

Abstract
Antibiotics targeting DNA gyrase have been a clinical success story for the past half-century, and the emergence of bacterial resistance has fueled the search for new gyrase inhibitors. In this paper we demonstrate that a new class of gyrase inhibitors, the gyramides, are bacteriostatic agents that competitively inhibit the ATPase activity of Escherichia coli gyrase and produce supercoiled DNA in vivo. E. coli cells treated with gyramide A have abnormally localized, condensed chromosomes that blocks DNA replication and interrupts chromosome segregation. The resulting alterations in DNA topology inhibit cell division through a mechanism that involves the SOS pathway. Importantly, gyramide A is a specific inhibitor of gyrase and does not inhibit the closely related E. coli enzyme topoisomerase IV. E. coli mutants with reduced susceptibility to gyramide A do not display cross-resistance to ciprofloxacin and novobiocin. The results demonstrate that the gyramides prevent bacterial growth by a mechanism in which the topological state of chromosomes is altered and halts DNA replication and segregation. The specificity and activity of the gyramides for inhibiting gyrase makes these compounds important chemical tools for studying the mechanism of gyrase and the connection between DNA topology and bacterial cell division.
Many bacterial processes require the local unwinding of duplex DNA to replicate and transcribe genetic information. To resolve subsequent DNA supercoils, knots, and recombination intermediates, bacteria have evolved a class of enzymes referred to as topoisomerases that relieve the adverse effects of overwound and entangled chromosomes.1 Two classes of topoisomerases reverse supercoiling by creating breaks in one (type I) or both strands of duplex DNA (type II), manipulating the cleaved strand with respect to the other (intact) strand and attaching the strands of DNA together.2,3
DNA gyrase (“gyrase”) is among the most widely studied member of the type IIA topoisomerases. Gyrase is an A2B2 tetramer consisting of GyrA and GyrB subunits and uses the energy of ATP hydrolysis to introduce negative supercoils and relieve mechanical stress in positively supercoiled chromosomes. The mechanism of gyrase activity involves multiple conformational states in which DNA is bound and cleaved (i.e., the G-segment), another DNA strand (i.e., the T segment) is passed through the cleaved duplex, and the DNA ends are ligated together.3 Inhibiting gyrase using small molecules has been an important mechanism for treating infectious diseases.4
Fluoroquinolones (e.g., ciprofloxacin, 1 in Figure 1) are among the most potent families of small molecule gyrase inhibitors available and produce double-stranded DNA (dsDNA) breaks by stabilizing the gyrase-DNA cleavage complex.5,6 This family of gyrase inhibitors produces dsDNA breaks that trigger the SOS response and eventually lead to cell death.6 Quinolones and fluoroquinolones are not specific inhibitors of gyrase, and their promiscuity for binding other targets varies between bacterial species. For example, the primary target of quinolones in Staphylococcus aureus is topoisomerase IV (Topo IV), which is a member of the type II family of bacterial topoisomerases.7
Figure 1.

Chemical structures of DNA gyrase inhibitors: ciprofloxacin (1), novobiocin (2), and gyramide A (3).
Aminocoumarins are another class of gyrase inhibitors that use a mechanism that is distinct from the quinolones. The aminocoumarins are competitive inhibitors of ATP hydrolysis and inhibit DNA supercoiling activity.8 As with the quinolones and fluoroquinolones, the aminocoumarins target both gyrase and Topo IV. Limited activity against Gram-negative bacteria and issues with mammalian cytotoxicity have resulted in the approval of only one member of this family, novobiocin (2, Figure 1) for the treatment for human infections.8 The inhibition of human type IIa topoisomerases by quinolone, fluoroquinolone, and aminocoumarin antibiotics has motivated the repurposing of these drugs as anticancer therapeutics.9 Despite widespread clinical success, quinolones and aminocoumarins suffer from rapid and widespread acquisition of resistance among clinical isolates.5 For example, the emergence of resistance to gyrase inhibitors has been recently reported in clinical strains of Streptococcus pneumococci and Haemophilus influenza involved in upper respiratory tract infections.10,11
Small molecule inhibitors of type II topoisomerase have been used as chemical biological tools to provide preliminary insight into the physiological activity of gyrase and Topo IV in Escherichia coli cells. For example, the role of gyrase in replication was first identified by the characterization of mutations in gyrA and gyrB that conferred bacterial resistance to fluoroquinolones and aminocoumarins.12,13 The role of gyrase and Topo IV in maintaining the superhelical density of DNA, replication initiation, and chromosome segregation has been studied using these small molecules.14−16 Compounds 1 and 2 have broad application as antibiotics, yet significant limitations as chemical biological probes due to their promiscuity among type II topoisomerases.17 This limitation makes it difficult to unwind downstream physiological changes in bacteria that arise from inhibiting supercoiling of gyrase or Topo IV.
Several new gyrase inhibitors have been reported recently. GSK299423 is an inhibitor of Topo IIA, produces single-stranded DNA breaks, and circumvents fluoroquinolone resistance in S. aureus.18,19 NXL101 is a gyrase inhibitor that is active against fluoroquinolone-resistant bacteria and has progressed to human clinical trials.18,20 Two natural products, amycolamicin21,22 and kibdelomycin,23 have been identified as inhibitors of the ATPase activity of Topo II. A new class of gyrase inhibitors, the gyramides (3, Figure 1), block bacterial division through an inhibitory mechanism that is distinct from that of 1.24,25 Compound 3 inhibits the supercoiling activity of gyrase in vitro and filaments E. coli cells.24 Although the cellular phenotype of 1 and 3 are similar, 3 does not stabilize the formation of dsDNA breaks in vitro,24 which differentiates it from the biological mechanism of the quinolones.
In this manuscript, we characterize the mechanism by which 3 inhibits gyrase, interrupts cell division, and terminates cell growth. We demonstrate that 3 inhibits gyrase and does not inhibit Topo IV. Compound 3 inhibits the ATPase activity of gyrase, traps the chromosome in a supercoiled state, halts DNA replication, and affects chromosome segregation in E. coli. Compound 3 represents a unique chemical tool for unraveling the relationship between gyrase activity, chromosome topology, and bacterial cell division. Long-term studies of this compound may lead to potent broad-spectrum therapeutic agents for treating infections of clinical strains resistant to 1 and 2.
