

Abstract
We have used a chemically diverse panel of kinase inhibitors to assess the chemical similarity of the ATP-binding sites of cyclin-dependent kinase (CDK) subfamily members in a range of activation states. Using this approach, we find that different activation states of a particular CDK may differ from each other as much as different CDKs in the same activation state. We also find that inhibitors discriminate more effectively among CDK family members in their monomeric state than in their cyclin-bound state, providing direct evidence for the belief that selective binding to inactive kinase states might be more readily achieved than selective binding to active states.
Protein kinases represent attractive targets for therapeutic intervention against a wide variety of human diseases, most notably cancer1,2 and chronic inflammatory diseases.3 A number of drugs that selectively target the protein kinase ATP binding site have been successfully introduced into the clinic, and many more are in clinical trials.2,4 However, a significant requirement for the further development of therapeutically useful compounds is a more complete understanding of the factors that dictate inhibitor selectivity across the protein kinase family.5,2,6
The eukaryotic serine/threonine and tyrosine protein kinase family is characterized by a conserved fold in which residues from both the N- and C-terminal lobes contribute to the active site.7,8 The identities of the residues that line the ATP binding pocket and the structural plasticity of the protein kinase fold constitute two key elements that together determine the inhibitor-binding profile of a protein kinase. Both of these elements have been successfully exploited to generate clinically useful drugs.1,2
The cyclin-dependent kinases (CDKs) constitute a subfamily of ca. 13 members in humans9 that play important roles in both the control of cell cycle progression (CDKs 1, 2, 3, 4, and 6) and in the regulation of transcription (CDKs 7, 8, 9, 12, and 13).10−12 CDK2 has provided a structural paradigm for the CDK family and has been widely exploited for structure-aided CDK inhibitor design.13,14 The prevailing structural model for CDK regulation by cyclin and CDK inhibitor (CKI) binding and by phosphorylation has been elaborated through a series of structures of CDK2/cyclin A complexes.15,16 Monomeric CDK2 is inactive as a result of the disposition of active site residues, in turn dependent on the pose of the C-helix, and the conformation of the activation segment.17 Cyclin A binding and Thr160 phosphorylation within the activation segment rearrange the CDK2 active site to orientate key ATP binding and catalytic residues and create the peptide substrate binding site.15,18 This model for the mechanism of regulation appears not to apply across the entire CDK subfamily. The determination of structures for CDK4/cyclin D319 and CDK4/cyclin D1 phosphorylated on Thr172 (pCDK4/D)20 revealed that CDK4 adopts an inactive C-helix out conformation despite being cyclin-bound. Two further examples are CDK521 and CDK822 that both adopt active conformations upon p25 and cyclin C binding, respectively, in the absence of activation loop phosphorylation.
Differential scanning fluorimetry (DSF) can be used to characterize inhibitor binding.23 Here we define ΔTm as the difference between the apparent melting temperature (Tm) of the protein in the presence of the ligand and the Tm of the protein alone. The value is related to the concentration and binding affinity of the ligand for the protein and to thermodynamic properties of the ligand binding event.23 The profile of ΔTm values measured for a protein in the presence of a sufficiently diverse range of inhibitors is characteristic of the inhibitor binding properties of that protein and therefore represents a fingerprint of the physicochemical properties of the active site.24 We have used such fingerprints to assess the chemical similarity of a number of CDK subfamily members, namely, CDK2, CDK4, CDK7, and CDK9. Additionally, we have explored the inhibitor binding profiles of CDKs in different activation states to assess the extent to which CDK activation affects inhibitor binding.
Results and Discussion
The discriminative power of this fingerprinting approach relies on including a sufficiently diverse panel of inhibitors to sense the chemical diversity apparent in the family of kinase active sites. To validate the use of our selected inhibitor set (Figure 1a and Supplementary Table 1) for this purpose, we confirmed that our set is able to target representative kinases from across the kinome. This analysis was achieved by mapping each inhibitor onto those kinases in the kinome phylogenetic tree that they are best documented to target (Supplementary Methods and Figure 1b). The approximately uniform distribution obtained confirms ours to be a relatively unbiased set of inhibitors.
