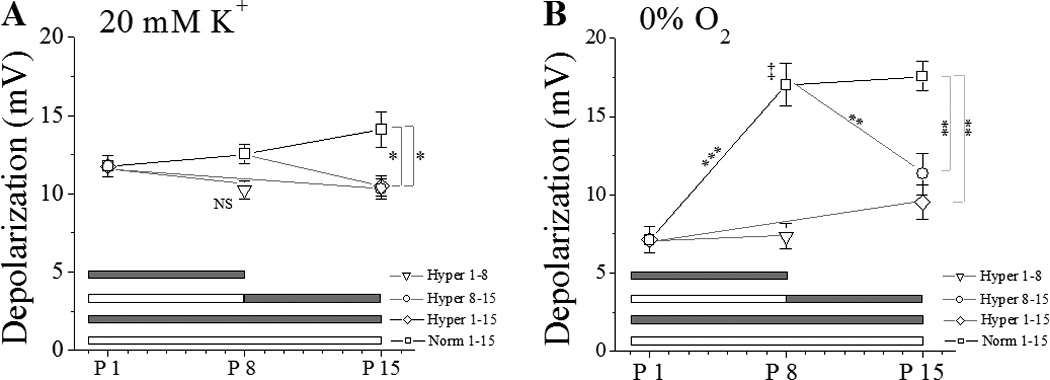
Membrane depolarization (mean±SEM) caused by 20 mM KCl (A) or 0% O2 (B) in glomus cells from hyperoxia-exposed (60% O2) or normoxia-reared rat pups. White bar = normoxia, gray bar = hyperoxia exposure. Statistical significance: p<0.05 (*), p<0.01 (**), p<0.001 (***), Hyperoxia P1-P8 different from normoxia group at P8 (P<0.01, ‡).
3.2.2 Effect of hyperoxia
One week of hyperoxia treatment starting at birth (Hyper 1–8) did not significantly affect 20 mM K+ induced depolarization (Fig. 2A), but hyperoxia treatment both for 1 week starting at P7 (Hyper 7–15) and 2 weeks starting at birth (Hyper 1–15) were associated with a slight but statistically significant reduction in the depolarization response to 20 mM K+ (Fig. 2).
Hyperoxia exposure profoundly reduced hypoxia-induced glomus cell membrane depolarization (Fig 2B). When started at birth, hyperoxia exposure abolished the normal postnatal increase in the depolarization response to acute hypoxia challenge, measured at P8 and P15 (Fig 2B). Even when the onset of hyperoxia exposure was delayed until P8, a time when the depolarization response to hypoxia appears to be fully developed in the control group, hyperoxia for a week (P7 to P15) markedly reduced the glomus cell depolarization response to acute hypoxia challenge (Fig. 2B). Hyperoxia exposure also reduced glomus cell membrane depolarization responses to anoxia, compared to same-age normoxia controls, to 11.01±1.38 mV after Hyper 1–8 (n=12, p<0.001), 13.60±1.22 after Hyper 7–15 (n=23, p<0.001) and 16.71±1.27 mV after Hyper 1–15 (n=18, p<0.01).
4. Discussion
The main conclusions from the present study are that environmental hyperoxia reduces the magnitude of glomus cell depolarization in response to acute hypoxia without affecting normalized resting conductance of CB glomus cells. Exposure to hyperoxia from birth prevented the normal postnatal increase in the glomus cell depolarization response to acute hypoxia, which normally takes place during the first two weeks of life. Hyperoxia exposure starting at P7, when the depolarization response to acute hypoxia was already mature in controls, reduced the glomus cell depolarization response by P15 to a level not significantly different from P1. These results demonstrate profound environment-driven plasticity in postnatal glomus cell maturation, at a level in the transduction cascade upstream to the Vm depolarization induced by acute hypoxia.
