

Abstract
Structure based drug design was used to develop a compound library of novel 2,5,6- and 2,5,7-trisubstituted benzimidazoles. Three structural analogs, SB-P1G10, SB-P8B2 and SB-P3G2 were selected from this library based on previous studies for advanced study. In vitro studies revealed that SB-P8B2 and SB-P3G2 had sigmoidal kill-curves while in contrast SB-P1G10 showed a narrow zonal susceptibility. The in vitro studies also demonstrated that exposure to SB-P8B2 or SB-P3G2 was bactericidal, while SB-P1G10 treatment never resulted in complete killing. The dose curves for the three compounds against clinical isolates were comparable to their respective dose curves in the laboratory strain of M. tuberculosis. SB-P8B2 and SB-P3G2 exhibited antibacterial activity against non-replicating bacilli under low oxygen conditions. SB-P3G2 and SB-P1G10 were assessed in acute short-term animal models of tuberculosis, which showed that SB-P3G2 treatment demonstrated activity against M. tuberculosis. Together, these studies reveal an in vitro- in vivo relationship of the 2,5,6-trisubstituted benzimidazoles that serves as a criterion for advancing this class of cell division inhibitors into more resource intensive in vivo efficacy models such as the long-term murine model of tuberculosis and Pre-IND PK/PD studies. Specifically, these studies are the first demonstration of efficacy and an in vitro–in vivo activity relationship for 2,5,6-trisubstituted benzimidazoles. The in vivo activity presented in this manuscript substantiates this class of cell division inhibitors as having potency and efficacy against M. tuberculosis.
Keywords: Mycobacterium tuberculosis, benzimidazole, in vivo efficacy, FtsZ inhibitor, murine model tuberculosis, in vitro activity
BACKGROUND
It is estimated that a third of the world’s population is infected with Mycobacteria tuberculosis, the causative agent of tuberculosis (TB), and between 5–10% of those infected individuals will develop active disease over their lifetime. As a result TB is the second leading cause of death from an infectious disease and the leading cause of death from a bacterial infection worldwide.1 In addition, to the global burden, the increasing rates of multi-drug resistant strains (MDR-TB) that require extensive treatment regimens with second line drugs impacts disease management.2, 3 Therefore, there is a need to develop new therapeutics with unique modes of action that can be co-administered with existing antitubercular drugs.
Cell division has been proposed as an attractive target for the development of new chemotherapeutics against M. tuberculosis.4 Of all the components involved in bacterial cell division, two proteins FtsZ and FtsI are the best characterized and are therefore receiving the most attention with regards to drug discovery.5–7 Although, FtsZ and tubulin share structural and functional homology, we have demonstrated that specificity for the mycobacterial FtsZ can be obtained through medicinal chemistry efforts.8, 9 Importantly, this affords the opportunity to use known pharmacophores such as, pyridopyrazine, pteridine and benzimidazole, as starting points for SAR optimization.6, 7 Specifically, selected 2,5,6- and 2,5,7-trisubstituted benzimidazoles developed through rational drug design, have demonstrated potency with low or negligible cytotoxicity. 9
In drug development, it is often difficult to apply appropriate criteria to prioritize and advance drugs through the drug discovery pipeline. Most often, the advancement of lead compounds into animal models of infection to assess efficacy is based primarily on minimal inhibitory concentration and cytotoxicity.10 Often, lack of efficacy is attributed to poor bioavailability or drug exposure. However, there is historical evidence that the bactericidal activity of some antimicrobials might not increase with additional concentrations of drug 11. Rather, these compounds display a multi-modal kill-curve characterized by a narrow range of concentrations that result in effective bactericidal activity. A more recent study described that drug treatment can instigate a tolerant phenotype by eliciting stress responses, which is consistent with this phenomenon, and the common observation of tolerance to treatment.12, 13
Previously, eleven 2,5,6- and 2,5,7-trisubstituted benzimidazoles were identified with varying potency against M. tuberculosis.9 In this study, we evaluated in more detail the three 2,5,6-trisubstituted benzimidazoles, SB-P1G10, SB-P3G2 and SB-P8B2, for activity against clinical strains and non-replicating bacilli, and determined the kill-curve characteristics of the compounds. SBP1G10 and SBP3G2 were assessed for efficacy in murine models of tuberculosis. These studies revealed an in vitro–in vivo activity relationship of the anti-TB benzimidazoles exhibiting efficacy in a murine model of tuberculosis. Importantly, the identification of an in vitro-in vivo activity relationship can serve as a criterion for advancing next generation cell division inhibitors into more resource intensive efficacy models such as the long-term murine model of tuberculosis and Pre-IND PK/PD studies.