Figure 1.

Explored chemical space. (a) Representative inhibitor scaffolds. Examples of the inhibitor scaffolds included in the panel are shown. All inhibitor structures are provided in the Supporting Information (Supplementary Table 1). (b) Chemical space explored by the panel of inhibitors used in this study. The target(s) in the human kinome for each of the inhibitors of the panel are identified by a red rectangle on the kinome tree. This figure was adapted from the phylogenetic tree published in ref (9).
We included four members of the CDK subfamily in the study. CDK2 was prepared in four different conformational states: monomeric unphosphorylated (CDK2), monomeric phosphorylated on Thr160 (pCDK2), unphosphorylated in complex with human cyclin A175–432 (CDK2/A), and Thr160-phosphorylated in complex with human cyclin A175–432 (pCDK2/A). CDK4 was characterized both as an inactive, nonphosphorylated monomer (CDK4) and as the fully activated binary complex (CDK4/cyclin D3 phosphorylated on Thr172, pCDK4/D). CDK7 and CDK9 were prepared in either a monomeric unphosphorylated (CDK7) or a cyclin-bound phosphorylated (CDK9/cyclin T1 phosphorylated on T186, pCDK9/T) state, respectively (Figure 2a). The identities of the CDKs together with their activation states define a search space within the functional kinome that has been explored by this study and is summarized in Figure 2b. CDK2, CDK7, CDK4, and CDK9 are highly homologous across their entire sequence (Supplementary Figure 1), and this similarity is particularly marked at the catalytic site (Figure 2c,d).
Figure 2.

CDK space explored in this study. (a) Representative melting curves for cyclin-dependent kinase (CDK)/cyclin complexes used to derive ΔTm values in the presence of an ATP-competitive inhibitor. The shift in Tm upon staurosporine binding (red) is illustrated for each of the CDK/cyclin complexes in this study. Black curves, CDK/cyclin complexes in the absence of added inhibitor. Curves with closed triangle symbols correspond to pCDK9/T, closed diamonds to CDK7, closed circles to CDK4, closed squares to pCDK4/D, open squares to CDK2/A, open diamonds to CDK2, and open triangles to pCDK2. (b) The full set of different phosphorylated and/or cyclin-associated states of the four CDKs characterized in this study are shown, with those members for which measurements were made highlighted by shaded boxes. Color intensity for these protein species reflects activation state (more saturated, more active). (c) The ATP binding site of CDKs. The CDK2 ATP-binding site residues from a pCDK2/A structure (PDB code: 1QMZ) are colored according to the degree of sequence conservation between the 4 CDK sequences, calculated as described in the Supplementary Methods, from blue (highly conserved) to red (highly variable). The ATP molecule is represented in ball-and-stick mode for context. (d) The ATP-binding site pocket is highly conserved in CDK2, CDK7, CDK4, and CDK9. Residues that constitute the first shell of the CDK2 ATP-binding site (CDK2 numbering) were identified as defined in Supplementary Methods and used to construct a pseudosequence alignment. This alignment is displayed together with the associated consensus sequence, with X representing any residue, and ϕ a hydrophobic amino acid. Sequence motifs that are highly conserved across the protein kinase family are identified by numbered bars. Bar 1, the glycine-rich loop (residues 11–16 in CDK2) that defines the general consensus sequence GXGXXG that recognizes the phosphate moieties of ATP; bar 2, the “hinge” sequence composed of residues 80–84 that links the N- and C-terminal lobes and contributes to the ATP adenine binding site; bar 3, the “DFG motif” that defines the start of the activation segment. Asterisks below E51 and D127 highlight the conserved glutamate in the C-helix and the catalytic aspartate, respectively. A full sequence alignment is presented in Supplementary Figure 1.