Results and Discussion
Compound 3 Inhibits DNA Supercoiling and Competitively Inhibits ATPase Activity
We previously demonstrated that 3 inhibits the DNA supercoiling activity of recombinant gyrase in vitro.24 To determine whether gyramides inhibit gyrase in vivo, we treated wild-type (WT) E. coli transformed with pUC19 plasmid DNA with 3 and measured the supercoiled state of pUC19. WT E. coli cells are less susceptible to 3 due to the presence of drug pumps that cause drug efflux.24 Previous research has shown that polar effects on gene expression arising due to tolC gene deletions can alter the membrane structure and osmotic barrier of E. coli and impact plasmid supercoiling.26 Therefore, we chose to use Phe-Arg-β-naphthylamine (PAβN), a small molecule inhibitor of the drug efflux pump TolC, to increase the susceptibility of WT E. coli to 3. We treated E. coli with 3, harvested plasmid DNA at different time points, and separated pUC19 DNA by gel electrophoresis (Figure 2a). The supercoiled state of pUC19 in E. coli cells treated with 3 was reduced compared to untreated cells (DMSO control). DMSO control cells and cells treated with PAβN contained pUC19 that was completely supercoiled and catenated. Treatment of cells with 3 produced supercoiled pUC19 in various topological states. Compound 3 inhibited DNA gyrase in vivo and altered the state of DNA supercoiling. These results are consistent with our in vitro experiments using recombinant gyrase.24
Figure 2.
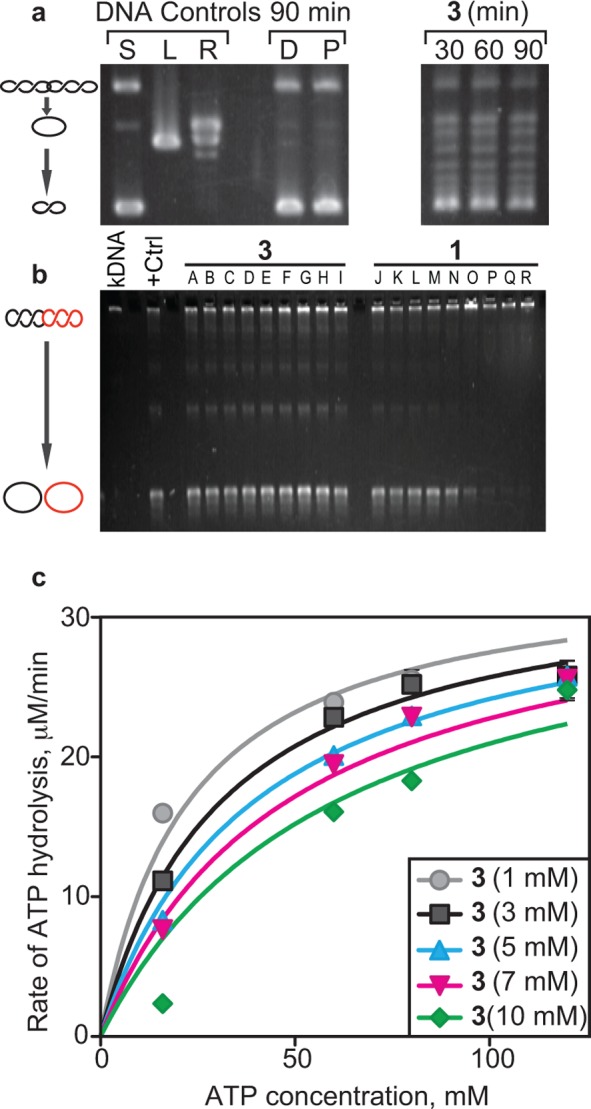
Enzyme activity of DNA gyrase in the presence of 3. (a) In vivo DNA supercoiling activity when treated with 3 for the indicated times. Treatment with 3 reduced the amount of negative supercoiling compared to solvent (D = DMSO) and TolC inhibitor (P = PAβN) controls. (b) Topo IV activity assay using kDNA substrates at different concentrations of 3 and 1. +Ctrl indicates the reaction without any inhibitors. (A) Solvent control (DMSO), (B) 0.078 μM, (C) 0.313 μM, (D) 1.25 μM, (E) 5 μM, (F) 20 μM, (G) 80 μM, (H) 160 μM, and (I) 320 μM 3. (J) Solvent control (50:50 [v/v] of 0.1 M HCl/MeOH), (K) 0.019 μM, (L) 0.078 μM, (M) 0.313 μM, (N) 1.25 μM, (O) 5 μM, (P) 20 μM, (Q) 80 μM, (R) 160 μM 1. (c) In vitro DNA-dependent ATP hydrolysis in the presence of different concentrations of 3. Km, Ki, and Vmax were calculated from the Michaelis–Menten plot shown. ATP assays were performed in triplicate, and error bars indicate standard deviation from the mean. Km varies with increasing concentration of 3; however, Vmax remains unchanged, which is indicative of competitive inhibition.
To determine the specificity of 3 against gyrase, we tested its activity against recombinant E. coli Topo IV in vitro using catenated plasmids referred to as kinetoplast DNA (kDNA) as substrates. We initiated the reaction by adding Topo IV to solutions of 3 and 1 or quenched the reaction after 30 min. Compound 1 inhibited the decatenation of DNA by Topo IV; however, 3 did not (Figure 2b). This result demonstrates that the inhibitory mechanism of 3 is specific for gyrase and not other members of the topo II family. The specificity of gyrase inhibition led us to explore the mechanism by which 3 inhibits the enzyme. Spontaneous point mutations in gyrase identified previously24 that conferred reduced susceptibility to 3 did not overlap with known mutations that conveyed resistance to 1 and 2, suggesting that 3 may bind gyrase and inhibit it through a mechanism that differs from the fluoroquinolones and aminocoumarins.
Gyrase is an ATPase that couples the hydrolysis of ATP to the relative motion of two DNA strands during the introduction of negative supercoiling. Inhibiting the ATPase activity of gyrase blocks the introduction of negative supercoils in DNA and traps the chromosome in a positively supercoiled state that may have a downstream impact on cell physiology and division.14 We determined whether 3 is an inhibitor of the ATP hydrolysis activity (i.e., akin to 2) of recombinant E. coli gyrase by continuously measuring the reduction of NADH using a coupled enzyme system consisting of pyruvate kinase and lactate dehydrogenase (Figure 2c). We found that 3 is a competitive inhibitor of ATP hydrolysis. Using a steady state approximation, we determined that Vmax and Km were 37.47 ± 0.0013 μM min–1 U–1 and 27.82 ± 2.83 mM, respectively (Figure 2c). The inhibition constant, Ki, of 3 is 4.35 ± 1.34 mM and Ki ≪ Km, thereby indicating competitive inhibition of the ATPase activity of gyrase by 3.
