

Abstract
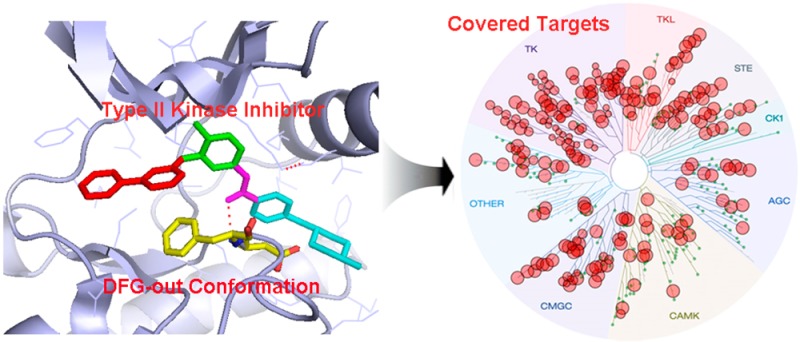
The ATP site of kinases displays remarkable conformational flexibility when accommodating chemically diverse small molecule inhibitors. The so-called activation segment, whose conformation controls catalytic activity and access to the substrate binding pocket, can undergo a large conformational change with the active state assuming a ‘DFG-in’ and an inactive state assuming a ‘DFG-out’ conformation. Compounds that preferentially bind to the DFG-out conformation are typically called ‘type II’ inhibitors in contrast to ‘type I’ inhibitors that bind to the DFG-in conformation. This review surveys the large number of type II inhibitors that have been developed and provides an analysis of their crystallographically determined binding modes. Using a small library of type II inhibitors, we demonstrate that more than 200 kinases can be targeted, suggesting that type II inhibitors may not be intrinsically more selective than type I inhibitors.
Kinases represent one of the largest enzyme families whose three-dimensional structures are largely conserved from simple prokaryotes to humans. By functioning within complex and intercommunicating networks, kinases serve as a fundamental building block that allows cells to compute an appropriate biological response to a particular stimulation.1 It is therefore not surprising that deregulation of kinases as a result of transcriptional, mutational, or post-translational modifications can lead to inappropriate cellular responses and ultimately to disease. Over the past two decades, intensive drug discovery efforts, particularly those targeting mutationally activated kinases or oncogenic kinase fusion proteins such as BRAF, PI3K, BCR-ABL, EML4-ALK, EGFR, and c-KIT, have resulted in the approval of 23 ATP competitive kinase inhibitor drugs with a large number of additional compounds advancing in clinical and preclinical development.2,3 Estimates suggest that deregulation of about 180 kinases is associated with disease, particularly cancer, and approximately 40 kinases are being actively pursued by drug discovery programs.4
The majority of small molecule kinase inhibitors bind in or around the nucleotide binding cleft, thereby preventing ATP from binding. The large number of available X-ray crystal structures in the public domain have revealed a multitude of different ways of small molecules can be recognized in and around the ATP-binding cleft. Recently, van Linden et al. have built up a database for kinase–ligand interaction (KLIFS) with the published 3-D structures and have provided comprehensive analysis of the binding modes.5 Historically these binding modes have been classified as type I, type II, type III, and type IV depending on whether the compounds bind competitively with ATP using the ‘DFG-in’ (type I) conformation or the ‘DFG-out’ (type II) conformation or noncompetitively by binding distal to the ATP-binding pocket (type III and type IV). Although it would be more appropriate to characterize inhibitors according to how they impact the kinetic parameters of the kinase (kcat, KM for ATP and substrate) and whether they display competitive, noncompetitive, or mixed inhibition profiles, this approach would have several practical limitations. First, this type of enzymological analysis is almost never reported for new kinase inhibitors, making it impossible to be used for classification. Second, kinases are typically subject to complex post-translational and regulatory interactions in the cell that make it very difficult to determine a form of the enzyme that might serve as a faithful representative of its intracellular state. In contrast, there are a large and ever increasing number of inhibitor-kinase co-crystal structures that have been determined using X-ray crystallography that can be used to classify binding conformations. However, type I inhibitor-bound structures constitute the majority of co-crystal structures, and structural data for type II inhibitor complexes, especially for serine/threonine kinases, are largely lacking. Crystallographically determined structures provide an invaluable guide to medicinal chemists who seek to understand how changes they make to the small molecule impact its molecular recognition across the kinase family. Key to successful implementation of this approach is the careful correlation of changes in biochemical and cellular function with observed structural changes.
Review of Binding Modes
The most commonly observed binding mode, called ‘type I’, involves the inhibitor binding to the ATP-site with the activation loop assuming a conformation conducive to phosphate transfer. Typically the inhibitor will possess some heterocyclic structure, exploit the hydrophobic adenine binding pocket, and form zero to three hydrogen bonds to the kinase ‘hinge segment’, which serves to connect the N- and C-terminal kinase lobes. A classic example is the EGFR inhibitor Gefitinib, where the quinazoline core occupies the adenine binding area and makes one hydrogen bond with Met793 in the hinge and two water-mediated hydrogen bonds with Thr790 and Thr854, respectively (Figure 1A, B).6,7
Figure 1.

Representative X-ray structures and demonstration of binding modes. (A) ATP complexes with IRK (PDB ID: 1IR3) in type I binding. (B) Gefitinib complexes with EGFR (PDB ID: 2ITY) in type I binding. (C) Imatinib complexes with Abl (PDB ID: 1IEP) in type II binding. (D) GNF-2 complexes with Abl (PDB ID: 3K5V) type IV binding. (E) Akt allosteric inhibitor complexes with Akt (PDB: 3O96); (F) PD318088 complexes with MEK1 (PDB: 1S9J) in type III binding. (G) Dasatinib complexes with BTK (PDB ID: 3OCT). (H) PF-00215955 complexes with p38 (PDB ID: 3K3I).