METHODS
Mycobacteria tuberculosis strains and drug treatment conditions
Mycobacterium tuberculosis H37Rv and Erdman (TMCC 107) strains are drug sensitive laboratory reference strains used in the MIC studies,9, 14 and the tuberculosis animal models of infection.15–17 The clinical isolates W210, NHN20, NHN382 and TN587 have varying drug susceptibility profiles.4,18 For the in vitro studies all strains were grown in Difco™ 7H9 Middlebrook liquid media (BD Biosciences, 271310) supplemented with 10% Middlebrook OADC Enrichment (VWR, 9000-614), 0.05% Tween (G-Biosciences, 786-519), and 0.2% Glycerol at 37 °C. M. tuberculosis was grown on Difco™ Middlebrook 7H11 agar (BD Biosciences, 283810) supplemented with 1% Asparagine and antibiotics for colony forming unit (CFU) assays from animal studies. Antibiotics used were carbenicillin 50 mg/L (Sigma, C1389) and cycloheximide 10 mg/L (Sigma, C7698). Antibiotics and asparagine were not included in agar plates used for CFU’s from M. tuberculosis in vitro assays. Three 2,5,6-trisubstituted benzimidazoles, SB-P1G10, SB-P3G2 and SB-P8B2, were synthesized as described previously.9
In vitro growth and bactericidal assays
MIC values were determined for the benzimidazoles using a modified 96-well microplate Alamar Blue assay (MABA).9, 19 For time dose studies, bacterial growth in the presence of compound was monitored by the O.D. 600 nm. Each compound was assessed at a number of concentrations relative to the MIC in triplicate every 24 hours over 5 days by O.D. 600 nm and confirmed by plating at days 0, 1, 3, 5 of drug exposure. For the dose response curves, the bacterial viability in presence of different concentrations of each compound was determined using the MABA assay. Bacteria were treated for 7 days before assessing viability. The values were plotted against time using GraphPad (www.graphpad.com).
In vitro low oxygen studies
The low oxygen studies were performed similar to those described previously.20 Briefly, M. tuberculosis H37Rv were diluted to an O.D. 600 nm of 0.003 in 7H9 media containing Methylene Blue (1.5 μg/mL) and were grown in 16 X 125 mm Hungate tubes with rubber septa (VWR cat# 89167-170). The tubes, with 11 mL of the diluted cultures were incubated at 37 °C under agitation (150 rpm). Methylene blue was monitored for color change indicating that oxygen was depleted from the system. One week after the reduction of methylene blue; approximately 28 days from the start, drug treatment was initiated with the test and control drugs. Prior to treatment, an aliquot was removed and plated for a CFU count to serve as a control. Isoniazid and metronidazole were used in the studies as comparative controls at two concentrations 4 μg/ml and 45 μg/ml. Recovery of aliquots for plating and the addition of drugs were conducted through septa using syringes to maintain the low-oxygen conditions. Ninety- six hours post-treatment, 1 mL of the culture was removed from the oxygen depleted cultures, the aliquot was serial diluted and the dilutions were plated. Log10 CFU’s were calculated for the different treatments and concentrations. Percentage growth reduction from the controls was calculated and reported.
In vitro drug combination studies
A checkerboard 96 well plate method was used to examine possible antagonism or synergy between the 2,5,6-trisubstituted benzimidazoles and rifampicin (RIF). Drugs were distributed in 2-fold dilutions in a checkerboard pattern in a 96 well plate starting with 2X MIC for each drug tested in combination. M. tuberculosis H37Rv cells were added to the plate to a final well volume of 200 μL. No growth and positive growth controls were included in the assay. After 6 days incubation at 37 °C, Alamar Blue was added. Fractional inhibitory concentrations (FIC) were calculated to determine synergy in the drug combinations as described.21 FIC is defined as the MIC of a drug in combination divided by the MIC of that drug alone. The fractional inhibitory index (FICI) is the sum of the FIC’s (ΣFIC) for the drugs tested in the combination. Drugs are considered synergistic when the ΣFIC is less than 0.5, indifferent when the ΣFIC is between 0.5 and 4, and antagonistic when the ΣFIC is greater than 4.