DNA gyrase is found preceding replication forks in DNA that are initiated by helicase and are utilized by DNA polymerase III. As helicase separates the DNA double helix to create a replication fork, the fixed position of the DNA ends cause dsDNA preceding the fork to become positively supercoiled and strained. Gyrase is positioned in front of the replication fork to reduce positive supercoiling and release superhelical tension.1 Our observation of E. coli cells filamenting in the presence of 3 and its inhibition of the ATPase activity of gyrase led us to hypothesize that the altered DNA topology activates the SOS response, produced through a noncanonical mechanism in which DNA is not damaged, or inhibits transcription of the genome.
Compound 3 Induces E. coli Cell Filamentation through the SOS Response
We observed that the chromosome in E. coli cells treated with 1 was diffusely localized, while cells treated with 3 displayed a compact chromosome (Figure 3). Untreated cells contained DNA that was diffuse and regularly spaced on either side of the division plane, while 3 altered the shape and location of the chromosome. When treated with 3, cells of mutants with reduced susceptibility to 3(24) had chromosomes that were similar in appearance to those of untreated cells (i.e., shape, length, chromosome position, and chromosome volume) (Supplementary Figure S1). dsDNA breaks in E. coli chromosomes produced by 1 locally unwind the double helix, release strain arising from supercoiling, and produce diffusely positioned nucleoids.27 Filamentation of E. coli cells by 1 has been attributed to the activation of the SOS response and the downstream production of the cell division inhibitor, SulA, that binds stochiometrically to the division protein FtsZ.28
Figure 3.

Chromosome organization in E. coli BW25113 ΔtolC cells treated with 1 and 3. (a, c) Treatment of cells with both compounds produced filamentous cells. (b, d) DAPI labeled cells demonstrate that treatment with 3 produced compact chromosomes, whereas treatment with 1, known to induce dsDNA breaks, produced diffuse chromosomes. This experiment indicates differences in the mechanism of DNA gyrase inhibition by these two compounds.
Replication blocks have also been shown to inhibit cell division through a SulA-independent pathway that depends on derepression of LexA and subsequent transcriptional control of the ftsQAZ genes.29 To determine if cell filamentation in response to 3 is a result of the SOS response, we treated cells of E. coli strain MG1655 ΔsulA lexA (ind-) with 3 and quantified the lengths of cells. Cells of E. coli strain MG1655 ΔsulA lexA (ind-) treated with 3 were significantly shorter compared to wild type E. coli cells after exposure to 3 (Figure 4c–g), suggesting that E. coli cell filamentation in response to 3 involves the induction of the SOS response. As discussed previously, gyrase is responsible for relieving positive supercoils in front of active replication forks. We hypothesized that inhibition of the DNA-dependent ATPase activity of gyrase by 3 stalls replication forks by preventing the release of mechanical stress on the chromosome and induces the SOS response. Consequently, we expect that treating cells with 3 reduces the amount of DNA in cells, stalls DNA replication, and interrupts chromosome segregation.
Figure 4.
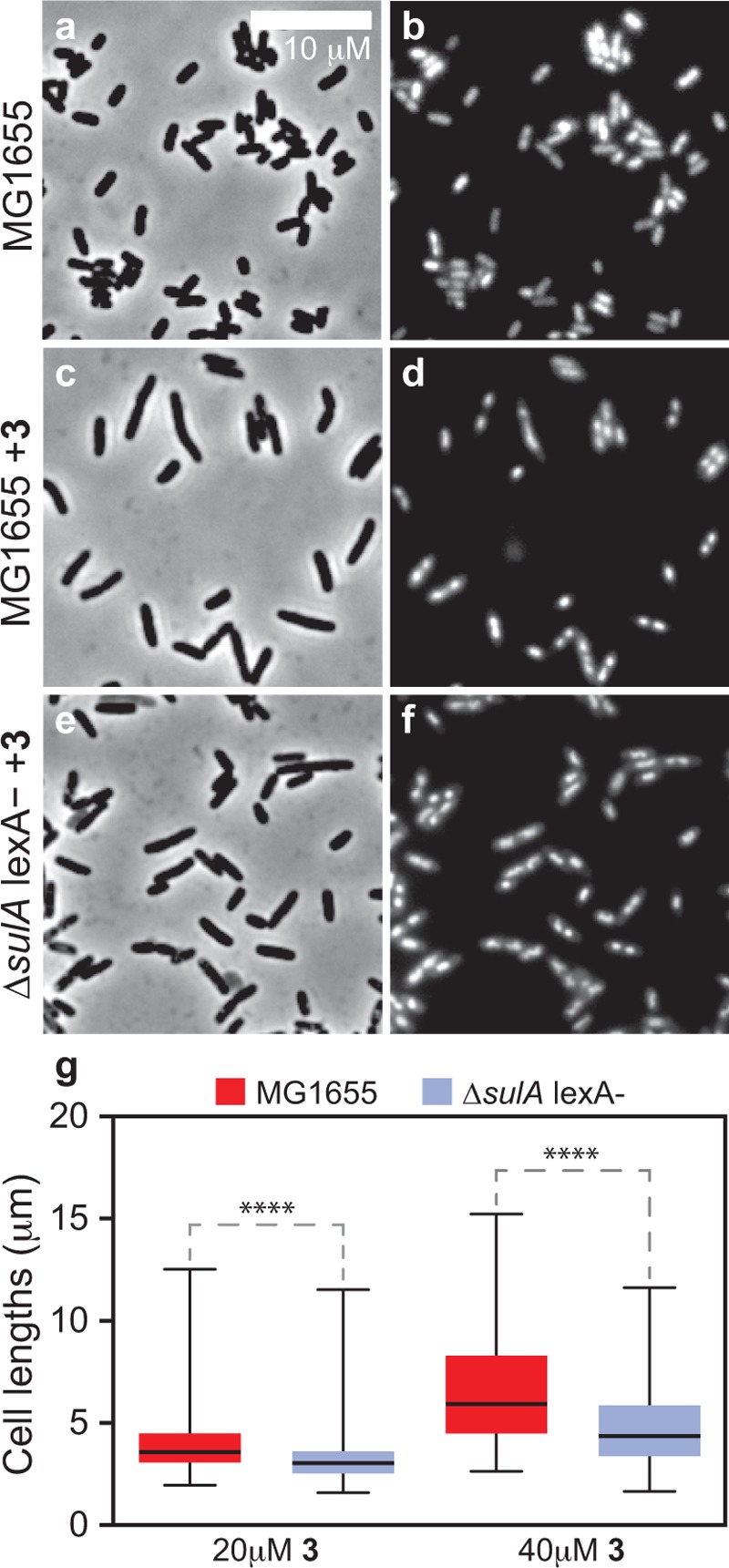
SulA-dependent inhibition of cell division occurs after treating cells with 3. (a, b) Images displaying the phenotype of untreated WT MG1655 E. coli cells. (c, d) Images demonstrating that WT MG1655 E. coli cells treated with 20 μM 3 for 2 h become filamented. (e, f) Images displaying cells of E. coli MG1655 ΔsulAlexA(ind-) after treatment with 20 μM 3 for 2 h. These cells have a phenotype similar to that of untreated WT cells. (g) Plot quantifying cell lengths after treating E. coli MG1655 WT and ΔsulAlexA(ind-) cells with different concentrations of 3. Mutant cells have a larger distribution of short cells (p-value < 0.0001) when compared to WT, which indicates that cell filamentation to 3 is a result of the SOS response. Error bars represent standard deviations of the mean of 250–300 cells; **** indicates a statistical significance of <0.0001.