We have adopted the linear correlation coefficient of the inhibitory profiles of two kinases as a measure of the chemical similarity of those kinases (Tm values tabulated in Supplementary Table 1, Supplementary Figure 2). By this criterion, we find that the set of kinases studied in the monomeric, unphosphorylated state (CDK2, CDK4, and CDK7) demonstrate significantly different inhibitory profiles, with a lowest correlation coefficient of 0.39 (CDK4 vs CDK7) and a highest correlation coefficient of 0.74 (CDK2 vs CDK7) (Figure 3a). That the inhibitor binding fingerprint discriminates so effectively between highly related CDK subfamily members confirms that it provides a sensitive measure of active site similarity.
Figure 3.

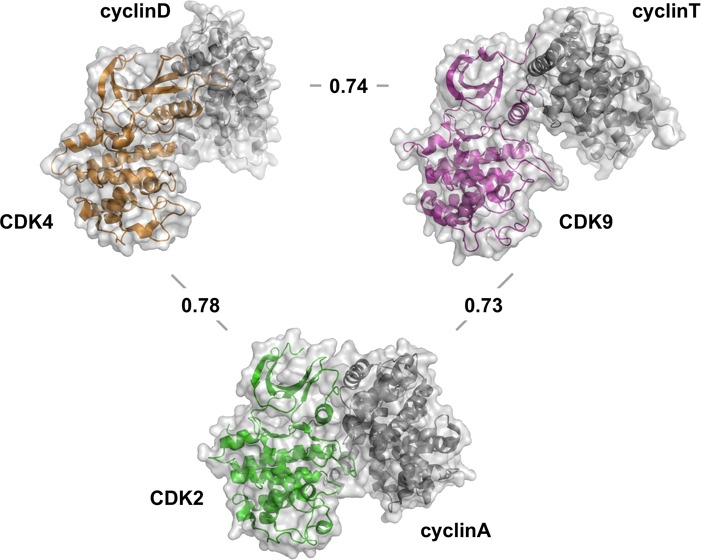
Comparison of the inhibitor-binding profiles of various cyclin-dependent kinase (CDK) subfamily members in different states of activation. Each block represents a CDK in a particular activation state: CDK (monomeric, unphosphorylated), pCDK (monomeric, phosphorylated), CDK/[A or D or T] (unphosphorylated binary complex between a CDK and the corresponding cyclin), and pCDK/[A or D or T] (phosphorylated binary complex between a CDK and the corresponding cyclin). The internal consistency of inhibitory fingerprints for each CDK/state pair, evaluated over two or three repeat measurements, is indicated by a value within the corresponding block. The correlation of fingerprints between different CDK/state pairs is indicated by a value on the line that connects them. (a) Comparison of the inhibitor fingerprints of CDK subfamily members in the monomeric form. (b) Comparison of the inhibitor fingerprints of CDK subfamily members in the phosphorylated binary forms. (c) Comparison of CDK2 inhibitor fingerprints in different activation states. Color intensity as in Figure 2b reflects activation state (more saturated, more active).
One application of this measure is to assess the chemical similarity of the active sites of orthologous proteins from different species. For example, the correlation coefficients obtained for the comparison of human monomeric, unphosphorylated CDKs with the closest CDK orthologue from Plasmodium falciparum, protein kinase 5 (PfPK5), range from 0.42 (PfPK5 vs CDK4) to 0.69 (PfPK5 vs CDK2). This result demonstrates that the active sites of orthologous proteins from highly diverged species may be more similar to each other than are the active sites of close paralogues within a species.
For context, the equivalent comparison of each of the monomeric, unphosphorylated CDKs with an “unrelated” kinase, P. falciparum protein kinase 7 (PfPK7),25 yields correlation coefficients in the range 0.33 (PfPK7 vs CDK2) to 0.31 (PfPK7 vs CDK2/A).