Rapid murine models of tuberculosis used for efficacy screening
Compound efficacy was assessed in two murine models of infection based on the typical [short term] rapid model described previously and the standard mouse model.16 All aerosol infections were performed using a Middlebrook aerosol generation devise (Glas-Col, Terre Haute, IN). In the first infection short term model, immune incompetent C57BL/6-Ifngtm1ts gamma interferon gene disruption (GKO) mice were infected with M. tuberculosis H37Rv. Animals were dosed q.d. IP beginning day 12 post-infection with 150 mg/kg of compound in 38% Ethanol and 10% Solutol for 9 consecutive days. In the second animal model, immune competent C57BL/6 mice were infected with M. tuberculosis Erdman (TMCC 107). Animals were dosed b.i.d. IP with 100 mg/kg compound in 25 % Ethanol and 25% Solutol beginning day 5 post-infection for 10 consecutive days. Isoniazid was used as a comparative positive control and was delivered q.d. at 25 mg/kg as a positive control. Different delivery vehicles were used for each model to accomidate the difference in drug dosing volumes and number of dosings per animal per studiy. In both studies, animals were sacrificed and necropsied, bacterial burden in lungs or spleens were quantified on 7H11 agar containing carbenicillin (Sigma, C1389) and cycloheximide (Sigma, C7698) and upon incubation for three weeks bacterial colony forming units were enumerated. Outliers were identified by the Grubbs’ Test and one-way t-test at a 95% confidence interval was used to compare treatment groups and controls, to calculate p-values, and to produce bar graphs and scatter plots GraphPad Prism (www.graphpad.com).
RESULTS
In vitro potency of benzimidazoles against Mtb H37Rv and Erdman strains
We previously reported the discovery of a novel class of benzimidazoles that demonstrated antibacterial activity against M. tuberculosis and representative clinical isolates with various drug susceptibility profiles.9 For these studies, the MIC of the candidates, SB-P1G10, SB-P3G2 and SB-P8B2 (Figure 1) was confirmed against M. tuberculosis H37Rv and Erdman strains. This revealed that SB-P8B2 and SB-P3G2 had similar MIC ranges of 0.39–0.78 μg/mL and 0.78–1.5 μg/mL, respectively, and SB-P1G10 exhibited a slightly higher MIC range of 3–6.25 μg/mL. Therefore, the experimental MIC ranges of these compounds are comparable based on typical CLSI drug guidelines, and as previously reported,9 the benzimidazoles SB8B2 (1b-G1,9), SBP3G2 (1a-G7,9) and SBP1G10 (1a-G3,9) did not exhibit appreciable cytotoxicity against standard Vero cell line resulting in therapeutic indices of greater than 512 to 32, respectively.
Figure 1. Drug structures.
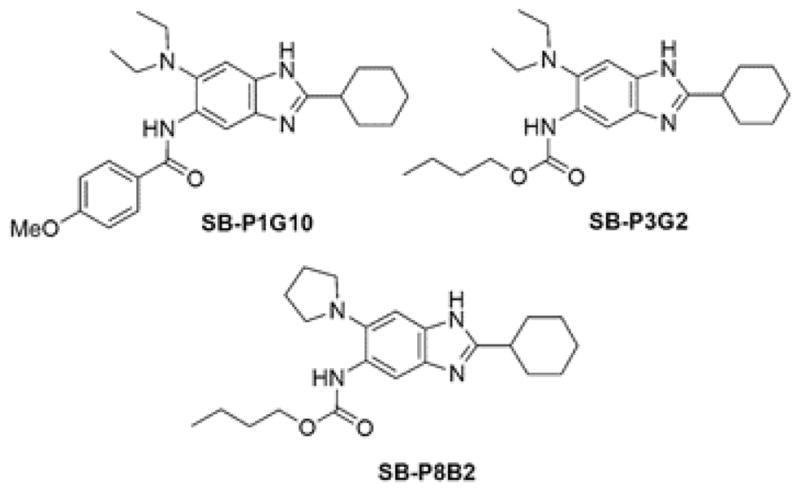
Chemical structures for the three 2,5,6-trisubstituted benzimidazoles discussed in the text.
Combination of SB-P8B2 with rifampicin
Since compounds whose mode of action is inhibition of cell division have not been evaluated for potential antagonistic interactions with currently used first line TB drugs, the compound with the lowest MIC, SB-P8B2, was used in combination with rifampicin as a representative of the mode of action of this novel class of compounds. This was assessed in a checkerboard in vitro growth inhibition assay (Table 1). The potency of both rifampicin and SB-P8B2 was enhanced by the presence of the other, resulting in a ΣFIC of 0.63. Specifically, SB-P8B2 was 8 times more potent in the presence of rifampicin and SB-P8B2 doubled the activity of rifampicin, thus indicating that there was enhanced killing using a combination of SB-P8B2 and the first line anti-TB drug rifampicin.
