

Abstract
Proteins with expanded polyglutamine (polyQ) segments cause a number of fatal neurodegenerative disorders, including Huntington’s disease (HD). Previous high-throughput screens in cellular and biochemical models of HD have revealed compounds that mitigate polyQ aggregation and proteotoxicity, providing insight into the mechanisms of disease and leads for potential therapeutics. However, the structural diversity of natural products has not yet been fully mobilized toward these goals. Here, we have screened a collection of ~11 000 natural product extracts for the ability to recover the slow growth of ΔProQ103-expressing yeast cells in 384-well plates (Z’ ~ 0.7, CV ~ 8%). This screen identified actinomycin D as a strong inhibitor of polyQ aggregation and proteotoxicity at nanomolar concentrations (~50–500 ng/mL). We found that a low dose of actinomycin D increased the levels of the heat-shock proteins Hsp104, Hsp70 and Hsp26 and enhanced binding of Hsp70 to the polyQ in yeast. Actinomycin also suppressed aggregation of polyQ in mammalian cells, suggesting a conserved mechanism. These results establish natural products as a rich source of compounds with interesting mechanisms of action against polyQ disorders.
Keywords: heat shock protein 70, high throughput screening, Huntington’s disease, molecular chaperones
Nine related neurodegenerative diseases, including Huntington’s disease (HD), are caused by genetic instability and expansion of polyglutamine (polyQ) segments in key proteins (1,2). In these diseases, the length of the polyQ segment determines the severity and age-of-onset of symptoms (3). For example, polyQ regions of ~40 residues in the huntingtin protein lead to adult onset HD, while segments longer than 100 are observed in juvenile cases. These elongated polyQ segments destabilize the disease-associated proteins, such as huntingtin or the androgen receptor, leading to misfolding, mislocalization and aggregation (3-5). In turn, oligomers of the misfolded, polyQ-containing protein are thought to disrupt numerous essential cellular processes, including transcription and protein turnover (6-9). Because of the numerous pathways impacted by polyQ aggregation, it is thought that preventing the initial formation of oligomers might be the most promising way to mitigate disease.
Expression of proteins with elongated polyQ segments leads to the formation of proteotoxic aggregates in cultured cells and animals, providing models for what occurs in HD patients. These disease models have been particularly useful for performing genetic screens (10-12), and many of these screens have identified molecular chaperones, such as heat-shock protein 70 (Hsp70) and heat-shock protein 90 (Hsp90), as key regulators of polyQ aggregation (13-15). For example, early studies in Saccharomyces cerevisae showed that overexpression of Hsp70 suppressed aggregation of an elongated polyQ fragment containing 103 glutamines (Q103; 16-18). Similar studies in other model systems, such as flies and mice, have confirmed this relationship (19-25). In addition to a role for molecular chaperones, these genetic screens have also pointed to important roles for the endocytosis and protein turnover pathways (22,26-29). Together, these studies in HD models have helped clarify the mechanisms of polyQ aggregation and proteotoxicity (10,30).
Another use of polyQ models is in high-throughput chemical screens. For example, Baldo et al. (31) recently used an engineered mouse hippocampal cell line (HN10) to screen approximately two million Novartis compounds for those that reduce the levels of a Q72 fragment. They reported that inhibitors of Hsp90 substantially reduced the levels of Q72 in embryonic stem cells and neurons, suggesting a path toward clinical use of Hsp90 inhibitors in HD. A conceptually similar screen was performed in a S. cerevisae model; a library of commercial 16 000 compounds was screened for those that restore the slow growth of cells expressing Q103 (32). Other screens have identified molecules that directly inhibit aggregation of polyQ fragments, using in vitro formats (33,34) and cell-based fluorescence resonance energy transfer (FRET; 35) and high-content screening (HCS; 36) assays. Together, these studies have provided promising lead compounds for drug discovery in HD and other polyQ disorders (18,32,37,38). Moreover, these chemical screens have also provided unexpected insights into the mechanisms of polyQ toxicity. For example, the Novartis screen found that Hsp90 inhibitors reduced Q72 levels without a requirement for a stress response (31), while the FRET screen identified a ROCK/p160 inhibitor as a potent suppressor of polyQ (36).
Natural product collections have historically provided structurally diverse and bioactive lead structures for the treatment of many diseases (39). It has been estimated that 60% of current FDA-approved drugs have origins in natural products, illustrating the power of these compounds in drug discovery. Surprisingly, a screen of natural product extracts in polyQ models has not been reported. Accordingly, we have taken advantage of a newly developed yeast model (40) to screen a collection of ~11 000 prefractionated natural product extracts in 384-well plates. This assay required optimization of the growth conditions to achieve good screening statistics, and the resulting protocol provided a robust ‘live-dead’ HTS methodology (Z’ factor 0.7; CV 8%). Moreover, this screen identified actinomycin D as a molecule that disrupts polyQ aggregation and reverses growth defects at low nanomolar concentrations. Initial mechanistic studies suggest that this molecule might suppress polyQ, in part, by increasing chaperone levels and enhancing binding to Hsp70.
Materials and Methods
Yeast strains
The yeast strain used in this study was YKO (pdr5Δ; MATa/MATα orf∷kanMX4/orf∷kanMX4 ura3Δ0/ura3Δ0 leu2Δ0/leu2Δ0 his3Δ1/his3Δ1 met15Δ0/MET15 lys2Δ0/LYS2). The pdr5Δ strain was used to enhance compound potency for the HTS efforts. The plasmids and methods used were originally developed by the Lindquist’s group and reported in recent manuscripts (16,18,40-42). Briefly, these plasmids enable galactose-inducible (GAL1) expression of ΔProQ25-CFP, ΔProQ103-CFP, Q25-GFP or Q103-GFP.
Antibodies
Antisera against Hsp26 and Hsp104 were a kind gift from the laboratories of Johannes Buchner and Stefan Walter. Antibodies to Hsp70 (3A3) and Hsp90 were purchased from Santa Cruz Biotechnology.