The type II binding mode was serendipitously discovered when Imatinib was co-crystallized with Abl.8 In contrast to the presumed U-shaped binding mode that would be required for the compound to be accommodated in the DFG-in conformation, the crystal structure demonstrated that the inhibitor bound in an elongated conformation with the benzamide moiety of the inhibitor displacing the phenylalanine of the DFG-motif. A subsequently determined structure of Sorafenib with c-Raf revealed an overall similar binding mode, which was termed a ‘type II’ binding mode.9 Imatinib forms four critical hydrogen bonds with Abl, which include Met318 to the pyridine N, Thr315 to aniline NH, Glu286 from the C-Helix to the amide oxygen, and Asp381 from the ‘DFG-motif’ to the amide NH (Figure 1C).10 Hydrogen bonds to Glu286 located in the C-Helix and Asp381 from the ‘DFG-motif’ are formed by most type II inhibitors. These conserved interactions have been used to define a pharmacophore model that allows for the rational design of compounds that have the potential to act as type II inhibitors.11
A third category of kinase inhibitors is allosteric, sometimes called ‘type IV’. To date, there are only a few well-characterized allosteric inhibitors, most of which bind remotely from the ATP-site such as the rapalogs,12 GNF-2 and GNF-5,13 as well as AKT inhibitors such as MK2206.14−16 GNF-2 and GNF-5 are inhibitors of ABL that bind to the myristoyl-binding pocket and facilitate an inhibitory latching of the SH3 and SH2 domains onto the kinase domain13 (Figure 1D). The non-ATP competitive AKT inhibitor exerts its inhibitory function via disrupting the interaction between the PH domain and kinase domain14 (Figure 1E) . Another well characterized class of inhibitors called ‘type III’ are the MEK1 and 2 kinase inhibitors such as PD318088,17 CI-1040,18 AZD6244,19 TAK733,20 GSK1120212,21 and AS703026.22 PD318088 binds to a hydrophobic pocket directly adjacent to the ATP-binding site and makes a hydrogen bond to the γ-phosphate of ATP (Figure 1F). PD318088 traps MEK1 kinase in a conformation where the c-Helix and activation loop are in a noncatalytic conformation.17 Other examples of allosteric binding have been observed for JNKs, PAK1, CHK1, CK2, GSK3-β, PKC-δ, and p38, and a discussion of their binding modes can be found in a recent comprehensive review.23
Some inhibitors have been observed to bind to conformations that are intermediate between the fully DFG-in and DFG-out conformations, For instance, dasatinib, typically classified as a type I inhibitor, binds to BTK (PDB ID: 3OCT) with both Asp539 and Phe540 of the ‘DFG motif’ assuming a conformation that is intermediate between the fully active and inactive states24 (Figure 1G). In addition, some of the inhibitors such as p38 kinase inhibitor PF-00215955 stabilize the inactive (DFG-out) conformation without occupying the adenine binding pocket near the hinge area stabilizing the binding through two hydrogen bonds with residues in the c-Helix and DFG motif and the binding pocket created by DFG-out conformation25 (Figure 1H).
Among the approved 23 kinase inhibitors that directly bind to the kinase catalytic domain, 16 of them adopt a type I binding mode to their primary targets, 6 of them adopt the type II binding mode, and 1 utilizes a type III binding mode (Figure 2). In addition, there are over two dozen well-characterized type II kinase inhibitors and among them at least 16 are under clinical development for different tumor indications (Figure 3 and Table 1).
Figure 2.

FDA-approved kinase inhibitors with binding modes.
Figure 3.

Examples of known type II kinase inhibitors.
Table 1. Example of Type II Kinase Inhibitors under Clinical Evaluation.
| drug name | drug targets | clinical development | company |
|---|---|---|---|
| Nilotinib (AMN-107) | BCR-Abl, c-Kit, LCK,EphA3, EphA8 | approved | Novartis |
| Sorafenib (Nexavar) | VEGFR, GFR, Raf | approved | Bayer/Onyx |
| Imatinib (Gleevec) | BCR-Abl, c-Kit | approved | Novartis |
| Ponatinib | BCR-Abl | approved | Ariad |
| Regorafenib | c-Kit, VEGFR-2 | approved | Bayer |
| XL-184 | MET, VEGFR-2, RET | approved | Exelixis |
| OSI-930 | c-kit, VEGFR-2 | phase I | Astellas Pharma Inc. |
| AMG-900 | Aurora | phase I | Amgen |
| KX2-391 | Src | phase II | Kinex Pharma Inc. |
| AC-220 | Flt-3 | phase II | Ambit Biosciences |
| BMS777607 | MET | phase II | Bristol-Myers Squibb |
| GSK-1363089 | MET, VEGFR-2, KDR | phase II | GlaxoSmithKline |
| AB1010 | c-Kit, PDGFRs, FGFR3 | phase III | AB Science |
| Apatinib (YN968D) | VEGFR-2 | phase III | FhengRui Medicine Co. |
Correlation of Selectivity and Kinase Conformation
Achieving high selectivity for kinase inhibitors is very challenging given the fact that the ATP-binding pocket’s spatial structure is highly conserved among all of the protein kinases in the catalytically active conformation. However, despite this similarity, highly selective inhibitors have been developed that exploit subtle differences in sequence and 3-dimensional structure of the ATP-binding cleft. Type I inhibitors typically have a heterocyclic motif that makes 1–3 hydrogen bonds to the hinge segment and that possess side-chains that introduce contacts with less conserved regions of the ATP-binding pocket. Several excellent reviews have been written about type I inhibitors and they will not be further discussed here.26−29 Type III inhibitors are usually the most selective as they exploit binding pockets and regulatory mechanisms that are unique to a particular kinase. Unfortunately, discovery of allosteric inhibitors is largely an empirical exercise, and relatively few allosteric kinase inhibitors have been discovered to date.
A prevailing view was that type II inhibitors are intrinsically more selective than type I inhibitors, which has served to spur many type II medicinal chemistry campaigns. One rationale for this commonly held belief is based on the observation that inactive conformations of kinase are more dynamic than the catalytically active conformations. This is humorously summarized by Nobel et al. who state, “All active kinases are alike but an inactive kinase is inactive after its own fashion.”30 For example, residues that surround the hydrophobic pocket created by the DFG-out flip are not as highly conserved as those in the ATP-binding pocket, leading to the suggestion that type II inhibitors might have a greater propensity for kinase selectivity relative to type I inhibitors.11 In addition, whether the DFG-out inactive conformation can be generally accessed for all kinases is an outstanding question. On the basis of the enumeration of the currently solved kinase co-structures one might assume that type II inhibition might be a relative rare phenomena.31 It has been postulated that type II binding modes might be most accessible for kinases possessing a small ‘gatekeeper’ residue such as a threonine.32 Recently, however it has become clear that kinases bearing medium-sized gatekeeper residues could also adopt a type II binding mode.33−35 Furthermore, why some kinases are more prone to type II inhibitors is not well understood as some of the crystalized apo inactive conformations have not been targeted by type II inhibitors yet such as CDK6. Molecular dynamics simulation studies with Abl suggest that binding mode selection is dependent upon pH. However, whether this is generally the case is not known.36 Sequence and mutagenesis studies of MAPK kinases have suggested that the gatekeeper residue and the identity of the amino acid proceeding the DFG motif are critical factors controlling the active and inactive conformations.37 It is therefore important to experimentally determine the chemical features of the inhibitors and their target kinases that make them susceptible to type II inhibition.