Compound 3 Affects Chromosome Number and Segregation in E. coli Strain ΔtolC
We studied the replication of E. coli chromosomes to determine whether replication progression and/or chromosome segregation are affected by inhibiting the ATPase activity of gyrase using 3. A fluorescence-based assay for quantifying the DNA content of cells has been described previously using flow cytometry and the fluorescent DNA probe Picogreen.30 As a control, we filamented E. coli cells using cephalexin to a length that was comparable to cells treated with 3 and quantified the DNA content of these cells using the flow-cytometry-based fluorescence assay. The control provided a baseline of DNA content against which to compare cells treated with 3. We quantified the number of chromosomes and their segregation in cells under normal and replication “run out” conditions.30 Treating cells with both cephalexin and rifampin arrests cell division and protein synthesis and creates a condition referred to as replication “run out”. This set of experimental conditions enabled us to accurately determine the number of chromosomes in cells in which new DNA replication rounds are halted and previously replicated chromosomes have finished segregating.
Cells treated with 3 were divided into populations of normal length and filamentous cells by gating the flow cytometry data; in these experiments we used forward scattering to approximate cell length. Normal and filamentous cell populations were compared with untreated cells or cells treated with cephalexin to resemble the length of filamentous cells, respectively. We observed irregular chromosome segregation in cells of both lengths treated with 3, which was characterized by poorly defined peaks in the flow cytometry data (Figure 5a,b,d) compared to untreated cells. After 2 h of incubation, we washed 3 out of cells and observed the cells recover and produce well-defined fluorescence profiles resembling those of the untreated cells, indicating that chromosome segregation had resumed (Figure 5c,e). An increase in the height of the peak corresponding to no fluorescence intensity in these cells (indicated by arrows) is characteristic of cells lacking chromosomes and may have arisen due to chromosomal replication or segregation defects.
Figure 5.
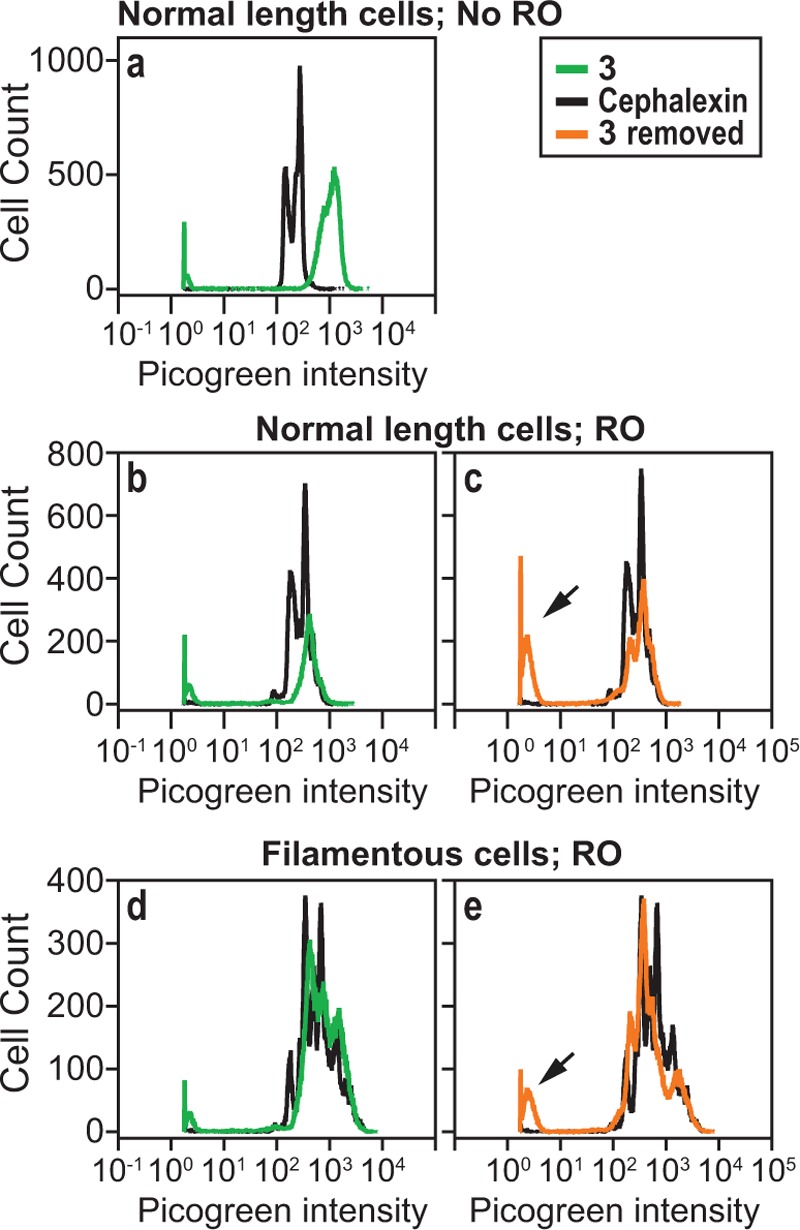
Analysis of DNA content in E. coli ΔtolC after treatment with 3. (a) A comparison of fluorescence intensity (FI) of Picogreen (x-axis) against total number of cells (y-axis) after treatment with 3 (green) and cephalexin (black). “Normal length” refers to cells that have not filamented after treatment with 1 or 3. (b) A comparison of the Picogreen FI of normal length cells treated with 3 (green) and cephalexin (black) with “run-out” (RO) conditions. (c) Picogreen FI of normal length cells in which 3 was washed away after treatment for 2 h, followed by growing cells under RO conditions for 4 h. This data is overlaid against the picogreen FI obtained for cephalexin (black). Picogreen FI of filamented cells (d) after treatment with 3 and RO conditions and (e) in which 3 was washed away after 2 h of treatment and cells were then grown for 4 h under RO conditions. Arrows in panels c and e indicate an increase in the height of the peak corresponding to very low fluorescence intensity, which is characteristic of anucleate cells.