Isolation of extracts
Strain 07-15-H3 was isolated from a Papua New Guinea marine sediment as described (43). The initial extracts were generated by growing the microbe in 100 mL of X1 media (0.25 g dextrose, 0.25 g yeast extract, 0.64 g malt extract, 30 g sodium chloride, 1 L H2O) in a 250-mL baffled flask on a rotary shaker at 200 rpm for 20 days. These cultures were centrifuged and the fermentation medium was extracted with amberlite XAD-16 resin. The resin was washed and then extracted with methanol (to produce SID#13569), acetone and ethanol. For active compound identification, the same microbe was grown in 2.5 L of ISP2 (4 g dextrose, 4 g yeast extract, 10 g malt extract, 30 g sodium chloride and 1 L d-H2O) in a 6-L flask at 200 rpm on a rotary shaker for 20 days, followed by the same extraction procedure.
High-throughput screen
Cultures harboring ΔProQ103 plasmid where grown overnight under selection (SC His(−) media with 2% glucose), as described elsewhere (41). The following morning, the cells were centrifuged (500 ×g), washed with dH2O and resuspended in SC His(−) media plus 2% raffinose. After 3 h at 30 °C, the OD600 was determined and a 2X inoculum culture was prepared by diluting to OD600 = 0.12 in SC His(−) media plus 2% raffinose and 2% galactose. Finally, assay plates were prepared by adding 20 μL of SC His(−) media plus 2% raffinose and 2% galactose to sterile, clear, medium binding 384-well plates (Corning 3680) using a Multidrop 384 dispenser (Thermo Labsystems, Waltham, MA, USA). Wells containing SC His(−) media and 4% raffinose were served as the positive control. Natural product extracts (15 mg/mL) were added in 0.2 μL of DMSO using a Beckman Biomek FX with a 384 HDR pintool. An aliquot (20 μL) of the 2X yeast culture was then added using the Multidrop 384 dispenser, and the plates were centrifuged for 1 min at 250 ×g to start the experiment. These plates were incubated in a humidified chamber at 30 °C for 48 h. Cell growth was determined by OD600 using a BMG Pherastar plate reader. The retesting of samples for progrowth activity was performed using an identical protocol.
Fluorescence microscopy
Microscopy experiments were conducted as previously described (41,42). Images were collected 1.5 h after 2% galactose induction for the Q103-GFP-expressing cells or 3 h after 2% galactose induction for the ΔProQ103-expressing cells. Cells lacking clear puncta were scored from randomly selected fields and reported as a fraction of the total number of cells (n = 200).
Isolation of the active compound
The methanol extract SID#13569 was fractionated on a C18 LOBAR LiChroprep column using MeOH/water (7:3), and fraction 7 was further fractionated by HPLC on an XBridge Prep C18 column under the same solvent conditions. The chemical structure of the major compound in fraction 7 was determined by mass spectra and NMR (1H, 13C and 2D spectra). The test sample had an m/z value consistent with actinomycin D (1277.7 [M+Na]), and a comparison of the test sample with authentic actinomycin D (Sigma #A12110; lot #071M21028V, St. Louis, MO, USA) showed good correlation of the peaks (Figure S1). The mass spectra were collected on a Micromass Auto-Spec Ultima Magnetic spectrometer (Waters Corp, Pleasanton, CA, USA). The NMR spectra were collected in CD3OD on a Varian INOVA 600 MHz NMR spectrometer (Palo Alto, CA, USA), using TMS as an internal standard.
Immunoprecipitations
The co-immunoprecipitations and Western blotting procedures have been described previously (18). Results were quantified using NIH image j and normalized to an actin loading control (Figure 4A) or anti-FLAG blot (Figure 4C).
Figure 4.
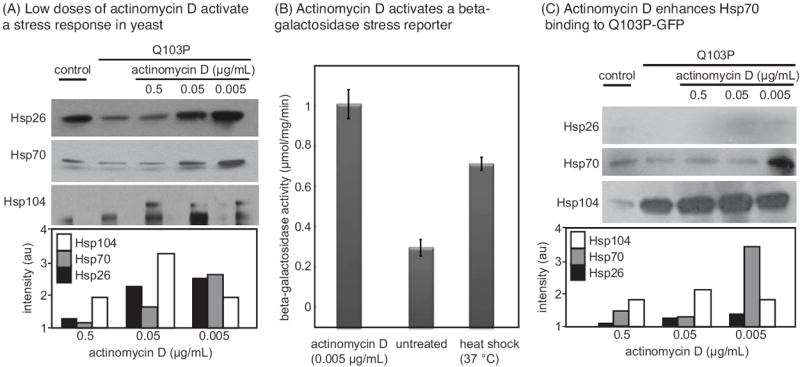
Suppression of polyQ toxicity and aggregation in yeast involves activation of a stress response and altered chaperone interactions. (A) Low doses of actinomycin D activate a stress response, as measured by Western blots for the stress markers Hsp26, Hsp70 and Hsp104 in treated yeast lysates. (B) Actinomycin D activates a stress response, as measured by a beta-galactosidase STRE reporter. Beta-galactosidase activity was measured by absorbance at 405 nm. Results are the average of triplicates, and error bars represent the standard error of the mean. (C) Treatment with low doses of actinomycin D enhances binding of Hsp104 and Hsp70 to Q103-GFP, as measured by co-immunoprecipiation of a FLAG epitope on Q103P-GFP.
Beta-galactosidase assays
For the β-galactosidase assay, cells were grown to mid-log phase in selective media followed by dilution to OD600 = 0.3. Cells were cultured for 3.5 h at 30 °C prior to sample preparation. The β-galactosidase assay was performed according to the manufacturer (Thermo Fisher Scientific, Waltham, MA, USA).
PolyQ aggregation assay in PC12 cells
Experiments were performed in inducible PC12 cells (14A2.5) expressing truncated huntingtin exon 1 containing 103Q repeats and fused to EGFP (36). Cells were maintained in DMEM, 10% horse serum, 5% fetal bovine serum, 1% penicillin/streptomycin with continual selection in 200 μg/ml geneticin. For the aggregation assay, cells were induced with 5 μm ponasterone A (Invitrogen, Carlsbad, CA, USA) for 30 h prior to treatment with compounds. Stock solutions of actinomycin D (Sigma A1410), tunicamycin (Sigma) and YM1 were prepared in DMSO. Cycloheximide (Sigma) was prepared in ddH2O at 1 mg/ml immediately prior to use. Compounds were added to culture media at the final concentrations indicated. Aggregates were assessed 48 h after polyQ induction by fluorescence microscopy on a Nikon Ti-E microscope equipped with a Coolsnap HQ2 CCD camera (Photometrics, Tuscon, AZ, USA). Aggregates were quantified with the spot detection feature in NIS-Elements software (Nikon, Tokyo, Japan). Greater than 300 cells, imaged from three fields, were analyzed per data point for each experiment.