Table 1.
Enhancement of drug activity (MIC) with combination treatment.
MIC was determined by MABA
ΣFIC is the sum of the FICs(MIC of a drug in combination/MIC of that drug alone) for all the drugs tested in the combination. Drugs are considered synergistic when the ΣFIC is less than 0.5 and antagonistic when the ΣFIC is greater than 4.
SB-P8B2, was used in combination with rifampicin as a representative of this novel class of FtsZ cell division inhibitors.
In vitro growth inhibition and bactericidal action
Bacterial growth was monitored in the presence of SB-P3G2, SB-P8B2 or SB-P1G10 at concentrations relative to the MIC for 5 days to evaluate the inhibition in bacterial growth rate (Figure 2). The growth curves for the three benzimidazoles examined were all dose-dependent and not time- dependent. The growth inhibition curves for SB-P3G2 and SB-P8B2 were similar, showing that inhibition of bacterial growth correlated with increased drug concentrations (Figure 2A). Bacterial growth was inhibited at concentrations greater than 0.62 μg/mL and 0.31 μg/mL for SB-P3G2 and SB-P8B2, respectively. The bacterial growth in the presence of 0.78 μg/mL, 1.56 μg/ml, 3.13 μg/mL, 6.26 μg/mL and 12.5 μg/mL of SB-P1G10 was characterized by different rates (Figure 2B). At 3.13 μg/ml SB-P1G10 by O.D. 600 nm data appeared to be cidal but the plating and enumeration of CFUs indicated that the bacteria were still viable (Figure 2C). Additional dose response studies confirmed this observation (data not shown).
Figure 2. Growth inhibition curves.
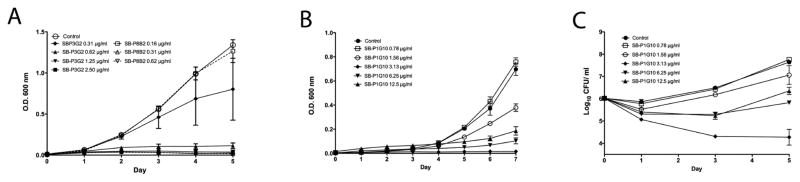
Bacterial growth was assessed in the presence of different concentrations of (A) SB-P3G2 and SB-P8B2 and (B) SB-P1G10 by optical density. (C) The growth inhibition of SB-P1G10 in panel B was validated by plating and enumeration of bacterial outgrowth (Log10 CFU/mL). The different drug concentrations were tested in triplicate at each time point and the mean and standard deviation of the values for the different doses was plotted.
To understand the kill characteristics of SB-P3G2, SB-P8B2 and SB-P1G10, the compounds were tested for bactericidal activity at different concentrations (Figure 3). Bacteria treated with either SB-P3G2 or SB-P8B2 demonstrated the typical sigmoidal bactericidal curve exhibited by the front line therapeutic INH, indicating that as the compound concentration increased the percent viable bacteria decreased to a minimal population at which no additional killing was observed (Figure 3A and B). In contrast, SB-P1G10 did not display a typical sigmoidal bactericidal curve. Rather, bactericidal activity was achieved in a very narrow concentration range of SB-P1G10, from 0.5–4 fold of MIC (Figure 3C). The resulting pronounced zonal susceptibility curve is characteristic of the Eagle-Musselman phenomenon where optimal effective killing is centered on the MIC, and at greater concentrations the bacteria remain viable in a bacteriostatic state.11 To assess whether the observed kill characteristics were applicable to clinical isolates, the dose response curves for SB-P3G2, SB-P8B2 and SB-P1G10 were generated against the clinical isolates W210, NHN20, NHN382 and TN587. Notably, not only are the MIC values of each of the compounds against the M. tuberculosis clinical strains comparable to the MIC for the H37Rv laboratory strain, but the dose curves generated for each compound against the clinical isolates examined in this study and H37Rv were remarkably similar (data not shown).
Figure 3. Drug concentration response curves.