Analysis of Kinases That Assume Type II Binding As Verified by X-ray Crystallography
In order to obtain an overview of which kinases can adopt the type II binding, we analyzed the available X-ray kinase structures (updated until June, 2013) in the protein data bank (PDB) and studied their binding modes. This analysis revealed 48 kinases adopted a type II (DFG-out) binding mode (Supplemental Table 1). All groups of the kinome are represented in this set including 27 of 90 kinases (30%) in the TK kinase group, 7 of 63 kinases (11%) in the CMGC group, 2 (4%) in the TKL group, 2 (2%) in the ‘other’ group, 3 (6%) in the STE group, 2 of 43 kinases (3%) in the AGC group, 4 of 74 kinases (5%) in the CAMK groups, and 1 of 12 kinases (8%) in the casein kinase (CK1) group (Figure 4A). Analysis of the gatekeeper residues among these 48 kinases demonstrated that 15 kinases (31.3%) possess methionine, 14 kinases (29.2%) possess threonine, 11 kinases (22.9%) possess phenylalanine, 3 kinases (6.3%) possess leucine, 3 kinases (6.3%) possess isoleucine, and 2 kinases (4.2%) possess valine (Figure 4B). In addition, we found that the activation loop can either direct toward the hinge (L, 37/48) or toward the αC-Helix (R, 11/48) (Figure 4C,D).
Figure 4.

Analysis of X-ray crystal structure confirmed type II conformation (DFG-out) kinases. (A) Treespot demonstration of type II conformation (DFG-out) kinase distribution. (B) Analysis of gate keeper residues. (C) Demonstration of DFG-out A-loop-R (direct to C-Helix) conformation (PDB ID: 1M52). (D) Demonstration of DFG-out A-loop-L (direct to Hinge) conformation (PDB ID: 1IEP).
Analysis of the chemical structures of the co-crystallized inhibitors revealed that many of them share common structural features that fit well with a previously articulated pharmacophore model for type II inhibitors11 (Supplemental Figure 1 and Figure 5). Similar to type I inhibitors, there usually is a ‘head’ hinge-binding moiety, typically a heterocycle that occupies the region normally occupied by the adenine of ATP. These head groups can be decorated with various substitutions that can take advantage of unique topological features of the kinase hinge, for example the presence of a proline. Next to the ‘hinge-binding moiety’ there is usually a ‘linker’ part segment that traverses the region adjacent to the kinase gatekeeper residue. This linker segment is usually 3–5 chemical bond lengths and can be either a highly flexible aliphatic chain or rigid aromatic ring systems. The linkers need to carefully accommodate the identity of the gatekeeper residue by either providing hydrogen bonding functionality for the gatekeeper threonine or by leaving enough space for kinases with large gatekeeper residues. Following the linker segment is a hydrogen-bonding moiety that is required for making conserved hydrogen bonds with the C-Helix and DFG motif. Typically this hydrogen-bonding moiety is an amide or urea. Kinase selectivity can be modulated by adjusting the ideal location of this moiety. In some rare cases there is only one hydrogen bond, and then this can be either an ether or an amine linkage. The final ‘tail’ segment typically consists of a hydrophobic moiety that occupies the pocket created by the DFG-out flip. This ‘tail’ sometimes contains a basic nitrogen that improves water solubility and sometimes engages in additional H-bonding interactions with the kinase. Despite the usefulness of this model in constructing new potential type II inhibitors, it is important to recognize a given inhibitor can adopt different binding modes to different kinases. For example imatinib binds to kinases such as Abl, c-Kit, and DDR1 in the classical extended conformation but assumes a U-shape conformation when bound to Syk (PDB ID: 1XBB).38 Indeed, a covalent version of imatinib, JNK-IN-8, co-crystallizes with JNK3 (PDB ID: 3V6S) as a type I inhibitor using a U-shaped conformation. (39)
Figure 5.
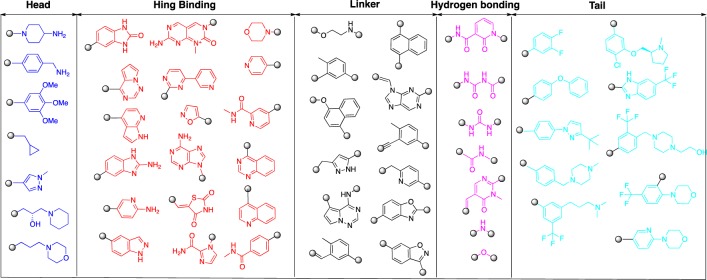
Schematic representation of type II kinase inhibitors constitution.
Analysis of Kinases That Are Predicted To Assume a Type II Binding Mode Based upon Pharmacological Profiling
To complement the enumeration of kinases based on experimentally confirmed type II binding, we also evaluated how many kinases could be potently targeted with a diverse assortment of 36 type II kinase inhibitors that were subject to profiling against panels of at least 400 kinases using DiscoveRx’s KinomeScan technology, which can detect most type I, type II, and other allosteric inhibitors.40 This includes 12 inhibitors (Figure 6, Supplemental Table 2) synthesized in our laboratory as well as 24 previously published type II inhibitors (Figure 3 and Supplemental Table 3).41 Analysis of the selectivity S(1) scores (the S score provides a measure of kinase selectivity with more selective inhibitors exhibiting smaller S scores) of 12 inhibitors reveals that the selectivity can range from highly selective (0.02) to poor selectivity (0.30) (Figure 6 and Supplemental Figure 2). We enumerated kinases that bound to inhibitors with an apparent Kd (or IC50 < 1 μM, or KinomeScan Score <1) of at least 1 μM based upon the KinomeScan technology. While we expect that the large majority of these kinases will be bound in the type II conformation (DFG-out) (indeed of the 9 available crystal structures with inhibitors in this set all bound as type II), there may be some that bind as type I or using other unanticipated binding modes. This analysis suggests that there may be up to 220 kinases that can be targeted even by this limited compound set, which is a considerably larger number than the 48 that have been crystallographically demonstrated to bind type II inhibitors to date (Table 2). Among these 220 kinases predicted to be sensitive to type II inhibitors, only 40 have been crystallographically validated to bind in the type II conformation (DFG-out) (Table 2, green highlight). Those 8 kinases that have been experimentally verified to have DFG-out conformation that were not revealed by this analysis is a consequence of these kinases not being targeted by the chemical diversity represented in this 36-member type-II kinase inhibitor set (Table 2, red highlight). The distribution of the 220 kinases spanned all the groups of the kinome including 80 of 90 TK family kinases (89%), 22 of 43 TKL family kinases (51%), 17 of 74 CAMK family kinases (23%), 39 of 63 CMGC family kinases (62%), 13 of 63 AGC family kinases (20%), 25 of 47 STE family kinases (53%), 4 of 12 CK1 family kinases (33%), and 20 of 81 other family kinases (25%) (Figure 7A). Sequence alignment revealed that there was broad tolerance for diverse gatekeeper residues with methionine accounting for 33%, threonine for 26%, phenylalanine for 17.7%, leucine for 12.7%, and valine for 5.5%. It is worth noting that kinases possessing very large gatekeeper residues such as isoleucine (2.3%), glutamic acid (1%), as well as tyrosine (1%) can also be targeted by type II inhibitors (Figure 7B). This follows almost the same trend as the gatekeeper residues distribution among the full kinome, suggesting that type II inhibitors do not display a strong bias for the identity of the gatekeeper residue (Figure 7C). In this inhibitor collection at least 10 type II inhibitors were capable of inhibiting: BLK, CSF1R, DDR1/2, EPHA8, FLT3, JNK2, KIT, LCK, LYN, LOK and MUSK, p38, PDGFRα/β, RET, SLK, SRC, TAK1, TIE2, TRKC, VEGFR2, and ZAK (Figure 7D,E and Supplemental Table 4). These kinases maybe be enriched in the set simply because several of them, including FLT3, KIT, p38, PDGFRα/β, RET, SRC, and VEGFR2, have been the subject of extensive drug discovery efforts that would result in an enrichment in chemotypes with activity on these and related kinases (Figure 7F). Further bioinformatical and structural analysis will be required to elucidate whether there are currently cryptic factors that predispose kinases to be prone to type II inhibition.