Next we investigated the apparent active-site similarity of a set of fully activated CDK subfamily members (pCDK2/A, pCDK4/D, and pCDK9/T). Although this comparison also yielded quantitatively different inhibitor fingerprints, our results suggest that the inhibitor-binding properties of the set of fully activated CDKs are more similar to each other than are those of the set of inactive monomeric forms (Figure 3b). The lowest correlation coefficient of 0.73 and the highest correlation coefficient of 0.78 were measured for comparisons of pCDK2/A vs pCDK9/T and pCDK2/A vs pCDK4/D, respectively. This result has two implications: first, it demonstrates that the inhibitor binding properties of CDK subfamily members depend not only on their respective sequence but also on the conformational state in which they are found. Second, it demonstrates that, in adopting an active conformation, two different CDKs assume more similar inhibitor binding properties. This latter point is most directly demonstrated by comparison of the correlations coefficients CDK2 vs CDK4 (CC = 0.69) and pCDK2/A vs pCDK4/D (CC = 0.78).
The inhibitor fingerprints of four different CDK2 activation states were recorded, and the resulting comparison is shown in Figure 3c. As expected from comparative structural studies,26,27 phosphorylation of the activation segment has little effect on the inhibitor-binding fingerprint (correlation coefficient for the comparison CDK2 vs pCDK2 is 0.94 and for the comparison of CDK2/A vs pCDK2/A is 0.96). Indeed, sequence and structural comparisons demonstrate that phosphorylation of CDK2 or CDK2/A on Thr160 introduces only minor changes to the identity and structure of the amino acids that line the extended inhibitor binding site (Supplementary Figure 3).
By contrast, when pCDK2 associates with cyclin A, the CDK2 inhibitor-binding fingerprint changes significantly (correlation coefficient for pCDK2 vs pCDK2/A is 0.79). Interestingly, the equivalent comparison for unphosphorylated CDK2 demonstrates somewhat less of a response: when cyclin A associates with the unphosphorylated form of CDK2, it perturbs its inhibitor binding profile to a lesser extent (correlation coefficient for CDK2 vs CDK2/A = 0.91). Comparison of the ATP binding site topology highlights a similar degree of conservation between the closest pairs considered, as summarized in Supplementary Figure 3.
For the CDKs analyzed in this study, the abundance of each conformational state within a cell will depend on a large number of factors that include CDK identity, cell type, cell environment, and cell cycle stage. Targeting a specific conformation of a protein kinase is a validated route to the design of selective and potent inhibitors and has been particularly successful in the design of tyrosine kinase inhibitors.28 Cell cycle CDKs of differing phosphorylation status and in monomeric and cyclin-bound forms can be detected in cells, suggesting that such states are accessible for targeting.29
In summary, by using DSF we have demonstrated that it is possible to discriminate between closely related CDKs by comparing their inhibitor-binding profiles. We have shown that activation state can have a significant effect on inhibitor-binding profile, with cyclin binding having more profound consequences than phosphorylation at least in the case of CDK2. Our results further suggest that the inhibitor binding properties of a set of fully activated, cyclin-bound CDKs are more similar to each other than are those of a set of inactive, monomeric forms. These data provide direct evidence that, as has been proposed elsewhere,13 protein kinases may be more amenable to selective inhibitor binding in their inactive states.
Methods
Inhibitor Set
A full description of the inhibitor set is provided in the Supporting Information.
CDK Expression and Purification
Human CDK4 and cyclin D3,19 CDK9 and cyclin T130 were all expressed in insect cells and purified as described. CDK2 phosphorylated on Thr160 in association with cyclin A2 (pCDK2/A),18 unphosphorylated CDK2 in association with cyclin A2 (CDK2/A), monomeric Thr160pCDK2 (pCDK2),26 monomeric unphosphorylated CDK2 (CDK2), and CDK7 were prepared as described.31 Monomeric CDK4 was expressed in Sf9 cells as a GST fusion and purified by affinity chromatography followed by 3C cleavage of the GST tag and subsequent size-exclusion chromatography. The proteins were concentrated to 2 mg mL–1 and flash frozen in liquid nitrogen prior to storage at −80 °C until further use.
Differential Scanning Fluorimetry
Thermal melting experiments were carried out using an Mx3005p Real Time PCR machine (Stratagene) essentially as described.32,33 A full description of the method is provided in the Supporting Information.