Gyramide Mutants Are Not Cross Resistant to Ciprofloxacin and Novobiocin
We measured the minimum bacterial concentration (MBC) of E. coli against 3 and determined the ratio between MBC and minimum inhibitory concentration (MIC) to be greater than 4, indicating the compound is a bacteriostatic agent. Using five mutants that developed spontaneous resistance to 3 (3R mutants),24 we determined the MIC of compounds 1, 2, and 3 to the 3R mutants (Supplementary Table S1). The MIC of 3 against all five mutants was >41 μg mL–1, which is at least an order of magnitude higher than the MIC against the parent E. coli strain BW25113 ΔtolC (4.1 μg mL–1). To determine the cross-resistance of 3, we measured the MIC of 1 and 2 against the five 3R mutants. The MICs of 3R mutants treated with 2 were identical to the parent E. coli strain (MIC, 0.77 μg mL–1). Treatment of 3R mutants with 1 produced MIC values that were <2-fold higher than the MIC of the parent E. coli strain (MIC, 0.002 μg mL–1). As the MIC values for 1 and 2 against 3R mutants are identical to or very close to the MIC against the parent E. coli strains, there appears to be minimal cross resistance.
Conclusion
Treating E. coli with 3 causes cell filamentation and abnormally organized, compact chromosomes, which ultimately halts cell growth. We established that cell filamentation is a result of SulA-dependent SOS response and that the gyramides inhibit ATP hydrolysis and subsequently inhibit the supercoiling activity of gyrase. The inhibition of the supercoiling activity causes the chromosome to reach a superhelical state that blocks replication forks, induces the SOS response, and eventually interrupts chromosome segregation.
In vitro experiments with 3 demonstrate it targets gyrase specifically and does not affect the function of TopoIV. The fluoroquinolones bind to the GyrA subunit of gyrase and stabilize double-stranded DNA breaks, thereby creating a linear form of DNA. We demonstrated previously that treating gyrase with 3 did not produce linear DNA.24 Aminocoumarins bind to the GyrB subunit in the ATP binding domain and competitively inhibit ATP hydrolysis. We found that the majority of the E. coli spontaneous mutants that have a reduced susceptibility to 3 contain point mutations centered in the GyrA subunit; surprisingly, the gyramides competitively inhibit ATP hydrolysis, which occurs in the GyrB subunit. The specificity of 3 for inhibiting DNA gyrase and the distinct point mutations that confer resistance to 3 described previously24 suggest that 3 possesses a unique mechanism of action from 1 and 2.
Our data suggest that the gyramides represent an important new class of chemical tools for studying the connection between DNA topology and bacterial cell division. ATP hydrolysis is coupled to the multiple conformational states of gyrase; however, the mechanisms by which ATPase activity influences the conformation of the protein gates are still not entirely understood.31,32 As mutations in 3R mutants are not located adjacent to the ATP binding domain, suggesting an allosteric mechanism of inhibition, the gyramides are a unique family of competitive gyrase ATPase inhibitors. Many of the 3R mutations are close to the DNA binding site in the A subunit of gyrase and suggest that 3 may inhibit the coupling of ATP hydrolysis to DNA binding by gyrase. Resistant mutants may help determine which ATP hydrolysis events are required for DNA binding. The application of 3 and structural analogs in combination with single molecule biophysical experiments may be useful in studying these mechanisms.32,33 Further studies will determine which ATP hydrolysis events are impaired by the gyramides. As 1 and 2 both bind gyrase and Topo IV, which is reported to make them particularly effective antibiotics, these compounds have limited utility for these types of chemical biology studies. Compound 3 and its structural analogs are not yet potent antibiotics; however, ongoing structural biology and structure–activity relationship studies will improve the potency and bioavailability of these inhibitors as chemical biological probes for studying bacteria and as therapeutic antimicrobial agents.
Methods
General Microscopy Sample Preparation
Overnight cultures of E. coli cells were diluted 1:100 in fresh LB media. The inoculum was incubated at 37 °C with shaking at 200 rpm until an absorbance of ∼0.1 (λ = 600 nm). After incubation, the appropriate concentration of 3 with or without PAβN-HCl (Sigma-Aldrich) was added to the inoculum. The treated cultures were incubated at 37 °C while shaking at 200 rpm for 2–2.5 h. Cultures were treated with equal volumes of DAPI (10 μg mL–1, Sigma-Aldrich), and 5 μL of labeled culture was transferred to the surface of a 1.5% (w/v) agarose pad. We imaged cells using both phase contrast and epifluorescence using a Nikon TE2000-E inverted microscope equipped with an Andor iXon EMCCD. Samples were illuminated with a 120 W mercury arc lamp (X-cite Series 120, EXFO), and images were acquired using a 100X oil objective and an additional 1.5X magnification (at the microscope body), a 500/20x excitation filter, and a 535/30 nm emission filter. All data was collected with the shutter off and a 100 ms exposure time controlled by the Metamorph software package (version 7.5.6.0 MDS Analytical Technologies).
Quantification of Cell Length Using MATLAB-Based MicrobeTracker
To quantify E. coli cell lengths, we used the MATLAB-based image analysis software MicrobeTracker.34 MicrobeTracker uses an edge detection algorithm and spline fitting to determine cell lengths. We performed a two-tailed t test of cell lengths in Graphpad Prism and report a significance of p < 0.0001 (****).