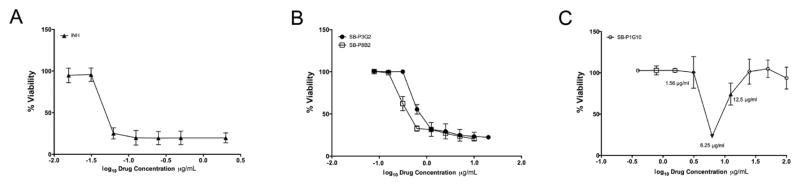
Drug concentration response curves were generated for (A) Isoniazid, (B) SB-P3G2 and SB-P8B2 and (C) SB-P1G10 by graphing the log10 drug concentrations (μg/mL) and % viability. Bacterial viability in the presence of different concentrations was determined by the microplate Alamar Blue assay (MABA).9, 19 Bacteria were treated for 7 days before assessing viability.
Activity of select benzimidazoles against non-replicating M. tuberculosis grown in low oxygen conditions
As part of our ongoing efforts on the development of FtsZ inhibitors as anti-TB agents, we extended the previous study by assessing the activity of benzimidazoles against non-replicating M. tuberculosis H37Rv strain in low oxygen conditions. Based on the confirmed MICs against M. tuberculosis H37Rv and the kill-curve characteristics, SB-P3G2 and SB-P8B2 were tested for potency against non-replicating bacteria in low oxygen conditions (Table 2). Metronidazole and INH were included as controls to confirm that the cultures were under low oxygen conditions during treatment. Both SB-P3G2 and SB-P8B2 showed greater activity against non-replicating bacilli than isoniazid, which is know to not have potent activity against non-replicating bacteria. SB-P8B2 and SB-P3G2 at 4 μg/mL reduced the survival of M. tuberculosis H37Rv by 59% and 72%, respectively (Table 2). Notably, SB-P3G2 at 4 μg/mL reduced the growth comparable to metronidazole at 4 μg/mL. SBP8B2 at 45.5 μg/mL reduced the growth of bacteria by almost 1 log10 CFU. In conclusion the substituted benzimidazoles SB-P3G2 and SB-P8B2 have activity against non-replicating M. tuberculosis resulting from low oxygen conditions.
Table 2.
In vitro potency of SB-P8B2 and SB-P3G2 against non-replicating M. tuberculosis grown under low oxygen conditions.
| Concentration μg/mL | Log10 (CFU+/−SD) | Log10 (CFU) Reduction | % Viable Reduction | |
|---|---|---|---|---|
| Control | 5.35 ± 0.01 | na | na | |
| a Isoniazid | 4 | 5.15 ± 0.2 | 0.2 | 36 |
| 45 | 5.01 ± 0.32 | 0.34 | 54 | |
| b Metronidazole | 4 | 4.83 ± 0.06 | 0.52 | 70 |
| 45 | 3.39 ± 0.11 | 1.96 | 99 | |
| c SB-P8B2 | 4 | 4.96 ± 0.18 | 0.39 | 59 |
| 45 | 4.38 ± 0.14 | 0.97 | 89 | |
| c SB-P3G2 | 4 | 4.87 ± 0.21 | 0.48 | 72 |
Isoniazid is not effective against non replicating bacteria.
Metronidazole is effective against non-replicating bacteria but has no efficacy against actively dividing bacteria.
Representative trisubstituted benzimidazoles.
In vitro-in vivo activity relationship
SB-P3G2 and SB-P1G10 were used in two different animal models of acute tuberculosis infection to investigate the in vitro-in vivo activity relationship between benzimidazoles with the two disparate kill-curve characteristics. These animal models are used in our drug discovery program because they afford the ability to evaluate if a drug candidate can reduce the bacterial load during an acute infection and inhibit dissemination from the site of infection to secondary sites. In the first acute infection animal study which uses immune incompetent GKO mice, SB-P3G2 reduced the bacterial load of M. tuberculosis H37Rv by 0.71 ± 0.17 log10 CFU in the lungs and 0.41 ± 0.36 log10 CFU in spleen (Figure 4A).16 Isoniazid reduced the bacterial load 1.8 log10 CFU in the lung and below the level of detection in the spleen. In the second dissemination model of infection using immune competent C57BL/6 mice, SB-P3G2 reduced the bacterial load of M. tuberculosis Erdman in the spleen by log10 1.6 ± 0.49 (Figure 4B). Isoniazid reduced the bacterial load in the lung and spleen below detectable levels. In the different animal models tested, SB-P3G2 always showed some level of detectable efficacy. Together, these animal studies show that SB-P3G2 has efficacy against an acute infection and can prevent dissemination to secondary sites. In contrast, treatment with SB-P1G10 never reduced the bacterial load in the lungs or spleen in the animal model of infection conducted (data not shown). This data indicates that two of the benzimidazoles with similar physicochemical and in vitro potency against M. tuberculosis, but different in vitro inhibition kill-curve characteristics demonstrate distinctly different efficacy outcomes.