Results and Discussion
Development of a high-throughput screen
In this study, we adapted a long polyQ construct (ΔProQ103) and a short polyQ construct (ΔProQ25) that were reported for use in yeast by the Lindquist group (40). These polyQ fragments are galactose inducible and expressed as fusions with the cyan fluorescent protein (CFP) to allow visualization of the proteins. Also, they lack the proline-rich domain of native huntingtin, which changes the aggregation propensity of ΔProQ103 protein (44) and exaggerates its growth defect phenotype in yeast (40). The shorter, inert ΔProQ25 protein also expresses at a comparable level to ΔProQ103, providing a good control for aggregation and proteotoxicity. Consistent with the previous findings, we confirmed that expression of ΔProQ103, but not the ΔProQ25 fragment, leads to slow growth of S. cerevisae (strain YKO; pdr5Δ) in both colony formation (Figure 1A) and liquid culture assays (Figure 1B). The pdr5Δ strain was used to maximize compound efficacy in anticipation of high-throughput chemical screens, and this strain background did not appear to diminish the polyQ-mediated growth effects.
Figure 1. Results of a yeast-based high-throughput screen (HTS) for suppression of polyQ growth defects and confirmation of two active extracts.
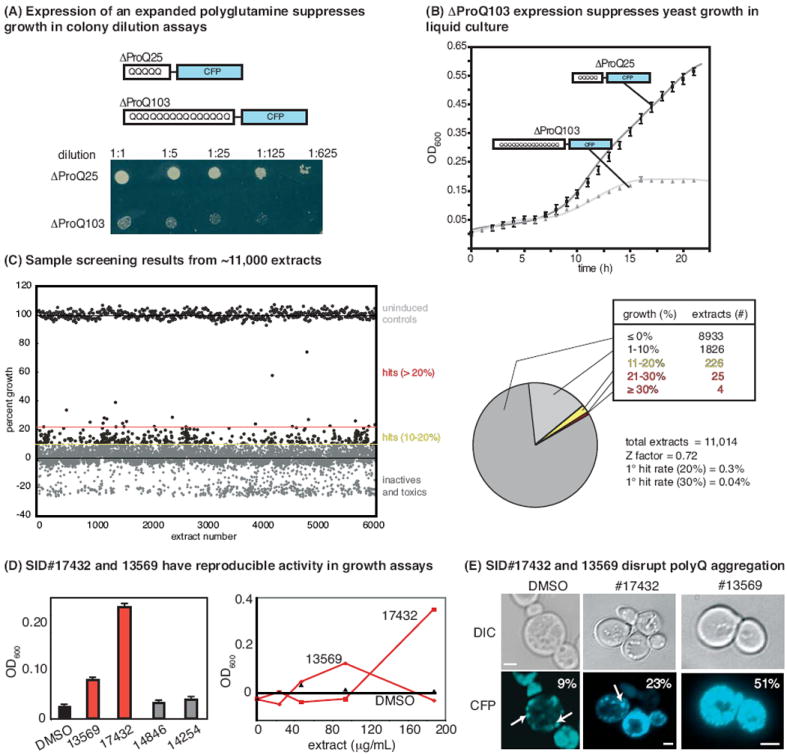
(A) Expression of an expanded polyQ fragment (ΔProQ103), but not the normal length ΔProQ25, in Saccharomyces cerevisae suppresses growth in colony dilution assays. Both constructs are expressed as cyan fluorescent protein (CFP) fusions under the control of a galactose-inducible promoter. (B) Expression of ΔProQ103, but not ΔProQ25, suppresses yeast growth in liquid culture. (C) Sample of the HTS results. A library of 11 000 prefractionated natural product extracts was screened in 384-well plates at approximately 100 μg/mL. Results are shown relative to uninduced controls (i.e., grown in 4% raffinose; no ΔProQ103 expression) and DMSO-treated controls. The screening statistics are shown. (C) The four most active extracts (>30% growth in the primary screen) were rescreened in triplicate (left) and tested at multiple dilutions (right). Two of the extracts were confirmed (SID #13569 and 17432). (D) By fluorescence microscopy, treatment with either of the active extracts disrupted the formation of puncta in some yeast. Aggregates are indicated by white arrows, and the percentage of cells with diffuse fluorescence is indicated (n = 200 cells). Scale bar is 2 μm.
Using this system, we envisioned that the liquid culture assay might be particularly suitable for high-throughput screening (HTS). Miniaturization of 96-well growth assays into HTS compatible, 384-well format often requires substantial reoptimization of conditions because of the differences in surface area at low volume. The goal of our particular optimization strategy was to produce the greatest difference between the growth of the ΔProQ25 and ΔProQ103 strains after induction of the polyQ fragments. Toward that goal, we systematically varied assay conditions, such as the initial inoculum density of the culture (between 0.03 and 0.3 OD600), incubation time (between 24 and 72 h), carbon source (varying ratios of raffinose and/or galactose) and total carbon concentration (total amount of raffinose and/or galactose; Figure S1). Using this approach, we determined that an initial inoculum density of 0.06, incubated for 48 h, in SC His(−) media with 2% raffinose and 2% galactose produced the optimum signal/background differential (see the Materials and Methods for details).
Using these conditions, we screened ~11 000 prefractionated natural product extracts at concentrations around 100 μg/mL. The extract collection was assembled from bacterial and fungal sources isolated from Papua New Guinea, Costa Rica and other locations, as previously described (45). Samples from this collection have been used to identify promising lead compounds in other screens (46), including unexpected uses for known molecules. In yeast expressing ΔProQ103, we identified 29 extracts that improved growth by more than 20% and four extracts that improved growth by at least 30% (Figure 1C). We also noted that a number of extracts decreased growth below the level of the mock-treated controls, suggesting that they might be cytotoxic. Overall, this assay yielded excellent screening characteristics (Z’ factor ~ 0.7, CV% ~ 8%). Moreover, it was inexpensive, required no detection reagents, and likely because active molecules must increase (rather than decrease) growth rates, the assay had a low ‘hit rate’ (>20% growth recovery = 0.3% actives). This modest rate is especially important in screens of prefractionated natural product extracts because the identification of the active component is often a time-intensive task.