Figure 6.

Chemical structures and S(1) score of 12 in-house generated/published inhibitors.
Table 2. Kinases That Bind Type II Kinase Inhibitors of the Tested Seta.
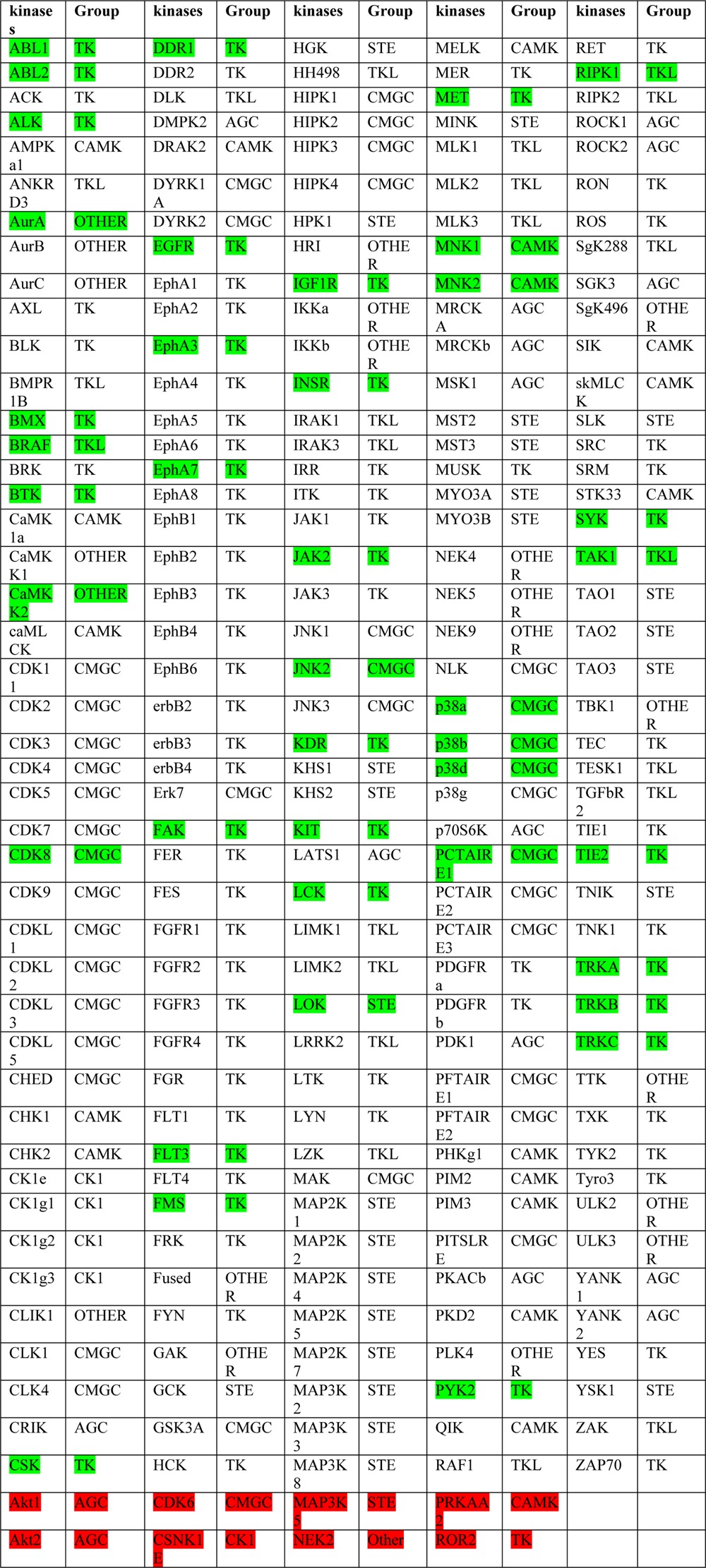
Green color indicates the targets revealed both by X-ray and inhibitor profiling analysis. Red color indicates those targets revealed only by X-ray.
Figure 7.

Analysis of selectivity profiling revealed type II binding kinases. (A) Treespot demonstration of kinase distribution among kinome. (B) Analysis of gatekeeper residues among the type II inhibitor targeted kinases. (C) Analysis of gatekeeper residues among the kinome. (D) Statistics of most targeted kinases by type II inhibitors. (E) Treespot demonstration of most targeted kinase distribution among kinome. (F) Analysis of gatekeeper residues of most targeted kinases by type II inhibitors.