Figure 4.
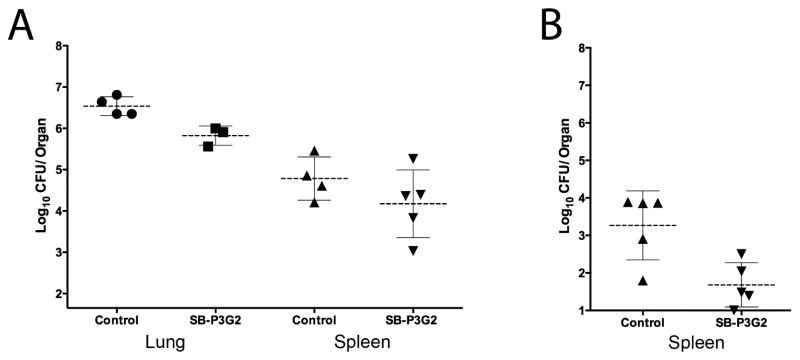
Efficacy in acute animal models of tuberculosis. (A) Scatter plots of the bacterial CFU counts from lungs (●) and spleen (▲) from untreated control animals and the lungs (■) and spleen (▼) of infected mice after treatment with SB-P3G2 delivered IP at 150 mg/kg for 9 consecutive days to immune incompetent GKO mice. SB-P3G2 reduced the bacterial load of M. tuberculosis H37Rv 0.71 ± 0.17 log10 CFU in the lungs and 0.41 ± 0.36 log10 CFU in spleen. (B) Scatter plots of the bacterial CFU counts from spleen (▲) from untreated control animals and the spleen (▼) of infected mice after treatment with SB-P3G2 delivered IP at 100 mg/kg BID for 10 consecutive days to immune competent C57BL/6 mice. SB-P3G2 reduced the bacterial load of M. tuberculosis Erdman in the spleen log10 1.6 ± 0.49. In both studies, no outliers were identified by the Grubbs’ Test.
DISCUSSION
The goal to reduce the global burden of tuberculosis has been hampered by difficult to treat chronic infections and the emergence of MDR strains of M. tuberculosis that are resistant to the frontline drugs or drug combinations needed to achieve durable cure. While drug discovery programs continue to make progress, there often remains a disconnect between a drug candidate’s in vitro potency and its observed efficacy in models of infection, which limits the number of lead candidates advancing to Pre-IND and clinical evaluation. We believe that this disconnect, in part, can be attributed to differences in a compound’s kill characteristics. The susceptibility of bacteria to a drug is primarily influenced by the concentration of the drug required to kill the bacteria and the rate at which the bacteria are killed at the appropriate concentration. This translates to efficacy because a drug that has a limited and pronounced zonal susceptibility and a slow killing rate will not demonstrate efficacy and therapeutic significance.
As part of our ongoing drug discovery program targeting FtsZ, three compounds were selected for more advanced in vitro characterization and efficacy testing. The in vitro-in vivo activity relationship of each of these compounds provides information about the drug exposure potential upon dosing in animal models of infection. MIC is the most common parameter used to prioritize a drug candidate’s progression into efficacy studies. However, MIC as a primary criterion has limitations, as it does not provide information about the rate of killing or the effective concentration of drug to achieve the rate of killing. Although the MIC of different drug candidates is similar, their kill-curve characteristics could be different resulting in observed difference in efficacy when delivered in vivo. The kill-curves generated for both SB-P3G2 and SB-P8B2 were concentration dependent and sigmoidal, indicating that as the concentration of the compound increases an inhibitory threshold is met and significant bactericidal activity is achieved. Importantly, the bactericidal activity is sustained despite significantly increased drug concentrations. This is consistent with the observed efficacy of SB-P3G2 in two different acute tuberculosis models of infection. These characteristics are typical of our lead compounds with efficacy in acute models of infection because at drug concentrations near the MIC the growth rate and number of viable bacteria is reduced, and the maximal effective dose range is large. This allows effective therapeutic concentrations to be maintained at the site of infection throughout the dosing interval. In contrast, the inhibition and dose response curves generated for SB-P1G10 showed a pronounced zonal susceptibility similar to what has been described as the Eagle-Musselman phenomenon.11 This type of kill-curve indicates that SB-P1G10 is bactericidal within a narrow range of concentrations centered on the MIC, but any increase in compound concentration greater than the optimal levels significantly reduces the extent of bacterial death. Therefore, the effective therapeutic concentration for SB-P1G10 within the narrow zone of susceptibility/bactericidality is maintained for a very short time during dosing. Together, the efficacy studies with SB-P3G2 and SB-P1G10 revealed the in vitro activity-efficacy relationship for these drug, and provide an explanation for the often observed disconnect between drug potency and treatment efficacy. This is further justified by a report, which demonstrates that bacterial tolerance is a result of drug exposure that has been shown to be the result of induction of stress responses.12 This observation is consistent with the notion that compounds with a narrow zone of susceptibility will lack significant efficacy because of the limited achievable effective therapeutic concentrations.