Focusing on the four most active extracts, we rescreened parent stocks at 100 μg/mL and found that SID#13569 and SID#17432 were reproducible inhibitors of the ΔProQ103 growth phenotype at multiple dilutions (Figure 1D). To ascertain whether these extracts impacted the aggregation of polyQ in the treated cells, we performed fluorescence microcopy studies. Consistent with recent reports, ΔProQ103-CFP fluorescence appeared as multiple, discrete puncta in mock-treated cells (Figure 1E). However, both SID#17432 and SID#13569 altered the fluorescence pattern, increasing the number of cells with diffuse fluorescence by approximately 2.5- and 5.5-fold, respectively (Figure 1E). Based on these results, the frozen spores of the original producer organisms were regrown under the original conditions and fresh extracts prepared (see the Materials and Methods). The producer organism for SID#17432 did not regrow (data not shown), but the fresh extract of SID#13569 retained activity and was further explored.
Bioactivity-guided fractionation of a promising extract
High-performance liquid chromatography (HPLC)-based crude separation of the SID#13569 extract resulted in ten fractions, which were then tested for bioactivity in the fluorescence microscopy-based assay. In these experiments, we switched from using the ΔProQ103 construct to a Q103-GFP fusion. The Q103-GFP fusion is less toxic and typically produces a single puncta per cell (16), making it easier to visually score the results. Using this approach, we found that a number of the fractions contained active compound (Figure 2). The early fractions (#2 through 4) appeared to contain a complex mixture of compounds, as estimated by HPLC (data not shown), so we focused on identifying the active component in fraction 7 that only contained a single peak (Figure 2A).
Figure 2.
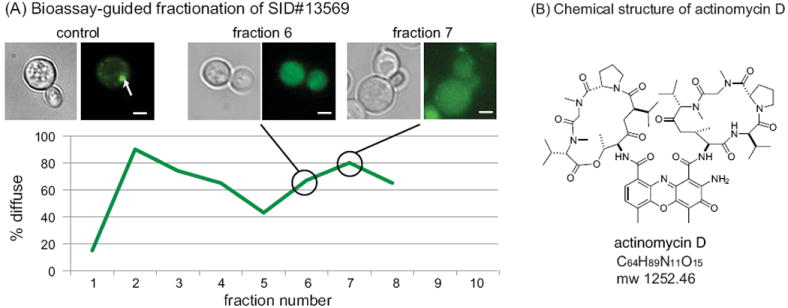
Bioassay-guided fractionation and identification of an active compound. (A) Using fluorescent puncta in yeast expressing Q103-GFP as a guide, the active HPLC fractions in crude extracts of SID#13569 were determined. Representative images are shown, and the arrows indicate puncta. Scale bar is 2 μm. The results were quantified by counting more than 200 yeast from random fields and scoring individual cells for the presence of well-resolved puncta. (B) The chemical structure of actinomycin D.
To chemically characterize the active component of fraction 7, we performed additional purification by HPLC (see the Materials and Methods). One pure fraction (0715-H3) contained a molecule with a molecular mass of 1277.7 (m/z), corresponding with the sodium form of actinomycin D ([M+Na] = 1277.7; Figure 2B; Figure S2). Proton NMR-based comparison with commercially available actinomycin D (Sigma #A12110; lot #071M21028V) further suggested that this compound might be the active component (Figure S2). Actinomycin D is a well known inhibitor of transcription that binds ssDNA in the transcription initiation complex (47).
Actinomycin D recovers yeast growth and suppresses aggregation
To test whether actinomycin D might be the active component of SID#13569, we measured the activity of commercially available compound (Sigma #A12110) in the yeast growth and fluorescence microscopy assays. These studies revealed that actinomycin D had a remarkable potency, recovering normal growth of ΔProQ103-expressing cells at ~40 nm (EC50 ~ 50 ng/mL; Figure 3A). At levels higher than 5 or 50 μg/mL, toxicity was observed, likely because of translational inhibition (47). In the fluorescence microscopy assay, actinomycin D also strongly suppressed polyQ aggregation at concentrations between ~0.5 and 0.0005 μg/mL, with activity reduced at both higher and lower levels (Figure 3B).
Figure 3.
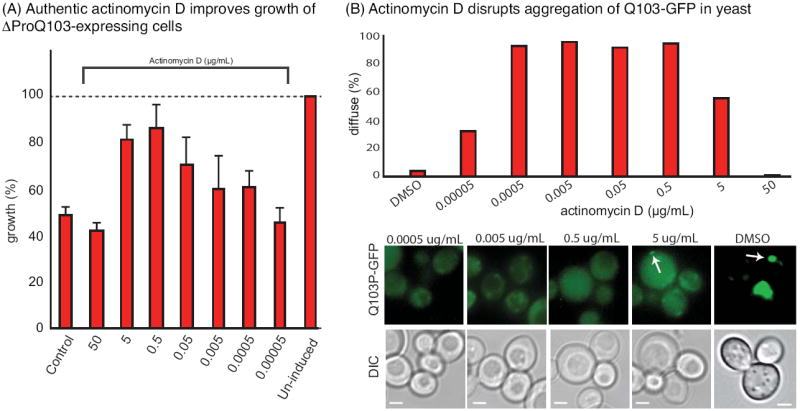
Confirmation of actinomycin D as an active compound. (A) Authentic actinomycin D (Sigma #A12110) improved growth of yeast expressing ΔProQ103. Results are representative of experiments performed in triplicate. Uninduced cells were grown in 4% raffinose. (B) Actinomycin D blocked the formation of puncta in cells expressing Q103-GFP. Representative images are shown, with puncta indicated by white arrows. Scale bar is 2 μm. For quantifications, more than 200 yeast cells were examined and scored for the presence of puncta.