Strategies To Develop New Type II Kinase Inhibitors
Several different strategies have been implemented to develop type II kinase inhibitors. The most common approach is rational design based upon the pharmacophore model described above in conjunction with molecular modeling.11,42−45 Frequently the starting chemical scaffold was serendipitously found to be cross-reactive with a new kinase of interest following a kinase profiling experiment.46 Another common approach is the ‘hybridization’ or ‘mix-n-match’ approach where validated fragments from one scaffold are grafted onto another.47,48 Several other interesting approaches have been described that are yet not widely adopted. For example, Erlanson et al. adopted the Well’s tethering approach where a cysteine was used to screen a cysteine-reactive thiol fragment library.49 This approach resulted in the discovery of a fragment that was subsequently elaborated to generate a highly selective type II PDK1 kinase inhibitor. (50) Another approach reported by Abagyan et al. has involved a computational in vitro screen against a model of a DFG-out conformation of the targeted kinase.32 High-throughput biochemical assays with highly activated recombinant enzymes is the typical approach to discover type I kinase inhibitors and is sometimes biased against type II compounds, which typically preferentially bind to the dephosphorylated, inactive kinase. Rauh et al. have developed an HTS compatible acrylodan-labeled kinase binding assay that can distinguish between type I and type II binding and demonstrated its utility to identify a highly selective p38α inhibitor.51,52 The same group also developed a fluorescent based high-throughput assay format that utilized a hydrophobic interaction between the P-loop and activation loop to identify both the active and inactive conformation kinase inhibitors.53 Laufer’s group has developed a conformation-specific fluorescent polarization binding assay that was implemented to identify p38 inhibitors that preferably bind to the inactive conformation of the kinase.54 Vogel et al. developed a time-resolved fluorescence resonance energy transfer (TR-FRET) based binding assay that allows the detection of both active or inactive conformation kinase inhibitors and has been exploited in several studies.55
Perspectives
Kinases display an impressive degree of conformational plasticity that is undoubtedly one major contributor to the surprising pharmacology that is frequently observed when kinase inhibitors are used in vivo. Kinase structural biology has provided a wealth of information as to different kinase binding modes, but the degree to which these snapshots are an accurate reflection of the bonafide intracellular inhibited kinase conformations remains an open question. For example, imatinib has been observed to crystallize with Abl and Src in virtually identical type II conformations (DFG-out) yet displays 100- to 1000-fold more potent biochemical and cellular inhibition of Abl versus Src. Medicinal chemists have surmounted these challenges with the empirical approach of correlating biochemical measurements with structural biology and cellular measurements, which frequently brings surprises. To date 6 of the 23 approved kinase inhibitors would be classified as type II inhibitors. In addition, theoretical analysis using entropy score as criteria revealed that type II inhibitors might be generally more selective than type I inhibitors.56 These have motivated the development of better methodologies that will improve our ability to rationally design such inhibitors with a desired selectivity profile. Our analysis of existent crystal structures reveals 48 kinases that have been observed in the DFG-out conformation. Using a collection of 36 type II inhibitors, we retrieved a set of 220 kinases that could be targeted. However, while the majority of these are likely to be targeted in the type II conformation (DFG-out), some may be recognized in type I or currently unanticipated manners, suggesting that more structural insight is needed into type II inhibitor binding modes in particular for kinases families that are poorly covered by the experimentally determined type II co-crystal structures. In addition, we are clearly heavily biased based upon the current selection of type II inhibitors, and the numbers will change as different chemotypes are added to the pharmacopeia. Nonetheless our analysis helps shape some general conclusions:
1. A significant fraction of the kinome is likely targetable by type II inhibitors, and there are a subset of kinases that seem to have a predisposition for binding currently available type II inhibitors.
2. The currently available type II inhibitors can range from highly selective to highly nonselective. There does not appear to be an inherent selectivity advantage to the type II binding mode.
3. There is a need to diversify the chemotypes being explored as type II inhibitors, especially in the linker and tail region where only a very limited number of pharmacophores have been explored.
Another interesting area of active research is investigating the differences in pharmacological effects that can be attained with type II versus type I inhibitors. For example, type I B-Raf kinase inhibitors such as PLX4032 have been documented to have only positive therapeutic effects on tumors harboring the B-Raf V600E mutation.57,58 In contrast, type II B-Raf inhibitors have been shown to block both the wild type and mutated B-Raf kinases, which is believed to be a consequence of type II binding preventing the transactivation that occurs as a result of B-Raf hetero dimerization.59 Type I and II inhibitors can also complement each other in terms of the spectrum of mutations that confer resistance to each. In addition, studies on c-Kit and FMS kinase have revealed that they might naturally populate the type II (DFG-out) conformation due to the JM domain enforcing this conformation in a cellular context.60 This raises the question whether the type II conformation (DFG-out) is a more native state for a particular subset of kinases. Therefore a careful examination of the pharmacology generated by both type I and type II inhibitors should be undertaken prior to deciding which mode is most advantageous to a given desired end point.61
Acknowledgments
S.K. is grateful for support by the SGC, a registered charity (number 1097737) that receives funds from the Canadian Institutes for Health Research, the Canada Foundation for Innovation, Genome Canada, GlaxoSmithKline, Pfizer, Eli Lilly, the Novartis Research Foundation, Takeda, the Ontario Ministry of Research and Innovation, and the Wellcome Trust.
Glossary
Keywords
- DFG motif
A three amino acids sequence (Asp-Phe-Gly) which is highly conserved among most of the kinases and located before the activation loop of kinase.
- Kinase inhibitors
Defined chemical entities that can block kinases’ catalytic activity.
- Kinases
A group of enzymes mediated variety of cellular signals through catalyzing the transfer of γ-phosphate from ATP to a Tyrosine, Serine or Threonine amino acids residues.
- Type II kinase inhibitors
A group of kinase inhibitors exert its inhibitory activity through stabilizing kinases inactive conformation that characterized as DFG-out conformation.