A new drug for tuberculosis should be co-administrable with currently used clinical drugs, and should have activity against clinical strains with differing drug susceptibilities and potency against non-replicating persistent bacilli. We assessed whether the inhibition of cell division with SB-P8B2 was antagonistic when used in combination with rifampicin. The FICI for the rifampicin: SB-P8B2 combination was 0.63 indicating that these two drugs are not antagonistic. In addition, the activity of SB-P8B2 against M. tuberculosis was enhanced by 8-fold in the presence of rifampicin. Only a modest 2-fold increase in the activity of rifampicin was observed. It is thought that because bacterial adaption to stress, including the bacterial response to drug exposure, occurs at the level of protein synthesis, the observed enhancement of SB-P8B2 activity is the result of rifampicin preventing the bacterial response to treatment with SB-P8B2. These drug candidates were shown to be equally active against clinical isolates and the laboratory strain, which is expected because the molecular target of this class of inhibitors is not a molecular target for current clinically, used compounds. It is well known that the frontline TB drug isoniazid is only active against replicating M. tuberculosis typical of acute infections, and does not effectively kill non-replicating persistent organisms. Therefore, to assess whether cell division inhibitors have activity against non-replicating persistent bacilli, we tested SB-P3G2 and SB-P8B2 and discovered that they were both effective against non-replicating bacteria. SB-P1G10 was not tested under these conditions because at higher concentrations the compound is bacteriostatic.
CONCLUSION
A new anti-TB drug needs to be active against clinical strains with various drug susceptibility profiles as well as non-replicating persistent organisms. In addition, it should be co-administrable to improve treatment of chronic TB infections. We have reported here the in vitro-in vivo activity relationship of benzimidazole efficacy in murine models of tuberculosis. In addition, we have shown that selected benzimidazoles with this mode of action have activity against clinical isolates, as well as non-replicating persistent bacilli, and can enhance current therapeutic regimens. Together, these results substantiate that the molecular target FtsZ and this class of compounds warrants further study as a clinically relevant target and a novel class of chemotherapeutic agents for the treatment of tuberculosis.
Acknowledgments
Funding
This work was supported by grants AIO78251 (IO, RAS) and AI082164 (RAS) from the National Institute of Health and New York State office of Science Technology and Academic Research (NYSTAR).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Anonymous. Tuberculosis Fact Sheet. World Health Organization; 2010. [Google Scholar]
- 2.Raviglione MC, Smith IM. XDR tuberculosis--implications for global public health. The New England journal of medicine. 2007;356:656–659. doi: 10.1056/NEJMp068273. [DOI] [PubMed] [Google Scholar]
- 3.Migliori GB, De Iaco G, Besozzi G, Centis R, Cirillo DM. First tuberculosis cases in Italy resistant to all tested drugs. Euro surveillance : bulletin europeen sur les maladies transmissibles = European communicable disease bulletin. 2007;12:E070517 070511. doi: 10.2807/esw.12.20.03194-en. [DOI] [PubMed] [Google Scholar]
- 4.Slayden RA, Knudson DL, Belisle JT. Identification of cell cycle regulators in Mycobacterium tuberculosis by inhibition of septum formation and global transcriptional analysis. Microbiology. 2006;152:1789–1797. doi: 10.1099/mic.0.28762-0. [DOI] [PubMed] [Google Scholar]
- 5.Slayden RA, Belisle JT. Morphological features and signature gene response elicited by inactivation of FtsI in Mycobacterium tuberculosis. J Antimicrob Chemother. 2009;63:451–457. doi: 10.1093/jac/dkn507. dkn507 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kumar K, Awasthi D, Berger WT, Tonge PJ, Slayden RA, Ojima I. Discovery of anti-TB agents that target the cell-division protein FtsZ. Future medicinal chemistry. 2010;2:1305–1323. doi: 10.4155/fmc.10.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang Q, Tonge PJ, Slayden RA, Kirikae T, Ojima I. FtsZ: a novel target for tuberculosis drug discovery. Current topics in medicinal chemistry. 2007;7:527–543. doi: 10.2174/156802607780059790. [DOI] [PubMed] [Google Scholar]