Actinomycin D activates a stress response at low concentrations
The simplest mechanistic explanation for these observations is that actinomycin D suppresses transcription of the polyQ fragment, relieving growth suppression by reducing the levels of polyQ protein. However, actinomycin D is only active as a transcriptional suppressor in S. cerevisae at ~50 μg/mL (40 μm; 48, 49). In contrast, we observed antipolyQ activity at concentrations that were at least 100-fold lower (see Figure 3). Further, there was not any significant change in the total amount of polyQ-CFP protein or RNA in cells treated with 50 ng/mL actinomycin D for 1.5 h, as estimated by CFP fluorescence intensity (~93 ± 8% of untreated) or RT-PCR for the CFP mRNA (91 ± 11% of untreated), respectively. Thus, although transcriptional attenuation is likely to be one contributing mechanism, there appeared to be an additional pathway leading to polyQ suppression in this system.
We hypothesized that low doses of actinomycin D might reduce polyQ aggregation and toxicity by activating a stress response. Cellular stress, such as exposure to high temperatures or heavy metals, causes upregulation of many protective heat-shock proteins, such as Hsp26, Hsp70 and Hsp104 (50). These chaperones are known to prevent proteotoxicity caused by expression of polyQ fragments (16,41,51). Further, pharmacological activation of a stress response has previously been reported to impact polyQ aggregation in other models (37,38,52). To test whether actinomycin D might cause a heat-shock response at low concentrations, we measured the levels of the stress markers, Hsp26, Hsp70 and Hsp104, by Western blots. At concentrations of 0.05 and 0.005 μg/mL, actinomycin D was a potent activator of heat-shock proteins, increasing the levels of Hsp104 by fourfold and the levels of Hsp70 and Hsp26 by at least twofold (Figure 4A). Similarly, a beta-galactosidase reporter of the stress response (STRE) confirmed that actinomycin D (0.005 μg/mL) was a potent activator (Figure 4B).
Hsp70 is known to be upregulated by the stress pathway, and this chaperone directly binds polyQ fragments to mitigate aggregation (18). Therefore, we tested whether actinomycin D treatment might enhance the binding of Hsp70 to ΔProQ103 by co-immunoprecipitation (co-IP). The results showed that Hsp70 binding was enhanced more than threefold after treatment with low doses of actinomycin D (Figure 4C). Binding of ΔProQ103 to Hsp104 was also increased (Figure 4C). While it is likely that other mechanisms are also involved, activation of a stress response and increased Hsp70 binding by low doses of actinomycin D appeared to be one contributing factor in suppressing polyQ aggregation and proteotoxicity.
Actinomycin D suppressed polyQ aggregation in mammalian cells
To investigate whether this mechanism might be conserved in mammalian cells, we tested the effects of actinomycin D on polyQ aggregation in a PC12 model (36). Similar to what was observed in yeast, low levels of actinomycin D (4–100 ng/mL) suppressed aggregation, while higher and lower concentrations were inactive (Figure 5A). These results suggest that the pathway for actinomycin D’s antipolyQ activity may be conserved.
Figure 5.
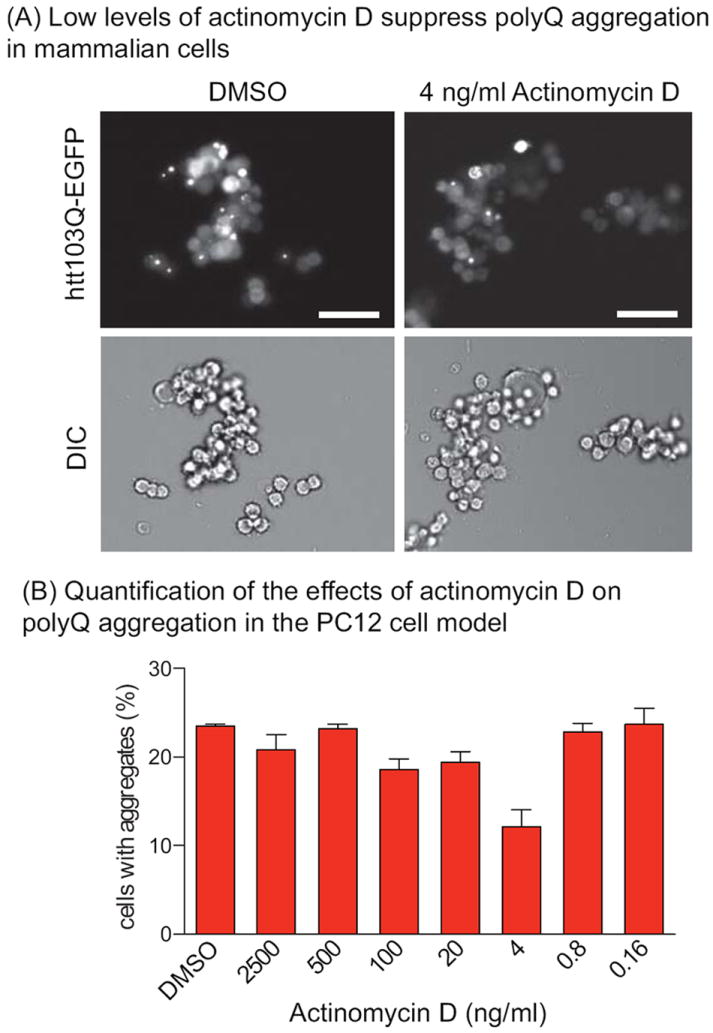
Suppression of polyQ aggregation in mammalian PC12 cells. (A) In PC12 cells, aggregation of htt103Q-EGFP into puncta is observed at 48 h after induction. Low concentrations of actinomycin D (4–100 ng/mL) reduced the number of cells with aggregates. Scale bar = 50 μm. (B) Quantification of the percentage of PC12 cells with aggregates averaged from three independent experiments ±SEM.
Conclusions
There is no cure for Huntington’s disease (HD) or any of the other polyQ disorders. Toward that goal, a number of HTS methods have been previously developed and used to identify promising leads (31-36). In this work, we focused on screening prefractionated natural product extracts as a potential source of molecules with antipolyQ activity. We were able to develop a robust, low-cost HTS protocol that was ideally suited for testing natural product collections because it readily removed cytotoxic fractions. We found that the key technical consideration was careful control over the yeast growth rate, especially by selecting the proper inoculation density and total carbon concentration.