Supporting Information Available
Supplemental figures and tables as described in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Adams J. A. (2001) Kinetic and catalytic mechanisms of protein kinases. Chem. Rev. 101, 2271–2290. [DOI] [PubMed] [Google Scholar]
- Barf T.; Kaptein A. (2012) Irreversible protein kinase inhibitors: balancing the benefits and risks. J. Med. Chem. 55, 6243–6262. [DOI] [PubMed] [Google Scholar]
- Fabbro D.; Cowan-Jacob S. W.; Mobitz H.; Martiny-Baron G. (2012) Targeting cancer with small-molecular-weight kinase inhibitors. Methods Mol. Biol. 795, 1–34. [DOI] [PubMed] [Google Scholar]
- Liu Q.; Sabnis Y.; Zhao Z.; Zhang T.; Buhrlage S. J.; Jones L. H.; Gray N. S. (2013) Developing irreversible inhibitors of the protein kinase cysteinome. Chem. Biol. 20, 146–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Linden O. P.; Kooistra A. J.; Leurs R.; de Esch I. J.; de Graaf C. (2014) KLIFS: a knowledge-based structural database to navigate kinase-ligand interaction space. J. Med. Chem. 57, 249–277. [DOI] [PubMed] [Google Scholar]
- Hubbard S. R. (1997) Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. EMBO J. 16, 5572–5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun C. H.; Boggon T. J.; Li Y.; Woo M. S.; Greulich H.; Meyerson M.; Eck M. J. (2007) Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell 11, 217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler T.; Bornmann W.; Pellicena P.; Miller W. T.; Clarkson B.; Kuriyan J. (2000) Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science 289, 1938–1942. [DOI] [PubMed] [Google Scholar]
- Wan P. T.; Garnett M. J.; Roe S. M.; Lee S.; Niculescu-Duvaz D.; Good V. M.; Jones C. M.; Marshall C. J.; Springer C. J.; Barford D.; Marais R. (2004) Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 116, 855–867. [DOI] [PubMed] [Google Scholar]
- Nagar B.; Bornmann W. G.; Pellicena P.; Schindler T.; Veach D. R.; Miller W. T.; Clarkson B.; Kuriyan J. (2002) Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571). Cancer Res. 62, 4236–4243. [PubMed] [Google Scholar]
- Liu Y.; Gray N. S. (2006) Rational design of inhibitors that bind to inactive kinase conformations. Nat. Chem. Biol. 2, 358–364. [DOI] [PubMed] [Google Scholar]
- Bayle J. H.; Grimley J. S.; Stankunas K.; Gestwicki J. E.; Wandless T. J.; Crabtree G. R. (2006) Rapamycin analogs with differential binding specificity permit orthogonal control of protein activity. Chem. Biol. 13, 99–107. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Adrian F. J.; Jahnke W.; Cowan-Jacob S. W.; Li A. G.; Iacob R. E.; Sim T.; Powers J.; Dierks C.; Sun F.; Guo G. R.; Ding Q.; Okram B.; Choi Y.; Wojciechowski A.; Deng X.; Liu G.; Fendrich G.; Strauss A.; Vajpai N.; Grzesiek S.; Tuntland T.; Liu Y.; Bursulaya B.; Azam M.; Manley P. W.; Engen J. R.; Daley G. Q.; Warmuth M.; Gray N. S. (2010) Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature 463, 501–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W. I.; Voegtli W. C.; Sturgis H. L.; Dizon F. P.; Vigers G. P.; Brandhuber B. J. (2010) Crystal structure of human AKT1 with an allosteric inhibitor reveals a new mode of kinase inhibition. PLoS One 5, e12913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai H.; Sootome H.; Nakatsuru Y.; Miyama K.; Taguchi S.; Tsujioka K.; Ueno Y.; Hatch H.; Majumder P. K.; Pan B. S.; Kotani H. (2010) MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol. Cancer Ther. 9, 1956–1967. [DOI] [PubMed] [Google Scholar]
- Barnett S. F.; Defeo-Jones D.; Fu S.; Hancock P. J.; Haskell K. M.; Jones R. E.; Kahana J. A.; Kral A. M.; Leander K.; Lee L. L.; Malinowski J.; McAvoy E. M.; Nahas D. D.; Robinson R. G.; Huber H. E. (2005) Identification and characterization of pleckstrin-homology-domain-dependent and isoenzyme-specific Akt inhibitors. Biochem. J. 385, 399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohren J. F.; Chen H.; Pavlovsky A.; Whitehead C.; Zhang E.; Kuffa P.; Yan C.; McConnell P.; Spessard C.; Banotai C.; Mueller W. T.; Delaney A.; Omer C.; Sebolt-Leopold J.; Dudley D. T.; Leung I. K.; Flamme C.; Warmus J.; Kaufman M.; Barrett S.; Tecle H.; Hasemann C. A. (2004) Structures of human MAP kinase kinase 1 (MEK1) and MEK2 describe novel noncompetitive kinase inhibition. Nat. Struct. Mol. Biol. 11, 1192–1197. [DOI] [PubMed] [Google Scholar]
- Mattingly R. R.; Kraniak J. M.; Dilworth J. T.; Mathieu P.; Bealmear B.; Nowak J. E.; Benjamins J. A.; Tainsky M. A.; Reiners J. J. Jr. (2006) The mitogen-activated protein kinase/extracellular signal-regulated kinase kinase inhibitor PD184352 (CI-1040) selectively induces apoptosis in malignant schwannoma cell lines. J. Pharmacol. Exp. Ther. 316, 456–465. [DOI] [PubMed] [Google Scholar]
- Huynh H.; Soo K. C.; Chow P. K.; Tran E. (2007) Targeted inhibition of the extracellular signal-regulated kinase kinase pathway with AZD6244 (ARRY-142886) in the treatment of hepatocellular carcinoma. Mol. Cancer Ther. 6, 138–146. [DOI] [PubMed] [Google Scholar]
- Dong Q.; Dougan D. R.; Gong X.; Halkowycz P.; Jin B.; Kanouni T.; O’Connell S. M.; Scorah N.; Shi L.; Wallace M. B.; Zhou F. (2011) Discovery of TAK-733, a potent and selective MEK allosteric site inhibitor for the treatment of cancer. Bioorg. Med. Chem. Lett. 21, 1315–1319. [DOI] [PubMed] [Google Scholar]
- Gilmartin A. G.; Bleam M. R.; Groy A.; Moss K. G.; Minthorn E. A.; Kulkarni S. G.; Rominger C. M.; Erskine S.; Fisher K. E.; Yang J.; Zappacosta F.; Annan R.; Sutton D.; Laquerre S. G. (2011) GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin. Cancer Res. 17, 989–1000. [DOI] [PubMed] [Google Scholar]