- 8.Huang Q, Kirikae F, Kirikae T, Pepe A, Amin A, Respicio L, Slayden RA, Tonge PJ, Ojima I. Targeting FtsZ for antituberculosis drug discovery: noncytotoxic taxanes as novel antituberculosis agents. Journal of medicinal chemistry. 2006;49:463–466. doi: 10.1021/jm050920y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kumar K, Awasthi D, Lee SY, Zanardi I, Ruzsicska B, Knudson S, Tonge PJ, Slayden RA, Ojima I. Novel trisubstituted benzimidazoles, targeting Mtb FtsZ, as a new class of antitubercular agents. Journal of medicinal chemistry. 2011;54:374–381. doi: 10.1021/jm1012006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mueller M, de la Pena A, Derendorf H. Issues in Pharmacokinetics and Pharmacodynamics of Anti-Infective Agents: Kill Curves versus MIC. Antimicrobial Agents and Chemotherapy. 2004;48:369–377. doi: 10.1128/aac.48.2.369-377.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eagle H, Musselman AD. The rate of bactericidal action of penicillin in vitro as a function of its concentration, and its paradoxically reduced activity at high concentrations against certain organisms. The Journal of experimental medicine. 1948;88:99–131. doi: 10.1084/jem.88.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller C, Thomsen LE, Gaggero C, Mosseri R, Ingmer H, Cohen SN. SOS response induction by beta-lactams and bacterial defense against antibiotic lethality. Science. 2004;305:1629–1631. doi: 10.1126/science.1101630. [DOI] [PubMed] [Google Scholar]
- 13.Balaban NQ, Merrin J, Chait R, Kowalik L, Leibler S. Bacterial persistence as a phenotypic switch. Science. 2004;305:1622–1625. doi: 10.1126/science.1099390. [DOI] [PubMed] [Google Scholar]
- 14.Camus JC, Pryor MJ, Medigue C, Cole ST. Re-annotation of the genome sequence of Mycobacterium tuberculosis H37Rv. Microbiology. 2002;148:2967–2973. doi: 10.1099/00221287-148-10-2967. [DOI] [PubMed] [Google Scholar]
- 15.Miyoshi-Akiyama T, Matsumura K, Iwai H, Funatogawa K, Kirikae T. Complete annotated genome sequence of Mycobacterium tuberculosis Erdman. Journal of bacteriology. 2012;194:2770. doi: 10.1128/JB.00353-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lenaerts AJ, Gruppo V, Brooks JV, Orme IM. Rapid in vivo screening of experimental drugs for tuberculosis using gamma interferon gene-disrupted mice. Antimicrobial agents and chemotherapy. 2003;47:783–785. doi: 10.1128/AAC.47.2.783-785.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kelly BP, Furney SK, Jessen MT, Orme IM. Low-dose aerosol infection model for testing drugs for efficacy against Mycobacterium tuberculosis. Antimicrob Agents Chemother. 1996;40:2809–2812. doi: 10.1128/aac.40.12.2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Slayden RA, Lee RE, Barry CE., 3rd Isoniazid affects multiple components of the type II fatty acid synthase system of Mycobacterium tuberculosis. Molecular microbiology. 2000;38:514–525. doi: 10.1046/j.1365-2958.2000.02145.x. [DOI] [PubMed] [Google Scholar]
- 19.Collins L, Franzblau SG. Microplate alamar blue assay versus BACTEC 460 system for high-throughput screening of compounds against Mycobacterium tuberculosis and Mycobacterium avium. Antimicrob Agents Chemother. 1997;41:1004–1009. doi: 10.1128/aac.41.5.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lenaerts AJ, Gruppo V, Marietta KS, Johnson CM, Driscoll DK, Tompkins NM, Rose JD, Reynolds RC, Orme IM. Preclinical testing of the nitroimidazopyran PA-824 for activity against Mycobacterium tuberculosis in a series of in vitro and in vivo models. Antimicrobial agents and chemotherapy. 2005;49:2294–2301. doi: 10.1128/AAC.49.6.2294-2301.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reddy VM, Einck L, Andries K, Nacy CA. In vitro interactions between new antitubercular drug candidates SQ109 and TMC207. Antimicrobial agents and chemotherapy. 2010;54:2840–2846. doi: 10.1128/AAC.01601-09. [DOI] [PMC free article] [PubMed] [Google Scholar]