Kinase Targets of the Molecules Contained in the Inhibitor Set
The kinase targeted by each of the inhibitors was identified from either the supplier’s information or by literature analysis, mainly via the work reported in refs (34−36.)
Computation
Residues corresponding to the first coordination shell of the ATP-binding sites were identified in ref (37). Sequence conservation scores were calculated using the ConSurf webserver (http://consurf.tau.ac.il).
Acknowledgments
We thank I. Taylor for technical assistance, E. Lowe and D. Staunton for crystallography and biophysics support, and TTP LabTech for help with inhibitor dispensing. We also thank R. Griffin and colleagues in the Department of Chemistry, Newcastle University and S. Knapp (SGC, Oxford) for providing reagents. We are grateful to S. Baumli (Merck Serono) for the gift of purified pCDK9/T and help with thermal denaturation data collection. This research was supported by Framework Program 6 (FP6) of the European Commission (PROKINASE Project, A.E.), and the MRC (J.A.E., A.H., and M.E.M.N.). A.J.H. was supported by a Wellcome Trust studentship (Grant 083113/Z/07/A).
Glossary
Abbreviations
- CDK
cyclin-dependent kinase
- DSF
differential scanning fluorimetry
- pCDK4/D
Thr172-phosphorylated CDK4/cyclin D3
- pCDK9/T
Thr186-phosphorylated CDK9/cyclin T1
- pCDK2/A
Thr160-phosphorylated CDK2/cyclin A2
Supporting Information Available
This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ Centre de Biochimie Structurale, INSERM-CNRS-UM1-UM2, 29 rue de Navacelles, 34090 Montpellier cedex, France.
Author Present Address
∥ 114 Milton Park, Milton, Abingdon, Oxfordshire OX14 4SA, U.K.
Author Present Address
⊥ Newcastle Cancer Centre, Northern Institute for Cancer Research, Newcastle University, Paul O’Gorman Building, Medical School, Framlington Place, Newcastle NE2 4HH, U.K.
The authors declare no competing financial interest.
Supplementary Material
References
- Zhang J.; Yang P. L.; Gray N. S. (2009) Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 9, 28–39. [DOI] [PubMed] [Google Scholar]
- Fabbro D.; Cowan-Jacob S. W.; Mobitz H.; Martiny-Baron G. (2012) Targeting cancer with small-molecular-weight kinase inhibitors. Methods Mol. Biol. (N. Y.) 795, 1–34. [DOI] [PubMed] [Google Scholar]
- Cohen P. (2009) Targeting protein kinases for the development of anti-inflammatory drugs. Curr. Opin. Cell Biol. 21, 317–324. [DOI] [PubMed] [Google Scholar]
- Janne P. A.; Gray N.; Settleman J. (2009) Factors underlying sensitivity of cancers to small-molecule kinase inhibitors. Nat. Rev. Drug Discovery 8, 709–723. [DOI] [PubMed] [Google Scholar]
- Karaman M. W.; Herrgard S.; Treiber D. K.; Gallant P.; Atteridge C. E.; Campbell B. T.; Chan K. W.; Ciceri P.; Davis M. I.; Edeen P. T.; Faraoni R.; Floyd M.; Hunt J. P.; Lockhart D. J.; Milanov Z. V.; Morrison M. J.; Pallares G.; Patel H. K.; Pritchard S.; Wodicka L. M.; Zarrinkar P. P. (2008) A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 26, 127–132. [DOI] [PubMed] [Google Scholar]
- Smyth L. A.; Collins I. (2009) Measuring and interpreting the selectivity of protein kinase inhibitors. J. Chem. Biol. 2, 131–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse M.; Kuriyan J. (2002) The conformational plasticity of protein kinases. Cell 109, 275–282. [DOI] [PubMed] [Google Scholar]