Previous studies indicate that hyperoxia from birth leads to loss of unmyelinated axons in the carotid sinus nerve (CSN), degenerative changes in petrosal neurons and marked CB hypoplasia, in part due to inhibition of glomus cell proliferation early in the exposure(Dmitrieff et al., 2012; Erickson et al., 1998). Previous studies from our laboratory showed that perinatal hyperoxia exposure impairs the spiking response of the remaining carotid chemoreceptor units, reduces hypoxia-induced catecholamine secretion, slows carotid sinus nerve conduction time and markedly blunts the glomus cell intracellular calcium response to acute hypoxia challenge (Donnelly et al., 2009; Donnelly et al., 2005). Despite these functional and structural changes, a recovery period in normoxia lasting 5–7d allows full recovery of single unit afferent nerve responses and intracellular calcium responses to acute hypoxia following return to normoxia (Bavis et al., 2011b). However, the axonal loss and glomus cell loss likely persist, accounting for the lower HVR. Thus, hyperoxia exposure exerts both pre-synaptic and post-synaptic actions to reduce the CB response to acute hypoxia.
Hypoxia causes glomus cell membrane depolarization, resulting in influx of extracellular calcium via voltage-gated calcium channels. The increased glomus cell [Ca2+]i in turn leads to release of a variety of neurotransmitters, including catecholamines, via exocytosis (Hansen, 1981). The results of the present study are consistent with the hypothesis that hyperoxia impairs glomus cell O2 responsiveness by blunting cell membrane depolarization in response to acute hypoxia, thereby preventing or reducing the rise in [Ca2+]i and its downstream effects (release of neurotransmitters, single unit spiking activity).
Major blunting of the glomus cell [Ca2+]i response to hypoxia would not necessarily require elimination of the depolarization response to hypoxia. As the rise in [Ca2+]i depends primarily on calcium influx via L-type voltage-gated calcium channels, a reduction of hypoxia-induced calcium influx may potentially be caused by a left shift in membrane potential (i.e., hyperpolarization), stabilization of membrane potential (e.g. activation of an anion current) or a reduced activation of calcium currents due to a shift in voltage dependence or expression level. Preliminary results using PCR showed a reduction in L-type calcium channel expression following hyperoxia exposure (Kim et al., 2012), but calcium influx caused by high K+ was not affected by 1 week of hyperoxia (Donnelly et al., 2009), suggesting no functional change in glomus cell calcium currents There are alternate possibilities to a reduction in the hypoxia-induced depolarizing current. For instance, expression of leak K+ channels or anion channels could stabilize the resting potential at hyperpolarized levels or an enhanced electrogenic Na+/K+ activity could shift the resting potential in a hyperpolarized direction. However, our results clearly show that hyperoxia exposure did not alter normalized conductance and only minimally affected glomus cell resting Vm, but did substantially reduce the magnitude of hypoxia-induced depolarization. At P8, after 1 week of hyperoxia, and P15, after 2 weeks of hyperoxia, the magnitude of hypoxia-induced depolarization was close to that observed in glomus cells from newborn rats, which also have a very small [Ca2+]i responses to hypoxia (Wasicko et al., 2006; Wasicko et al., 1999).
How does chronic hyperoxia exposure reduce the magnitude of depolarization caused by acute hypoxia challenge? Although the mechanism of hypoxia-induced depolarization is not well-understood, the present view is that rat glomus cells have resting potassium currents, believed to be largely carried by hypoxia-sensitive TASK-1/3 channels (Kim et al., 2009) as well as standing sodium current (Carpenter and Peers, 2001). The standing Na+ current is hypoxia insensitive and provides a constant depolarizing force, which drives cell-membrane depolarization when counterbalancing currents (eg., resting K+ currents) are inhibited (Carpenter and Peers, 2001). We did not measure the Na+ current, but our findings that resting Vm, normalized conductance and glomus cell depolarization in response to elevated extracellular K+ were either not affected or minimally altered by chronic hyperoxia, suggests that the standing Na+ current remains intact after hyperoxia treatment. In addition, a reduction in the standing Na+ current should result in a leftward (hyperpolarized) shift glomus cell membrane potential and increase input resistance; neither occurred. The membrane potential shifted slightly rightward (depolarized) after two weeks of hyperoxia treatment (Hyper 1–15, Table 1).