Characterization of the Supercoiling Activity of Gyrase in Vivo
E. coli strain BW25113 pUC19 was grown to saturation in the presence of 50 μg mL–1 ampicillin (Sigma-Aldrich). The cells were pelleted in a Beckman Coulter GH-3.8 rotor at 1500 × g for 10 min, resuspended in fresh LB to an absorbance of 2.0 (λ = 600 nm), and treated with 3. Control experiments were performed by incubating cultures for 90 min at 37 °C in media containing (1) DMSO (0.4%) or (2) PAβN (60 μM) as the adjuvant for 3. For determining supercoiling states of pUC19 DNA after treatment, 3 (40 μM) and PAβN (60 μM) were added, and the cultures were grown at 37 °C with 200 rpm shaking. This cocktail of small molecules inhibits >99% of growth of E. coli strain BW25113. Cells treated with 3 were harvested after incubating for 30, 60, or 90 min, and pUC19 was extracted using a Qiagen QIAprep spin miniprep kit (Qiagen); 100 ng of supercoiled, linearized, and relaxed pUC19 DNA was used as controls, and 175 ng of pUC19 extracted from treated cultures. The topological states of plasmid pUC19 extracted from E. coli BW25113 was separated by electrophoresis in 0.8% (w/v) agarose with 1X TBE buffer at 65 V for 3 h.
Characterization of the Decatenation Activity of Topo IV in Vitro
In vitro E. coli Topo IV relaxation activity assays were performed by incubating 1 unit of Topo IV (TopoGEN) with 170 ng of kDNA (TopoGEN) in 30 μL of reaction buffer. The reaction contained 50 mM HEPES-KOH (pH 8.0), 100 mM potassium glutamate, 10 mM magnesium acetate, 10 mM DTT, 50 μg mL–1 BSA, and 20 mM ATP. All reactions contained a final concentration of 3.3% (v/v) DMSO with different concentrations of 3 or 3.3% of 50:50 0.1 HCl: MeOH with different concentrations of 1. The reactions were incubated at 37 °C for 30 min and quenched by adding 6 μL of 15% (w/v) Ficoll, 4% (w/v) SDS with 0.24% (w/v) bromophenol blue to a final concentration of 0.5% (w/v) Ficoll, 0.13% (w/v) SDS. The DNA reaction mixtures were separated on a 0.8% (w/v) agarose gel with 1X TBE at a constant voltage at 75 V for 2.5 h. We stained gels with ethidium bromide and imaged them using a Computar H6Z0812 camera with FOTO/Analyst PC Image software.
Measuring the Gyrase ATPase Inhibitory Activity of 3
We used a spectrophotometric-coupled enzyme assay to measure the DNA-dependent ATPase activity of recombinant E. coli DNA gyrase (New England Biolabs) in the presence of increasing concentrations of 3. We coupled the regeneration of ATP from phosphoenolpyruvate (PEP) and ADP to the oxidation of NADH and measured the decrease in the absorbance of light at 37 °C using a TECAN M1000 (λem = 340 nm). Using the NADH extinction coefficient (6220 M–1 cm–1 at λ = 340 nm), we converted the oxidation of NADH to the hydrolysis of ATP. Reaction mixtures (80 μL) contained 35 mM Tris-HCl, 24 mM KCl, 4 mM MgCl2, 2 mM DTT, 5 mM spermidine, and 6.5% (v/v) glycerol at pH 7.5. We added 3.5 mM PEP (Alfa Aesar), pyruvate kinase (PK; 10 U) (MP Biomedicals), lactate dehydrogenase (LDH; 10 U) (MP Biomedicals), 1.5 mM NADH (Acros organics), and 1 U of DNA gyrase. ATP (Fisher Scientific) was added to start the reaction. For determining the Km and Vmax, we performed assays at different concentrations of ATP using 1 U of DNA gyrase. This experiment was repeated with 5 different concentrations of 3, and the Ki was determined by plotting ATP concentration on the x-axis against initial velocity on the y-axis. Equations for competitive, uncompetitive, and noncompetitive inhibition were fit to this plot using GraphPad Prism, and the inhibition model was determined by analyzing statistics using an F test.
Quantification of Chromosome Number Using Flow Cytometry
Overnight cultures of E. coli cells were diluted 1:100 in fresh LB media and incubated at 37 °C with shaking at 200 rpm until an absorbance (λ, 600 nm) of ∼0.1. Compound 3 (20 μM, 2X MIC) was added to 1 mL of a suspension of E. coli BW25113 ΔtolC cells that had an absorbance of ∼0.1 (λ, 600 nm) and incubated at 37 °C for 2 h. In a parallel experiment, cephalexin (3.75 μg mL–1, 1/2X MIC) (Sigma-Aldrich) was added to 1 mL of a culture of E. coli BW25113 ΔtolC cells that had an absorbance of ∼0.1 (λ = 600 nm) and incubated at 37 °C for 30 min. The cultures were centrifuged for 10 min at 2500 rpm. Cultures that had previously been treated with 3 were resuspended in LB media containing 20 μM 3, 5 μg mL–1 of rifampin (Fisher Scientific), and 0.5 μg mL–1 of cephalexin (Sigma-Aldrich) and incubated at 37 °C with shaking at 200 rpm for 4 h. We also determined if the phenotypes observed for 3 were reversible. For these experiments we removed 3 after treatment for 2 h by resuspending the pellets in fresh LB containing only rifampin and cephalexin at the above concentrations. After 4 h of incubation, the cultures were centrifuged for 10 min at 4 °C and 2500 rpm, and the pellet was resuspended in 1.0 mL of 1X PBS. Then 100 μL of the cell suspension was aliquoted into 900 μL of 70% (v/v) ethanol, mixed thoroughly, and incubated overnight at 4 °C. The mixture was centrifuged at 6000 rpm for 10 min at 25 °C, the ethanol was decanted, and the material was incubated for 30 min to evaporate residual ethanol. The cell pellet was resuspended in 100 μL of 1X PBS containing 20 μL of PicoGreen (Invitrogen) and incubated at 25 °C in the dark for at least 30 min. The samples were diluted with 880 μL of 1X PBS and analyzed using a Becton Dickinson FACSCalibur flow cytometer with a 488 nm wavelength argon laser. The fluorescence intensity of Picogreen was recorded using the FL1 channel at an emission of 530/30 nm, and the mode of acquisition was logarithmic to accommodate high fluorophore intensities corresponding to longer cells. We collect 50,000 events. Data were analyzed using FlowJo 9.3.3 (Tree Star, Inc.), and each sample was plotted as a histogram of the number of cells against the green fluorescence intensity (FL1-H).