- Kim K.; Kong S. Y.; Fulciniti M.; Li X.; Song W.; Nahar S.; Burger P.; Rumizen M. J.; Podar K.; Chauhan D.; Hideshima T.; Munshi N. C.; Richardson P.; Clark A.; Ogden J.; Goutopoulos A.; Rastelli L.; Anderson K. C.; Tai Y. T. (2010) Blockade of the MEK/ERK signalling cascade by AS703026, a novel selective MEK1/2 inhibitor, induces pleiotropic anti-myeloma activity in vitro and in vivo. Br. J. Hamaetol. 149, 537–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Z.; Grutter C.; Rauh D. (2013) Strategies for the selective regulation of kinases with allosteric modulators: exploiting exclusive structural features. ACS Chem. Biol. 8, 58–70. [DOI] [PubMed] [Google Scholar]
- Zuccotto F.; Ardini E.; Casale E.; Angiolini M. (2010) Through the ″gatekeeper door″: exploiting the active kinase conformation. J. Med. Chem. 53, 2681–2694. [DOI] [PubMed] [Google Scholar]
- Tecle H.; Feru F.; Liu H.; Kuhn C.; Rennie G.; Morris M.; Shao J.; Cheng A. C.; Gikunju D.; Miret J.; Coli R.; Xi S. H.; Clugston S. L.; Low S.; Kazmirski S.; Ding Y. H.; Cao Q.; Johnson T. L.; Deshmukh G. D.; DiNitto J. P.; Wu J. C.; English J. M. (2009) The design, synthesis and potential utility of fluorescence probes that target DFG-out conformation of p38alpha for high throughput screening binding assay. Chem. Biol. Drug Des. 74, 547–559. [DOI] [PubMed] [Google Scholar]
- Norman R. A.; Toader D.; Ferguson A. D. (2012) Structural approaches to obtain kinase selectivity. Trends Pharmacol. Sci. 33, 273–278. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Crespo A.; Fernandez A. (2008) Turning promiscuous kinase inhibitors into safer drugs. Trends Biotechnol. 26, 295–301. [DOI] [PubMed] [Google Scholar]
- Fedorov O.; Sundstrom M.; Marsden B.; Knapp S. (2007) Insights for the development of specific kinase inhibitors by targeted structural genomics. Drug Discovery Today 12, 365–372. [DOI] [PubMed] [Google Scholar]
- Knight Z. A.; Shokat K. M. (2005) Features of selective kinase inhibitors. Chem. Biol. 12, 621–637. [DOI] [PubMed] [Google Scholar]
- Noble M. E.; Endicott J. A.; Johnson L. N. (2004) Protein kinase inhibitors: insights into drug design from structure. Science 303, 1800–1805. [DOI] [PubMed] [Google Scholar]
- Jura N.; Zhang X.; Endres N. F.; Seeliger M. A.; Schindler T.; Kuriyan J. (2011) Catalytic control in the EGF receptor and its connection to general kinase regulatory mechanisms. Mol. Cell 42, 9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kufareva I.; Abagyan R. (2008) Type-II kinase inhibitor docking, screening, and profiling using modified structures of active kinase states. J. Med. Chem. 51, 7921–7932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M.; Nishigaki N.; Washio Y.; Kano K.; Harris P. A.; Sato H.; Mori I.; West R. I.; Shibahara M.; Toyoda H.; Wang L.; Nolte R. T.; Veal J. M.; Cheung M. (2007) Discovery of novel benzimidazoles as potent inhibitors of TIE-2 and VEGFR-2 tyrosine kinase receptors. J. Med. Chem. 50, 4453–4470. [DOI] [PubMed] [Google Scholar]
- Hodous B. L.; Geuns-Meyer S. D.; Hughes P. E.; Albrecht B. K.; Bellon S.; Caenepeel S.; Cee V. J.; Chaffee S. C.; Emery M.; Fretland J.; Gallant P.; Gu Y.; Johnson R. E.; Kim J. L.; Long A. M.; Morrison M.; Olivieri P. R.; Patel V. F.; Polverino A.; Rose P.; Wang L.; Zhao H. (2007) Synthesis, structural analysis, and SAR studies of triazine derivatives as potent, selective Tie-2 inhibitors. Bioorg. Med. Chem. Lett. 17, 2886–2889. [DOI] [PubMed] [Google Scholar]
- Cai Z. W.; Wei D.; Schroeder G. M.; Cornelius L. A.; Kim K.; Chen X. T.; Schmidt R. J.; Williams D. K.; Tokarski J. S.; An Y.; Sack J. S.; Manne V.; Kamath A.; Zhang Y.; Marathe P.; Hunt J. T.; Lombardo L. J.; Fargnoli J.; Borzilleri R. M. (2008) Discovery of orally active pyrrolopyridine- and aminopyridine-based Met kinase inhibitors. Bioorg. Med. Chem. Lett. 18, 3224–3229. [DOI] [PubMed] [Google Scholar]
- Shan Y.; Seeliger M. A.; Eastwood M. P.; Frank F.; Xu H.; Jensen M. O.; Dror R. O.; Kuriyan J.; Shaw D. E. (2009) A conserved protonation-dependent switch controls drug binding in the Abl kinase. Proc. Natl. Acad. Sci. U.S.A. 106, 139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hari S. B.; Merritt E. A.; Maly D. J. (2013) Sequence determinants of a specific inactive protein kinase conformation. Chem. Biol. 20, 806–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwell S.; Adams J. M.; Badger J.; Buchanan M. D.; Feil I. K.; Froning K. J.; Gao X.; Hendle J.; Keegan K.; Leon B. C.; Muller-Dieckmann H. J.; Nienaber V. L.; Noland B. W.; Post K.; Rajashankar K. R.; Ramos A.; Russell M.; Burley S. K.; Buchanan S. G. (2004) A novel mode of Gleevec binding is revealed by the structure of spleen tyrosine kinase. J. Biol. Chem. 279, 55827–55832. [DOI] [PubMed] [Google Scholar]
- Zhang T.; Inesta-Vaquera F.; Niepel M.; Zhang J.; Ficarro S. B.; Machleidt T.; Xie T.; Marto J. A.; Kim N.; Sim T.; Laughlin J. D.; Park H.; LoGrasso P. V.; Patricelli M.; Nomanbhoy T. K.; Sorger P. K.; Alessi D. R.; Gray N. S. (2012) Discovery of potent and selective covalent inhibitors of JNK. Chem. Biol. 19, 140–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian M. A.; Biggs W. H. 3rd; Treiber D. K.; Atteridge C. E.; Azimioara M. D.; Benedetti M. G.; Carter T. A.; Ciceri P.; Edeen P. T.; Floyd M.; Ford J. M.; Galvin M.; Gerlach J. L.; Grotzfeld R. M.; Herrgard S.; Insko D. E.; Insko M. A.; Lai A. G.; Lelias J. M.; Mehta S. A.; Milanov Z. V.; Velasco A. M.; Wodicka L. M.; Patel H. K.; Zarrinkar P. P.; Lockhart D. J. (2005) A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 23, 329–336. [DOI] [PubMed] [Google Scholar]