The finding that glomus cells were significantly depolarized following hyperoxia treatment from P1 to P15 suggests that prolonged hyperoxia treatment may affect expression of ion channels involved in determining resting Vm. Previously we reported that TASK1 and TASK3 mRNAs were significantly reduced following hyperoxia treatment (Bavis et al., 2011b; Kim et al., 2012) and a reduction in TASK1/TASK3 channel protein may explain the reduced depolarization to increased K+. As the TASK1/3 heterodimer appears to be the normally-expressed oxygen sensitive leak channel (Kim et al., 2009), expression of other leak-type channels may have compensated for a reduction in TASK1/TASK3 levels. This suggestion is supported by the observation that carotid bodies remain functional in mice lacking both TASK1 and TASK3 channels, so other leak channels must be assuming the roles of TASK1/TASK3 (Ortega-Saenz et al., 2010). Alternatively, mRNA levels may not reflect the amount of channel protein, which may remain unchanged. Thus, long-term hyperoxia exposure (days to weeks) would likely affect glomus cell oxygen sensor(s) and/or signaling between O2 sensor(s) and effectors (cell membrane K+ channels). At the present time, the identity of glomus cell O2 sensor(s) and signaling pathways are not resolved.
In the present study, a one week hyperoxia exposure resulted in complete absence of the normal, developmental increase in the cell membrane depolarization response seen in age-matched controls and two weeks of hyperoxia extended this effect (Fig. 2B). We previously reported that two weeks of hyperoxia exposure markedly blunted both nerve and [Ca2+]i responses to acute hypoxia (Donnelly et al., 2009; Donnelly et al., 2005). This effect seems to be independent of timing, at least within the perinatal period, as a one week hyperoxia exposure effected similar changes whether presented at P1 or P7 (Fig. 2B). Similarly, we previously reported that a one week hyperoxia exposure, beginning on P7, also markedly blunted both nerve and glomus cell [Ca2+]i responses to acute hypoxia (Donnelly et al., 2009; Donnelly et al., 2005). Thus, the observed timing of the effects of hyperoxia are entirely consistent with previous observations on single unit spiking activity and [Ca2+]i responses to hypoxia (Donnelly et al., 2009; Donnelly et al., 2005).
In conclusion, the present results demonstrate that suppression of peripheral chemoreceptor function by perinatal hyperoxia exposure is due to a decreased hypoxia-induced depolarizing current. Since baseline electrophysiological variables (Vm, Ro) were largely unchanged, this suggests that the site of impairment is between the oxygen sensor and leak K+ channel.
Highlights.
Perinatal hyperoxia exposure markedly reduces rat carotid body glomus cell membrane depolarization in response to hypoxia.
Hyperoxia during development has little effect on glomus cell baseline electrophysiological characteristics.
Hyperoxia during development has no effect on glomus cell resting conductance normalized to cell size.
Impairment of carotid body function by perinatal hyperoxia is exposure appears to be due to a decreased hypoxia-induced depolarizing current.