Calculation of the Minimum Bacterial Concentration (MBC) of 3
The antibacterial mode (i.e., bactericidal, bacteriostatic) of 3 was determined by calculating the quotient of the minimum bacterial concentration (MBC) and the minimum inhibitory concentration (MIC). If the quotient is greater than 4, the compound is bacteriostatic; if the quotient is lower than 4, the compound is bactericidal.35 We used a microdilution protocol to perform MIC and MBC experiments according to the NCCLS guidelines.36 To make a 2-fold dilution series for the determination of the MIC using the microdilution technique, we added 3 to the first well of 96-well plates to give the highest concentration and subsequently diluted it in a set of wells containing inoculated media. The final volume for each well was 100 μL. We prepared solvent controls and sterility controls using the same concentration of solvent as the wells with the highest concentration of antibiotic. The MIC end points were determined in triplicate by identifying the lowest concentration of 3 that completely inhibited growth by visual inspection. The total volume in the wells with no visual growth was plated on LB agar and incubated for 22 h. The MBC was determined to be the lowest concentration of antibiotic that did not produce visible colonies.
Cross Resistance Analysis of 1, 2, and 3 against 3R Mutants
Single-step mutants of E. coli BW25113 ΔtolC (AcrAB-TolC pump knockout) resistant to 3 were isolated as described previously.24 The sensitivity of 3R mutants to 1 (Fluka from Sigma-Aldrich), 2 (Sigma-Aldrich), and 3 was determined. MIC assays with 1, 2, and 3 were performed as explained in the MBC calculation protocol, according to recommendations from the CLSI for aerobic growth of bacteria.363R bacteria was grown in LB media at 37 °C for 16 h with shaking at 200 rpm. The MIC was determined as the lowest concentration that displayed no apparent turbidity.
Acknowledgments
This research was supported by the National Institutes of Health (1DP2OD008735-01) and the Human Frontiers Science Program (RGY0076/2013). Research in the J. Shaw lab was supported by NIH/NIAID (R01AI08093). We also acknowledge the University of Wisconsin Carbone Cancer Center Cancer Center Support Grant P30 CA014520 for flow cytometry instrumentation. M. Rajendram acknowledges a fellowship from the Wisconsin Alumni Research Foundation. K. Hurley acknowledges a fellowship from the American Foundation of Pharmaceutical Education. D. Weibel acknowledges a fellowship from the Alfred P. Sloan Foundation. We also acknowledge Y. Eun and N. Sorto for helpful insight and contribution.
Supporting Information Available
This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
PDB IDs: 3NUH, 2XCS [abstract art].
Author Contributions
⊥ These authors contributed equally to this work.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Wang J. C. (2002) Cellular roles of DNA topoisomerases: a molecular perspective. Nat. Rev. Mol. Cell Biol. 3, 430–440. [DOI] [PubMed] [Google Scholar]
- Champoux J. J. (2001) DNA topoisomerases: structure, function, and mechanism. Annu. Rev. Biochem. 70, 369–413. [DOI] [PubMed] [Google Scholar]
- Schoeffler A. J.; Berger J. M. (2008) DNA topoisomerases: harnessing and constraining energy to govern chromosome topology. Q. Rev. Biophys. 41, 41–101. [DOI] [PubMed] [Google Scholar]
- Collin F.; Karkare S.; Maxwell A. (2011) Exploiting bacterial DNA gyrase as a drug target: current state and perspectives. Appl. Microbiol. Biotechnol. 92, 479–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrega A.; Madurga S.; Giralt E.; Vila J. (2009) Mechanism of action of and resistance to quinolones. Microb. Biotechnol. 2, 40–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drlica K.; Zhao X. (1997) DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol. Mol. Biol. Rev. 61, 377–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng E. Y.; Trucksis M.; Hooper D. C. (1996) Quinolone resistance mutations in topoisomerase IV: relationship to the flqA locus and genetic evidence that topoisomerase IV is the primary target and DNA gyrase is the secondary target of fluoroquinolones in Staphylococcus aureus. Antimicrob. Agents Chemother. 40, 1881–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderle C.; Stieger M.; Burrell M.; Reinelt S.; Maxwell A.; Page M.; Heide L. (2008) Biological activities of novel gyrase inhibitors of the aminocoumarin class. Antimicrob. Agents Chemother. 52, 1982–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sissi C.; Palumbo M. (2003) The quinolone family: from antibacterial to anticancer agents. Curr. Med. Chem. Anticancer Agents 3, 439–450. [DOI] [PubMed] [Google Scholar]
- Vila J.; Ruiz J.; Sanchez F.; Navarro F.; Mirelis B.; de Anta M. T.; Prats G. (1999) Increase in quinolone resistance in a Haemophilus influenzae strain isolated from a patient with recurrent respiratory infections treated with ofloxacin. Antimicrob. Agents Chemother. 43, 161–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karchmer A. W. (2004) Increased antibiotic resistance in respiratory tract pathogens: PROTEKT US—an update. Clin Infect Dis. 39(Suppl 3), S142–150. [DOI] [PubMed] [Google Scholar]
- Gellert M.; Mizuuchi K.; O’Dea M. H.; Itoh T.; Tomizawa J. I. (1977) Nalidixic acid resistance: a second genetic character involved in DNA gyrase activity. Proc. Natl. Acad. Sci. U.S.A. 74, 4772–4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugino A.; Peebles C. L.; Kreuzer K. N.; Cozzarelli N. R. (1977) Mechanism of action of nalidixic acid: purification of Escherichia coli nalA gene product and its relationship to DNA gyrase and a novel nicking-closing enzyme. Proc. Natl. Acad. Sci. U.S.A. 74, 4767–4771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodursky A. B.; Peter B. J.; Schmid M. B.; DeRisi J.; Botstein D.; Brown P. O.; Cozzarelli N. R. (2000) Analysis of topoisomerase function in bacterial replication fork movement: use of DNA microarrays. Proc. Natl. Acad. Sci. U.S.A. 97, 9419–9424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung K. J.; Badarinarayana V.; Selinger D. W.; Janse D.; Church G. M. (2003) A microarray-based antibiotic screen identifies a regulatory role for supercoiling in the osmotic stress response of Escherichia coli. Genome Res. 13, 206–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falconer S. B.; Czarny T. L.; Brown E. D. (2011) Antibiotics as probes of biological complexity. Nat. Chem. Biol. 7, 415–423. [DOI] [PubMed] [Google Scholar]