- Davis M. I.; Hunt J. P.; Herrgard S.; Ciceri P.; Wodicka L. M.; Pallares G.; Hocker M.; Treiber D. K.; Zarrinkar P. P. (2011) Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 29, 1046–1051. [DOI] [PubMed] [Google Scholar]
- Badrinarayan P.; Srivani P.; Narahari Sastry G. (2011) Design of 1-arylsulfamido-2-alkylpiperazine derivatives as secreted PLA2 inhibitors. J. Mol. Model. 17, 817–831. [DOI] [PubMed] [Google Scholar]
- Kuglstatter A.; Ghate M.; Tsing S.; Villasenor A. G.; Shaw D.; Barnett J. W.; Browner M. F. (2010) X-ray crystal structure of JNK2 complexed with the p38alpha inhibitor BIRB796: insights into the rational design of DFG-out binding MAP kinase inhibitors. Bioorg. Med. Chem. Lett. 20, 5217–5220. [DOI] [PubMed] [Google Scholar]
- Xu M.; Yu L.; Wan B.; Yu L.; Huang Q. (2011) Predicting inactive conformations of protein kinases using active structures: conformational selection of type-II inhibitors. PLoS One 6, e22644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okram B.; Nagle A.; Adrian F. J.; Lee C.; Ren P.; Wang X.; Sim T.; Xie Y.; Wang X.; Xia G.; Spraggon G.; Warmuth M.; Liu Y.; Gray N. S. (2006) A general strategy for creating ″inactive-conformation″ abl inhibitors. Chem. Biol. 13, 779–786. [DOI] [PubMed] [Google Scholar]
- Goldstein D. M.; Gray N. S.; Zarrinkar P. P. (2008) High-throughput kinase profiling as a platform for drug discovery. Nat. Rev. Drug Discovery 7, 391–397. [DOI] [PubMed] [Google Scholar]
- Choi Y.; Syeda F.; Walker J. R.; Finerty P. J. Jr.; Cuerrier D.; Wojciechowski A.; Liu Q.; Dhe-Paganon S.; Gray N. S. (2009) Discovery and structural analysis of Eph receptor tyrosine kinase inhibitors. Bioorg. Med. Chem. Lett. 19, 4467–4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich J.; Hulme C.; Hurley L. H. (2010) The design, synthesis, and evaluation of 8 hybrid DFG-out allosteric kinase inhibitors: a structural analysis of the binding interactions of Gleevec, Nexavar, and BIRB-796. Bioorg. Med. Chem. Lett. 18, 5738–5748. [DOI] [PubMed] [Google Scholar]
- Erlanson D. A.; Hansen S. K. (2004) Making drugs on proteins: site-directed ligand discovery for fragment-based lead assembly. Curr. Opin. Chem. Biol. 8, 399–406. [DOI] [PubMed] [Google Scholar]
- Erlanson D. A.; Arndt J. W.; Cancilla M. T.; Cao K.; Elling R. A.; English N.; Friedman J.; Hansen S. K.; Hession C.; Joseph I.; Kumaravel G.; Lee W. C.; Lind K. E.; McDowell R. S.; Miatkowski K.; Nguyen C.; Nguyen T. B.; Park S.; Pathan N.; Penny D. M.; Romanowski M. J.; Scott D.; Silvian L.; Simmons R. L.; Tangonan B. T.; Yang W.; Sun L. (2011) Discovery of a potent and highly selective PDK1 inhibitor via fragment-based drug discovery. Bioorg. Med. Chem. Lett. 21, 3078–3083. [DOI] [PubMed] [Google Scholar]
- Simard J. R.; Grutter C.; Pawar V.; Aust B.; Wolf A.; Rabiller M.; Wulfert S.; Robubi A.; Kluter S.; Ottmann C.; Rauh D. (2009) High-throughput screening to identify inhibitors which stabilize inactive kinase conformations in p38alpha. J. Am. Chem. Soc. 131, 18478–18488. [DOI] [PubMed] [Google Scholar]
- Simard J. R.; Kluter S.; Grutter C.; Getlik M.; Rabiller M.; Rode H. B.; Rauh D. (2009) A new screening assay for allosteric inhibitors of cSrc. Nat. Chem. Biol. 5, 394–396. [DOI] [PubMed] [Google Scholar]
- Simard J. R.; Getlik M.; Grutter C.; Schneider R.; Wulfert S.; Rauh D. (2010) Fluorophore labeling of the glycine-rich loop as a method of identifying inhibitors that bind to active and inactive kinase conformations. J. Am. Chem. Soc. 132, 4152–4160. [DOI] [PubMed] [Google Scholar]
- Munoz L.; Selig R.; Yeung Y. T.; Peifer C.; Hauser D.; Laufer S. (2010) Fluorescence polarization binding assay to develop inhibitors of inactive p38alpha mitogen-activated protein kinase. Anal. Biochem. 401, 125–133. [DOI] [PubMed] [Google Scholar]
- Lebakken C. S.; Riddle S. M.; Singh U.; Frazee W. J.; Eliason H. C.; Gao Y.; Reichling L. J.; Marks B. D.; Vogel K. W. (2009) Development and applications of a broad-coverage, TR-FRET-based kinase binding assay platform. J. Biomol. Screening 14, 924–935. [DOI] [PubMed] [Google Scholar]
- Uitdehaag J. C.; Zaman G. J. (2011) A theoretical entropy score as a single value to express inhibitor selectivity. BMC Bioinformatics 12, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulikakos P. I.; Zhang C.; Bollag G.; Shokat K. M.; Rosen N. (2010) RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 464, 427–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzivassiliou G.; Song K.; Yen I.; Brandhuber B. J.; Anderson D. J.; Alvarado R.; Ludlam M. J.; Stokoe D.; Gloor S. L.; Vigers G.; Morales T.; Aliagas I.; Liu B.; Sideris S.; Hoeflich K. P.; Jaiswal B. S.; Seshagiri S.; Koeppen H.; Belvin M.; Friedman L. S.; Malek S. (2010) RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 464, 431–435. [DOI] [PubMed] [Google Scholar]
- Murphy E. A.; Shields D. J.; Stoletov K.; Dneprovskaia E.; McElroy M.; Greenberg J. I.; Lindquist J.; Acevedo L. M.; Anand S.; Majeti B. K.; Tsigelny I.; Saldanha A.; Walsh B.; Hoffman R. M.; Bouvet M.; Klemke R. L.; Vogt P. K.; Arnold L.; Wrasidlo W.; Cheresh D. A. (2010) Disruption of angiogenesis and tumor growth with an orally active drug that stabilizes the inactive state of PDGFRbeta/B-RAF. Proc. Natl. Acad. Sci. U.S.A. 107, 4299–4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mol C. D.; Dougan D. R.; Schneider T. R.; Skene R. J.; Kraus M. L.; Scheibe D. N.; Snell G. P.; Zou H.; Sang B. C.; Wilson K. P. (2004) Structural basis for the autoinhibition and STI-571 inhibition of c-Kit tyrosine kinase. J. Biol. Chem. 279, 31655–31663. [DOI] [PubMed] [Google Scholar]
- Wang X.; Kim J. (2012) Conformation-specific effects of Raf kinase inhibitors. J. Med. Chem. 55, 7332–7341. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.