- Cowan-Jacob S. W. (2006) Structural biology of protein tyrosine kinases. Cell. Mol. Life Sci. 63, 2608–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning G.; Whyte D. B.; Martinez R.; Hunter T.; Sudarsanam S. (2002) The protein kinase complement of the human genome. Science 298, 1912–1934. [DOI] [PubMed] [Google Scholar]
- Morgan D. O. (2007) The Cell Cycle, Principles of Control (Primers in Biology), New Science Press Ltd., London. [Google Scholar]
- Bartkowiak B.; Liu P.; Phatnani H. P.; Fuda N. J.; Cooper J. J.; Price D. H.; Adelman K.; Lis J. T.; Greenleaf A. L. (2010) CDK12 is a transcription elongation-associated CTD kinase, the metazoan ortholog of yeast Ctk1. Genes Dev. 24, 2303–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H. H.; Wong Y. H.; Geneviere A. M.; Fann M. J. (2007) CDK13/CDC2L5 interacts with L-type cyclins and regulates alternative splicing. Biochem. Biophys. Res. Commun. 354, 735–740. [DOI] [PubMed] [Google Scholar]
- Noble M. E.; Endicott J. A.; Johnson L. N. (2004) Protein kinase inhibitors: insights into drug design from structure. Science 303, 1800–1805. [DOI] [PubMed] [Google Scholar]
- Ali S.; Heathcote D. A.; Kroll S. H.; Jogalekar A. S.; Scheiper B.; Patel H.; Brackow J.; Siwicka A.; Fuchter M. J.; Periyasamy M.; Tolhurst R. S.; Kanneganti S. K.; Snyder J. P.; Liotta D. C.; Aboagye E. O.; Barrett A. G.; Coombes R. C. (2009) The development of a selective cyclin-dependent kinase inhibitor that shows antitumor activity. Cancer Res. 69, 6208–6215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavletich N. P. (1999) Mechanisms of cyclin-dependent kinase regulation: structures of Cdks, their cyclin activators, and Cip and INK4 inhibitors. J. Mol. Biol. 287, 821–828. [DOI] [PubMed] [Google Scholar]
- Echalier A.; Endicott J. A.; Noble M. E. (2010) Recent developments in cyclin-dependent kinase biochemical and structural studies. Biochim. Biophys. Acta 1804, 511–519. [DOI] [PubMed] [Google Scholar]
- De Bondt H. L.; Rosenblatt J.; Jancarik J.; Jones H. D.; Morgan D. O.; Kim S. H. (1993) Crystal structure of cyclin-dependent kinase 2. Nature 363, 595–602. [DOI] [PubMed] [Google Scholar]
- Brown N. R.; Noble M. E.; Endicott J. A.; Johnson L. N. (1999) The structural basis for specificity of substrate and recruitment peptides for cyclin-dependent kinases. Nat. Cell Biol. 1, 438–443. [DOI] [PubMed] [Google Scholar]
- Takaki T.; Echalier A.; Brown N. R.; Hunt T.; Endicott J. A.; Noble M. E. (2009) The structure of CDK4/cyclin D3 has implications for models of CDK activation. Proc. Natl. Acad. Sci. U.S.A. 106, 4171–4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day P. J.; Cleasby A.; Tickle I. J.; O’Reilly M.; Coyle J. E.; Holding F. P.; McMenamin R. L.; Yon J.; Chopra R.; Lengauer C.; Jhoti H. (2009) Crystal structure of human CDK4 in complex with a D-type cyclin. Proc. Natl. Acad. Sci. U.S.A. 106, 4166–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarricone C.; Dhavan R.; Peng J.; Areces L. B.; Tsai L. H.; Musacchio A. (2001) Structure and regulation of the CDK5-p25(nck5a) complex. Mol. Cell 8, 657–669. [DOI] [PubMed] [Google Scholar]
- Schneider E. V.; Bottcher J.; Blaesse M.; Neumann L.; Huber R.; Maskos K. (2011) The structure of CDK8/CycC implicates specificity in the CDK/cyclin family and reveals interaction with a deep pocket binder. J. Mol. Biol. 412, 251–266. [DOI] [PubMed] [Google Scholar]
- Lo M. C.; Aulabaugh A.; Jin G.; Cowling R.; Bard J.; Malamas M.; Ellestad G. (2004) Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal. Biochem. 332, 153–159. [DOI] [PubMed] [Google Scholar]