These HTS efforts identified actinomycin D as a potent inducer of the stress response that was able to block polyQ aggregation and growth defects in yeast and reduce the number of cells with puncta in mammalian cells. Although it isn’t yet clear how actinomycin D activates a stress response (see Figure 4), we speculate that low concentrations of the compound might modestly and nonspecifically inhibit transcription, leading to the accumulation of truncated mRNA and perhaps shortened gene products. Although the direct utility of actinomycin D as a drug lead for HD or other polyQ disorders is uncertain, these studies provide further support for the stress response as an important pathway impacting polyQ toxicity (20,37,53). Thus, these studies lend impetus to ongoing efforts targeting the stress response, such as compounds targeting the major stress transcription factor (20,54,55) and those targeting proteins in the unfolded protein system (46,56). Our results suggest that molecules identified by these efforts may upregulate Hsp70 and suppress polyQ proteotoxicity.
Acknowledgments
The authors thank Anuj Kumar for generous use of the fluorescence microscope and thank Tomoko Komiyama and Anne Gillies for experimental assistance. The polyQ constructs were kindly provided by Martin Duennwald. This work was supported by NIH Grant NS059690 to JEG. The authors are grateful to PNG BioNet, the University of Papua New Guinea and the PNG Department of the Environment and Conservation for permission to collect research samples.
Footnotes
The authors declare that they have no conflicts of interest.
References
- 1.Gatchel JR, Zoghbi HY. Diseases of unstable repeat expansion: mechanisms and common principles. Nat Rev Genet. 2005;6:743–755. doi: 10.1038/nrg1691. [DOI] [PubMed] [Google Scholar]
- 2.Shao J, Diamond MI. Polyglutamine diseases: emerging concepts in pathogenesis and therapy. Hum Mol Genet. 2007;16(Spec No. 2):R115–R123. doi: 10.1093/hmg/ddm213. [DOI] [PubMed] [Google Scholar]
- 3.Williams AJ, Paulson HL. Polyglutamine neurodegeneration: protein misfolding revisited. Trends Neurosci. 2008;31:521–528. doi: 10.1016/j.tins.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen S, Berthelier V, Hamilton JB, O’Nuallain B, Wetzel R. Amyloid-like features of polyglutamine aggregates and their assembly kinetics. Biochemistry. 2002;41:7391–7399. doi: 10.1021/bi011772q. [DOI] [PubMed] [Google Scholar]
- 5.Martindale D, Hackam A, Wieczorek A, Ellerby L, Wellington C, McCutcheon K, Singaraja R, Kazemi-Esfarjani P, Devon R, Kim SU, Bredesen DE, Tufaro F, Hayden MR. Length of huntingtin and its polyglutamine tract influences localization and frequency of intracellular aggregates. Nat Genet. 1998;18:150–154. doi: 10.1038/ng0298-150. [DOI] [PubMed] [Google Scholar]
- 6.Sanchez I, Mahlke C, Yuan J. Pivotal role of oligomerization in expanded polyglutamine neurodegenerative disorders. Nature. 2003;421:373–379. doi: 10.1038/nature01301. [DOI] [PubMed] [Google Scholar]
- 7.Satyal SH, Schmidt E, Kitagawa K, Sondheimer N, Lindquist S, Kramer JM, Morimoto RI. Polyglutamine aggregates alter protein folding homeostasis in caenorhabditis elegans. Proc Natl Acad Sci USA. 2000;97:5750–5755. doi: 10.1073/pnas.100107297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schaffar G, Breuer P, Boteva R, Behrends C, Tzvetkov N, Strippel N, Sakahira H, Siegers K, Hayer-Hartl M, Hartl FU. Cellular toxicity of polyglutamine expansion proteins: mechanism of transcription factor deactivation. Mol Cell. 2004;15:95–105. doi: 10.1016/j.molcel.2004.06.029. [DOI] [PubMed] [Google Scholar]
- 9.Takahashi T, Kikuchi S, Katada S, Nagai Y, Nishizawa M, Onodera O. Soluble polyglutamine oligomers formed prior to inclusion body formation are cytotoxic. Hum Mol Genet. 2008;17:345–356. doi: 10.1093/hmg/ddm311. [DOI] [PubMed] [Google Scholar]
- 10.Nollen EA, Garcia SM, van Haaften G, Kim S, Chavez A, Morimoto RI, Plasterk RH. Genome-wide rna interference screen identifies previously undescribed regulators of polyglutamine aggregation. Proc Natl Acad Sci USA. 2004;101:6403–6408. doi: 10.1073/pnas.0307697101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Suhr ST, Senut MC, Whitelegge JP, Faull KF, Cuizon DB, Gage FH. Identities of sequestered proteins in aggregates from cells with induced polyglutamine expression. J Cell Biol. 2001;153:283–294. doi: 10.1083/jcb.153.2.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y, Meriin AB, Costello CE, Sherman MY. Characterization of proteins associated with polyglutamine aggregates: a novel approach towards isolation of aggregates from protein conformation disorders. Prion. 2007;1:128–135. doi: 10.4161/pri.1.2.4440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chan HY, Warrick JM, Gray-Board GL, Paulson HL, Bonini NM. Mechanisms of chaperone suppression of polyglutamine disease: selectivity, synergy and modulation of protein solubility in drosophila. Hum Mol Genet. 2000;9:2811–2820. doi: 10.1093/hmg/9.19.2811. [DOI] [PubMed] [Google Scholar]
- 14.Robertson AL, Headey SJ, Saunders HM, Ecroyd H, Scanlon MJ, Carver JA, Bottomley SP. Small heat-shock proteins interact with a flanking domain to suppress polyglutamine aggregation. Proc Natl Acad Sci USA. 2010;107:10424–10429. doi: 10.1073/pnas.0914773107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rujano MA, Kampinga HH, Salomons FA. Modulation of polyglutamine inclusion formation by the hsp70 chaperone machine. Exp Cell Res. 2007;313:3568–3578. doi: 10.1016/j.yexcr.2007.07.034. [DOI] [PubMed] [Google Scholar]