Acknowledgements
This work was supported by NIH R01 HL084520.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bavis RW, Dmitrieff EF, Young KM, Piro SE. Hypoxic ventilatory response of adult rats and mice after developmental hyperoxia. Respir Physiol Neurobiol. 2011a;177:342–346. doi: 10.1016/j.resp.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bavis RW, Kim I, Pradhan N, Nawreen N, Dmitrieff EF, Carroll JL, Donnelly DF. Recovery of carotid body O2 sensitivity following chronic postnatal hyperoxia in rats. Respir Physiol Neurobiol. 2011b;177:47–55. doi: 10.1016/j.resp.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisgard GE, Olson EB, Jr, Wang ZY, Bavis RW, Fuller DD, Mitchell GS. Adult carotid chemoafferent responses to hypoxia after 1, 2, and 4 wk of postnatal hyperoxia. J Appl Physiol. 2003;95:946–952. doi: 10.1152/japplphysiol.00985.2002. [DOI] [PubMed] [Google Scholar]
- Buckler KJ, Vaughan-Jones RD. Effects of hypoxia on membrane potential and intracellular calcium in rat neonatal carotid body type I cells. J Physiol. 1994;476:423–428. doi: 10.1113/jphysiol.1994.sp020143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter E, Peers C. A standing Na+ conductance in rat carotid body type I cells. Neuroreport. 2001;12:1421–1425. doi: 10.1097/00001756-200105250-00025. [DOI] [PubMed] [Google Scholar]
- Carroll JL. Developmental plasticity in respiratory control. J Appl.Physiol. 2003;94:375–389. doi: 10.1152/japplphysiol.00809.2002. [DOI] [PubMed] [Google Scholar]
- Dmitrieff EF, Piro SE, Broge TA, Jr, Dunmire KB, Bavis RW. Carotid body growth during chronic postnatal hyperoxia. Respir Physiol Neurobiol. 2012;180:193–203. doi: 10.1016/j.resp.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly DF, Bavis RW, Kim I, Dbouk HA, Carroll JL. Time course of alterations in pre- and post-synaptic chemoreceptor function during developmental hyperoxia. Respir Physiol Neurobiol. 2009;168:189–197. doi: 10.1016/j.resp.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly DF, Kim I, Carle C, Carroll JL. Perinatal hyperoxia for 14 days increases nerve conduction time and the acute unitary response to hypoxia of rat carotid body chemoreceptors. J Appl Physiol. 2005;99:114–119. doi: 10.1152/japplphysiol.01009.2004. [DOI] [PubMed] [Google Scholar]
- Erickson JT, Mayer C, Jawa A, Ling L, Olson EB, Jr, Vidruk EH, Mitchell GS, Katz DM. Chemoafferent degeneration and carotid body hypoplasia following chronic hyperoxia in newborn rats. J Physiol. 1998;509(Pt 2):519–526. doi: 10.1111/j.1469-7793.1998.519bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen JT. Morphological aspects of secretion in the glomus cell paraneurons of the carotid body: evidence for calcium-dependent exocytosis. Cytobios. 1981;32:79–88. [PubMed] [Google Scholar]
- Kim D, Cavanaugh EJ, Kim I, Carroll JL. Heteromeric TASK-1/TASK-3 is the major oxygen-sensitive background K+ channel in rat carotid body glomus cells. J Physiol. 2009;587:2963–2975. doi: 10.1113/jphysiol.2009.171181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, Donnelly DF, Carroll JL. Postnatal hyperoxia iImpairs acute oxygen sensing of rat glomus cells by reduced membrane depolarization. Advances in Experimental Medicine and Biology. 2012;758:49–54. doi: 10.1007/978-94-007-4584-1_7. [DOI] [PubMed] [Google Scholar]
- Ortega-Saenz P, Levitsky KL, Marcos-Almaraz MT, Bonilla-Henao V, Pascual A, Lopez-Barneo J. Carotid body chemosensory responses in mice deficient of TASK channels. J Gen Physiol. 2010;135:379–392. doi: 10.1085/jgp.200910302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasicko MJ, Breitwieser GE, Kim I, Carroll JL. Postnatal development of carotid body glomus cell response to hypoxia. Respir Physiol Neurobiol. 2006;154:356–371. doi: 10.1016/j.resp.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Wasicko MJ, Sterni LM, Bamford OS, Montrose MH, Carroll JL. Resetting and postnatal maturation of oxygen chemosensitivity in rat carotid chemoreceptor cells. J Physiol. 1999;514(Pt 2):493–503. doi: 10.1111/j.1469-7793.1999.493ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]