- Foss M. H.; Eun Y.-J.; Weibel D. B. (2011) Chemical-biological studies of subcellular organization in bacteria. Biochemistry. 50, 7719–7734. [DOI] [PubMed] [Google Scholar]
- Shapiro A. B.; Andrews B. (2012) Allosteric inhibition of the DNA-dependent ATPase activity of Escherichia coli DNA gyrase by a representative of a novel class of inhibitors. Biochem. Pharmacol. 84, 900–904. [DOI] [PubMed] [Google Scholar]
- Bax B. D.; Chan P. F.; Eggleston D. S.; Fosberry A.; Gentry D. R.; Gorrec F.; Giordano I.; Hann M. M.; Hennessy A.; Hibbs M.; Huang J.; Jones E.; Jones J.; Brown K. K.; Lewis C. J.; May E. W.; Saunders M. R.; Singh O.; Spitzfaden C. E.; Shen C.; Shillings A.; Theobald A. J.; Wohlkonig A.; Pearson N. D.; Gwynn M. N. (2010) Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature 466, 935–940. [DOI] [PubMed] [Google Scholar]
- Black M. T.; Stachyra T.; Platel D.; Girard A. M.; Claudon M.; Bruneau J. M.; Miossec C. (2008) Mechanism of action of the antibiotic NXL101, a novel nonfluoroquinolone inhibitor of bacterial type II topoisomerases. Antimicrob. Agents Chemother. 52, 3339–3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawa R.; Takahashi Y.; Hashizume H.; Sasaki K.; Ishizaki Y.; Umekita M.; Hatano M.; Abe H.; Watanabe T.; Kinoshita N.; Homma Y.; Hayashi C.; Inoue K.; Ohba S.; Masuda T.; Arakawa M.; Kobayashi Y.; Hamada M.; Igarashi M.; Adachi H.; Nishimura Y.; Akamatsu Y. (2012) Amycolamicin: a novel broad-spectrum antibiotic inhibiting bacterial topoisomerase. Chemistry 18, 15772–15781. [DOI] [PubMed] [Google Scholar]
- Tohyama S.; Takahashi Y.; Akamatsu Y. (2010) Biosynthesis of amycolamicin: the biosynthetic origin of a branched alpha-aminoethyl moiety in the unusual sugar amycolose. J. Antibiot. (Tokyo) 63, 147–149. [DOI] [PubMed] [Google Scholar]
- Phillips J. W.; Goetz M. A.; Smith S. K.; Zink D. L.; Polishook J.; Onishi R.; Salowe S.; Wiltsie J.; Allocco J.; Sigmund J.; Dorso K.; Lee S.; Skwish S.; de la Cruz M.; Martin J.; Vicente F.; Genilloud O.; Lu J.; Painter R. E.; Young K.; Overbye K.; Donald R. G.; Singh S. B. (2011) Discovery of kibdelomycin, a potent new class of bacterial type II topoisomerase inhibitor by chemical-genetic profiling in Staphylococcus aureus. Chem. Biol. 18, 955–965. [DOI] [PubMed] [Google Scholar]
- Foss M. H.; Hurley K. A.; Sorto N.; Lackner L. L.; Thornton K. M.; Shaw J. T.; Weibel D. B. (2011) N-Benzyl-3-sulfonamidopyrrolidines are a new class of bacterial DNA gyrase inhibitors. ACS Med. Chem. Lett. 2, 289–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer C.; Janin Y. L. (2013) Non-quinolone inhibitors of bacterial type IIA topoisomerases: A feat of bioisosterism. Chem. Rev. 114, 2313–2342. [DOI] [PubMed] [Google Scholar]
- Dorman C. J.; Lynch A. S.; Bhriain N. N.; Higgins C. F. (1989) DNA supercoiling in Escherichia coli: topA mutations can be suppressed by DNA amplifications involving the tolC locus. Mol. Microbiol. 3, 531–540. [DOI] [PubMed] [Google Scholar]
- Tamayo M.; Santiso R.; Gosalvez J.; Bou G.; Fernandez J. L. (2009) Rapid assessment of the effect of ciprofloxacin on chromosomal DNA from Escherichia coli using an in situ DNA fragmentation assay. BMC Microbiol. 9, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee A.; Cao C.; Lutkenhaus J. (1998) Inhibition of FtsZ polymerization by SulA, an inhibitor of septation in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 95, 2885–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill T. M.; Sharma B.; Valjavec-Gratian M.; Smith J. (1997) sfi-independent filamentation in Escherichia coli Is lexA dependent and requires DNA damage for induction. J. Bacteriol. 179, 1931–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferullo D. J.; Cooper D. L.; Moore H. R.; Lovett S. T. (2009) Cell cycle synchronization of Escherichia coli using the stringent response, with fluorescence labeling assays for DNA content and replication. Methods 48, 8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nollmann M.; Crisona N. J.; Arimondo P. B. (2007) Thirty years of Escherichia coli DNA gyrase: from in vivo function to single-molecule mechanism. Biochimie 89, 490–499. [DOI] [PubMed] [Google Scholar]
- Basu A.; Schoeffler A. J.; Berger J. M.; Bryant Z. (2012) ATP binding controls distinct structural transitions of Escherichia coli DNA gyrase in complex with DNA. Nat. Struct. Mol. Biol. 19(538–546), S531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nollmann M.; Stone M. D.; Bryant Z.; Gore J.; Crisona N. J.; Hong S. C.; Mitelheiser S.; Maxwell A.; Bustamante C.; Cozzarelli N. R. (2007) Multiple modes of Escherichia coli DNA gyrase activity revealed by force and torque. Nat. Struct. Mol. Biol. 14, 264–271. [DOI] [PubMed] [Google Scholar]
- Sliusarenko O.; Heinritz J.; Emonet T.; Jacobs-Wagner C. (2011) High-throughput, subpixel precision analysis of bacterial morphogenesis and intracellular spatio-temporal dynamics. Mol. Microbiol. 80, 612–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankey G. A.; Sabath L. D. (2004) Clinical relevance of bacteriostatic versus bactericidal mechanisms of action in the treatment of Gram-positive bacterial infections. Clin. Infect. Dis. 38, 864–870. [DOI] [PubMed] [Google Scholar]
- CLSI (2000) Methods for Dilution Antimicrobial Susceptiblility Tests for Bacteria That Grow Aerobically; Approved Standard - Fifth Edition, Clinical and Laboratory Standards Institute, Wayne, PA, Vol. 20. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.