- 16.Krobitsch S, Lindquist S. Aggregation of huntingtin in yeast varies with the length of the polyglutamine expansion and the expression of chaperone proteins. Proc Natl Acad Sci USA. 2000;97:1589–1594. doi: 10.1073/pnas.97.4.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meriin AB, Zhang X, He X, Newnam GP, Chernoff YO, Sherman MY. Huntington toxicity in yeast model depends on polyglutamine aggregation mediated by a prion-like protein rnq1. J Cell Biol. 2002;157:997–1004. doi: 10.1083/jcb.200112104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walter GM, Smith MC, Wisen S, Basrur V, Elenitoba-Johnson KS, Duennwald ML, Kumar A, Gestwicki JE. Ordered assembly of heat shock proteins, hsp26, hsp70, hsp90, and hsp104, on expanded polyglutamine fragments revealed by chemical probes. J Biol Chem. 2011;286:40486–40493. doi: 10.1074/jbc.M111.284448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jana NR, Tanaka M, Wang G, Nukina N. Polyglutamine length-dependent interaction of hsp40 and hsp70 family chaperones with truncated n-terminal huntingtin: their role in suppression of aggregation and cellular toxicity. Hum Mol Genet. 2000;9:2009–2018. doi: 10.1093/hmg/9.13.2009. [DOI] [PubMed] [Google Scholar]
- 20.Katsuno M, Sang C, Adachi H, Minamiyama M, Waza M, Tanaka F, Doyu M, Sobue G. Pharmacological induction of heat-shock proteins alleviates polyglutamine-mediated motor neuron disease. Proc Natl Acad Sci USA. 2005;102:16801–16806. doi: 10.1073/pnas.0506249102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kobayashi Y, Kume A, Li M, Doyu M, Hata M, Ohtsuka K, Sobue G. Chaperones hsp70 and hsp40 suppress aggregate formation and apoptosis in cultured neuronal cells expressing truncated androgen receptor protein with expanded polyglutamine tract. J Biol Chem. 2000;275:8772–8778. doi: 10.1074/jbc.275.12.8772. [DOI] [PubMed] [Google Scholar]
- 22.Muchowski PJ, Schaffar G, Sittler A, Wanker EE, Hayer-Hartl MK, Hartl FU. Hsp70 and hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc Natl Acad Sci USA. 2000;97:7841–7846. doi: 10.1073/pnas.140202897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wacker JL, Zareie MH, Fong H, Sarikaya M, Muchowski PJ. Hsp70 and hsp40 attenuate formation of spherical and annular polyglutamine oligomers by partitioning monomer. Nat Struct Mol Biol. 2004;11:1215–1222. doi: 10.1038/nsmb860. [DOI] [PubMed] [Google Scholar]
- 24.Warrick JM, Chan HY, Gray-Board GL, Chai Y, Paulson HL, Bonini NM. Suppression of polyglutamine-mediated neurodegeneration in drosophila by the molecular chaperone hsp70. Nat Genet. 1999;23:425–428. doi: 10.1038/70532. [DOI] [PubMed] [Google Scholar]
- 25.Wyttenbach A, Sauvageot O, Carmichael J, Diaz-Latoud C, Arrigo AP, Rubinsztein DC. Heat shock protein 27 prevents cellular polyglutamine toxicity and suppresses the increase of reactive oxygen species caused by huntingtin. Hum Mol Genet. 2002;11:1137–1151. doi: 10.1093/hmg/11.9.1137. [DOI] [PubMed] [Google Scholar]
- 26.Gidalevitz T, Ben-Zvi A, Ho KH, Brignull HR, Morimoto RI. Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science. 2006;311:1471–1474. doi: 10.1126/science.1124514. [DOI] [PubMed] [Google Scholar]
- 27.Meriin AB, Zhang X, Miliaras NB, Kazantsev A, Chernoff YO, McCaffery JM, Wendland B, Sherman MY. Aggregation of expanded polyglutamine domain in yeast leads to defects in endocytosis. Mol Cell Biol. 2003;23:7554–7565. doi: 10.1128/MCB.23.21.7554-7565.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ortega Z, Diaz-Hernandez M, Maynard CJ, Hernandez F, Dantuma NP, Lucas JJ. Acute polyglutamine expression in inducible mouse model unravels ubiquitin/proteasome system impairment and permanent recovery attributable to aggregate formation. J Neurosci. 2010;30:3675–3688. doi: 10.1523/JNEUROSCI.5673-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet. 2002;11:1107–1117. doi: 10.1093/hmg/11.9.1107. [DOI] [PubMed] [Google Scholar]
- 30.Bowman AB, Yoo SY, Dantuma NP, Zoghbi HY. Neuronal dysfunction in a polyglutamine disease model occurs in the absence of ubiquitin-proteasome system impairment and inversely correlates with the degree of nuclear inclusion formation. Hum Mol Genet. 2005;14:679–691. doi: 10.1093/hmg/ddi064. [DOI] [PubMed] [Google Scholar]
- 31.Baldo B, Weiss A, Parker CN, Bibel M, Paganetti P, Kaupmann K. A screen for enhancers of clearance identifies huntingtin as a heat shock protein 90 (hsp90) client protein. J Biol Chem. 2012;287:1406–1414. doi: 10.1074/jbc.M111.294801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang X, Smith DL, Meriin AB, Engemann S, Russel DE, Roark M, Washington SL, et al. A potent small molecule inhibits polyglutamine aggregation in Huntington’s disease neurons and suppresses neurodegeneration in vivo. Proc Natl Acad Sci USA. 2005;102:892–897. doi: 10.1073/pnas.0408936102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heiser V, Engemann S, Brocker W, Dunkel I, Boeddrich A, Waelter S, Nordhoff E, Lurz R, Schugardt N, Rautenberg S, Herhaus C, Barnickel G, Bottcher H, Lehrach H, Wanker EE. Identification of benzothiazoles as potential polyglutamine aggregation inhibitors of huntington’s disease by using an automated filter retardation assay. Proc Natl Acad Sci USA. 2002;99(Suppl 4):16400–16406. doi: 10.1073/pnas.182426599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nagai Y, Tucker T, Ren H, Kenan DJ, Henderson BS, Keene JD, Strittmatter WJ, Burke JR. Inhibition of polyglutamine protein aggregation and cell death by novel peptides identified by phage display screening. J Biol Chem. 2000;275:10437–10442. doi: 10.1074/jbc.275.14.10437. [DOI] [PubMed] [Google Scholar]