- Najmanovich R. J.; Allali-Hassani A.; Morris R. J.; Dombrovsky L.; Pan P. W.; Vedadi M.; Plotnikov A. N.; Edwards A.; Arrowsmith C.; Thornton J. M. (2007) Analysis of binding site similarity, small-molecule similarity and experimental binding profiles in the human cytosolic sulfotransferase family. Bioinformatics 23, e104–109. [DOI] [PubMed] [Google Scholar]
- Merckx A.; Echalier A.; Langford K.; Sicard A.; Langsley G.; Joore J.; Doerig C.; Noble M.; Endicott J. (2008) Structures of P. falciparum protein kinase 7 identify an activation motif and leads for inhibitor design. Structure 16, 228–238. [DOI] [PubMed] [Google Scholar]
- Brown N. R.; Noble M. E.; Lawrie A. M.; Morris M. C.; Tunnah P.; Divita G.; Johnson L. N.; Endicott J. A. (1999) Effects of phosphorylation of threonine 160 on cyclin-dependent kinase 2 structure and activity. J. Biol. Chem. 274, 8746–8756. [DOI] [PubMed] [Google Scholar]
- Russo A. A.; Jeffrey P. D.; Pavletich N. P. (1996) Structural basis of cyclin-dependent kinase activation by phosphorylation. Nat. Struct. Biol. 3, 696–700. [DOI] [PubMed] [Google Scholar]
- Fang Z.; Grutter C.; Rauh D. (2013) Strategies for the selective regulation of kinases with allosteric modulators: exploiting exclusive structural features. ACS Chem. Biol. 8, 58–70. [DOI] [PubMed] [Google Scholar]
- Schachter M. M.; Merrick K. A.; Larochelle S.; Hirschi A.; Zhang C.; Shokat K. M.; Rubin S. M.; Fisher R. P. (2013) A Cdk7-Cdk4 T-loop phosphorylation cascade promotes G1 progression. Mol. Cell 50, 250–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumli S.; Lolli G.; Lowe E. D.; Troiani S.; Rusconi L.; Bullock A. N.; Debreczeni J. E.; Knapp S.; Johnson L. N. (2008) The structure of P-TEFb (CDK9/cyclin T1), its complex with flavopiridol and regulation by phosphorylation. EMBO J. 27, 1907–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lolli G.; Lowe E. D.; Brown N. R.; Johnson L. N. (2004) The crystal structure of human CDK7 and its protein recognition properties. Structure 12, 2067–2079. [DOI] [PubMed] [Google Scholar]
- Matulis D.; Kranz J. K.; Salemme F. R.; Todd M. J. (2005) Thermodynamic stability of carbonic anhydrase: measurements of binding affinity and stoichiometry using ThermoFluor. Biochemistry 44, 5258–5266. [DOI] [PubMed] [Google Scholar]
- Bullock A. N.; Debreczeni J. E.; Fedorov O. Y.; Nelson A.; Marsden B. D.; Knapp S. (2005) Structural basis of inhibitor specificity of the human protooncogene proviral insertion site in moloney murine leukemia virus (PIM-1) kinase. J. Med. Chem. 48, 7604–7614. [DOI] [PubMed] [Google Scholar]
- Bain J.; McLauchlan H.; Elliott M.; Cohen P. (2003) The specificities of protein kinase inhibitors: an update. Biochem. J. 371, 199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain J.; Plater L.; Elliott M.; Shpiro N.; Hastie C. J.; McLauchlan H.; Klevernic I.; Arthur J. S.; Alessi D. R.; Cohen P. (2007) The selectivity of protein kinase inhibitors: a further update. Biochem. J. 408, 297–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies S. P.; Reddy H.; Caivano M.; Cohen P. (2000) Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 351, 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt D. J.; Bentley J.; Jewsbury P.; Boyle F. T.; Endicott J. A.; Noble M. E. (2006) Dissecting the determinants of cyclin-dependent kinase 2 and cyclin-dependent kinase 4 inhibitor selectivity. J. Med. Chem. 49, 5470–5477. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.