- 35.Pollitt SK, Pallos J, Shao J, Desai UA, Ma AA, Thompson LM, Marsh JL, Diamond MI. A rapid cellular fret assay of polyglutamine aggregation identifies a novel inhibitor. Neuron. 2003;40:685–694. doi: 10.1016/s0896-6273(03)00697-4. [DOI] [PubMed] [Google Scholar]
- 36.Apostol BL, Kazantsev A, Raffioni S, Illes K, Pallos J, Bodai L, Slepko N, Bear JE, Gertler FB, Hersch S, Housman DE, Marsh JL, Thompson LM. A cell-based assay for aggregation inhibitors as therapeutics of polyglutamine-repeat disease and validation in drosophila. Proc Natl Acad Sci USA. 2003;100:5950–5955. doi: 10.1073/pnas.2628045100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Waza M, Adachi H, Katsuno M, Minamiyama M, Sang C, Tanaka F, Inukai A, Doyu M, Sobue G. 17-aag, an hsp90 inhibitor, ameliorates polyglutamine-mediated motor neuron degeneration. Nat Med. 2005;11:1088–1095. doi: 10.1038/nm1298. [DOI] [PubMed] [Google Scholar]
- 38.Zhang YQ, Sarge KD. Celastrol inhibits polyglutamine aggregation and toxicity though induction of the heat shock response. J Mol Med. 2007;85:1421–1428. doi: 10.1007/s00109-007-0251-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Newman DJ, Cragg GM. Natural products as sources of new drugs over the last 25 years. J Nat Prod. 2007;70:461–477. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]
- 40.Duennwald ML, Jagadish S, Muchowski PJ, Lindquist S. Flanking sequences profoundly alter polyglutamine toxicity in yeast. Proc Natl Acad Sci USA. 2006;103:11045–11050. doi: 10.1073/pnas.0604547103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chafekar SM, Wisen S, Thompson AD, Echeverria A, Walter GM, Evans CG, Makley LN, Gestwicki JE, Duennwald ML. Pharmacological tuning of heat shock protein 70 modulates polyglutamine toxicity and aggregation. ACS Chem Biol. 2012;7:1556–1564. doi: 10.1021/cb300166p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wisen S, Bertelsen EB, Thompson AD, Patury S, Ung P, Chang L, Evans CG, Walter GM, Wipf P, Carlson HA, Brodsky JL, Zuiderweg ER, Gestwicki JE. Binding of a small molecule at a protein-protein interface regulates the chaperone activity of hsp70-hsp40. ACS Chem Biol. 2010;5:611–622. doi: 10.1021/cb1000422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hirsch CF, Christensen DL. Novel method for selective isolation of actinomycetes. Appl Environ Microbiol. 1983;46:925–929. doi: 10.1128/aem.46.4.925-929.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Darnell G, Orgel JP, Pahl R, Meredith SC. Flanking polyproline sequences inhibit beta-sheet structure in polyglutamine segments by inducing ppii-like helix structure. J Mol Biol. 2007;374:688–704. doi: 10.1016/j.jmb.2007.09.023. [DOI] [PubMed] [Google Scholar]
- 45.Cruz PG, Auld DS, Schultz PJ, Lovell S, Battaile KP, MacArthur R, Shen M, Tamayo-Castillo G, Inglese J, Sherman DH. Titration-based screening for evaluation of natural product extracts: identification of an aspulvinone family of luciferase inhibitors. Chem Biol. 2011;18:1442–1452. doi: 10.1016/j.chembiol.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fribley AM, Cruz PG, Miller JR, Callaghan MU, Cai P, Narula N, Neubig RR, et al. Complementary cell-based high-throughput screens identify novel modulators of the unfolded protein response. J Biomol Screen. 2011;16:825–835. doi: 10.1177/1087057111414893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sobell HM. Actinomycin and DNA transcription. Proc Natl Acad Sci USA. 1985;82:5328–5331. doi: 10.1073/pnas.82.16.5328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Parsons AB, Lopez A, Givoni IE, Williams DE, Gray CA, Porter J, Chua G, et al. Exploring the mode-of-action of bioactive compounds by chemical-genetic profiling in yeast. Cell. 2006;126:611–625. doi: 10.1016/j.cell.2006.06.040. [DOI] [PubMed] [Google Scholar]
- 49.Battaner E, Vazquez D. Inhibitors of protein synthesis by ribosomes of the 80-s type. Biochim Biophys Acta. 1971;254:316–330. doi: 10.1016/0005-2787(71)90840-9. [DOI] [PubMed] [Google Scholar]
- 50.Santoro MG. Heat shock factors and the control of the stress response. Biochem Pharmacol. 2000;59:55–63. doi: 10.1016/s0006-2952(99)00299-3. [DOI] [PubMed] [Google Scholar]
- 51.Muchowski PJ, Wacker JL. Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci. 2005;6:11–22. doi: 10.1038/nrn1587. [DOI] [PubMed] [Google Scholar]
- 52.Herbst M, Wanker EE. Small molecule inducers of heat-shock response reduce polyq-mediated huntingtin aggregation. A possible therapeutic strategy. Neurodegener Dis. 2007;4:254–260. doi: 10.1159/000101849. [DOI] [PubMed] [Google Scholar]
- 53.Tanaka M, Machida Y, Niu S, Ikeda T, Jana NR, Doi H, Kurosawa M, Nekooki M, Nukina N. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of huntington disease. Nat Med. 2004;10:148–154. doi: 10.1038/nm985. [DOI] [PubMed] [Google Scholar]
- 54.Neef DW, Turski ML, Thiele DJ. Modulation of heat shock transcription factor 1 as a therapeutic target for small molecule intervention in neurodegenerative disease. PLoS Biol. 2010;8:e1000291. doi: 10.1371/journal.pbio.1000291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Calamini B, Silva MC, Madoux F, Hutt DM, Khanna S, Chalfant MA, Saldanha SA, Hodder P, Tait BD, Garza D, Balch WE, Morimoto RI. Small-molecule proteostasis regulators for protein conformational diseases. Nat Chem Biol. 2012;8:185–196. doi: 10.1038/nchembio.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wiseman RL, Zhang Y, Lee KP, Harding HP, Haynes CM, Price J, Sicheri F, Ron D. Flavonol activation defines an unanticipated ligand-binding site in the kinase-rnase domain of ire1. Mol Cell. 2010;38:291–304. doi: 10.1016/j.molcel.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]