

ABSTRACT
Clostridium difficile infection is one of the most common health care-associated infections, and up to 40% of patients suffer from recurrence of disease following standard antibiotic therapy. Recently, fecal microbiota transplantation (FMT) has been successfully used to treat recurrent C. difficile infection. It is hypothesized that FMT aids in recovery of a microbiota capable of colonization resistance to C. difficile. However, it is not fully understood how this occurs. Here we investigated changes in the fecal microbiota structure following FMT in patients with recurrent C. difficile infection, and imputed a hypothetical functional profile based on the 16S rRNA profile using a predictive metagenomic tool. Increased relative abundance of Bacteroidetes and decreased abundance of Proteobacteria were observed following FMT. The fecal microbiota of recipients following transplantation was more diverse and more similar to the donor profile than the microbiota prior to transplantation. Additionally, we observed differences in the imputed metagenomic profile. In particular, amino acid transport systems were overrepresented in samples collected prior to transplantation. These results suggest that functional changes accompany microbial structural changes following this therapy. Further identification of the specific community members and functions that promote colonization resistance may aid in the development of improved treatment methods for C. difficile infection.
IMPORTANCE
Within the last decade, Clostridium difficile infection has surpassed other bacterial infections to become the leading cause of nosocomial infections. Antibiotic use, which disrupts the gut microbiota and its capability in providing colonization resistance against C. difficile, is a known risk factor in C. difficile infection. In particular, recurrent C. difficile remains difficult to treat with standard antibiotic therapy. Fecal microbiota transplantation (FMT) has provided a successful treatment method for some patients with recurrent C. difficile infection, but its mechanism and long-term effects remain unknown. Our results provide insight into the structural and potential metabolic changes that occur following FMT, which may aid in the development of new treatment methods for C. difficile infection.
INTRODUCTION
Clostridium difficile infection (CDI) is the leading cause of health care-associated diarrhea, and rates of CDI have significantly increased during the past decade (1). It is estimated that there are more than 500,000 cases of CDI in the United States annually, and estimates of the financial burden attributed to CDI range from $1.3 billion to $3.4 billion (2, 3). The manifestations of CDI can range from cramps, diarrhea, and colitis potentially to more severe outcomes, including toxic megacolon and death (1). Of significant concern is the rate of recurrence following an initial CDI episode; 20 to 40% of patients do not resolve their infection following standard CDI treatment (metronidazole or vancomycin) and experience a recurrence of disease, which is associated with increased morbidity and mortality (1, 4). Once recurrence has occurred, the risk of subsequent recurrences is increased further (5).
The gut microbiota plays an important part on the pathogenesis of CDI. A healthy gut microbiota is thought to limit colonization by pathogens, a function of the microbiota known as colonization resistance. A major risk factor for CDI is antibiotic use, which disrupts the normal structure of the gut microbiota and leads to the loss of colonization resistance (6). The difficulty in acquiring patient samples prior to the development of CDI has limited the identification of specific members of the microbiota that are capable of providing colonization resistance against CDI in humans. While it is hypothesized that recurrent CDI occurs due to the inability of the indigenous microbiota to recover following antibiotic use (7), it is still unknown whether a specific microbial community is more prone to recurrence than others. Comparison of the fecal microbiota in patients developing nosocomial diarrhea with and without C. difficile colonization revealed few community differences to indicate specific community structures susceptible to CDI (8), although prior studies have demonstrated that a small number of patients with recurrent CDI exhibited a unique microbiota community structure (9). More than likely, the structural differences observed following antibiotics reflect changes in the functional, or metabolic, environment that contribute to CDI susceptibility (10).
In recent years, a nonantibiotic treatment method for CDI that is capable of restoring colonization resistance to C. difficile has reemerged. Fecal microbiota transplantation (FMT), the introduction of intestinal microbiota from a healthy donor to restore the indigenous gut microbiota, has been successfully used to treat recurrent cases of CDI when standard therapy fails (5, 11–13). Although the use of FMT has a reported 90% success rate for recurrent CDI (12), the specific mechanisms by which FMT restores colonization resistance remains undetermined. Recovery of colonization resistance to C. difficile has been described for both human (14) and mouse (15, 16) studies following FMT therapy, but few studies have explored the functional changes following FMT. One recent study using targeted metabolites observed an increase in secondary bile salts following FMT, suggesting that recovery of a microbiota community capable of restoring bile salt metabolism is important in limiting recurrent CDI (17). It is likely that a complex set of metabolites is required for full recovery of colonization resistance.
The primary goal of this study was to follow structural changes of the microbiota following successful FMT to elucidate the means by which FMT restores colonization resistance. This study investigated changes in the composition and structure of the fecal microbiota before and after FMT for treatment of CDI. Using 16S rRNA-encoding gene surveys, we compared the microbiota community from pre- and post-FMT fecal samples to that in their respective donor sample. In addition to identifying the types of organisms capable of colonizing the gut following FMT, we aimed to understand the functional contribution of the organisms introduced, using a predictive metagenomic tool that imputes the metagenomic potential based on the 16S rRNA community present.
RESULTS
Patient description and data acquisition.
To compare compositional changes following fecal microbiota transplantation (FMT), we used 16S rRNA sequencing with fecal samples collected before and after FMT from 14 recipient subjects. For 10 recipients, donor stool was available for recipient-donor comparison. All recipients had a history of at least two recurrent C. difficile infections (CDI) following an initial infection and failed standard therapy (vancomycin, metronidazole, and/or fidaxomicin) (Fig. 1; see also Table S1 in the supplemental material). Healthy donors were defined as household members or known contacts without a history of acute or chronic gastrointestinal disease and were previously screened for several pathogens prior to the procedure (see Materials and Methods).
FIG 1 .

Timeline of sample collection for each recipient-donor pair. Relative sample collection time, history of antibiotic use, and results for C. difficile clinical diagnostic tests (Illumigene assay) for the time of FMT for each recipient are indicated.
Total DNA was extracted from the fecal samples, and PCR amplicons spanning the 16S rRNA V3-V5 region were sequenced to compare the microbial communities before and after FMT. Following removal of low-quality or chimeric sequences, 569,880 high-quality sequences were available for analysis (an average of 14,243 sequences/sample). To characterize the microbial communities, sequences were aligned to the SILVA reference alignment and clustered into operational taxonomic units (OTUs) (cutoff = 0.02) using the software program mothur (v.1.31) (18). Sequences were taxonomically classified, or phylotyped, using the RDP database (v9) (19). A total of 1,796 unique OTUs were detected within all samples, belonging to 164 genera (see Table S2 in the supplemental material).
Fecal community structure and diversity in donor-recipient samples following FMT.
Marked differences at both the phylum and genus levels were observed among pre-FMT, post-FMT, and donor samples (Fig. 2; see also Fig. S1 in the supplemental material). The majority of sequences within pre-FMT samples were classified as Proteobacteria (48.2%), whereas the relative abundance of these sequences were significantly lower in post-FMT (11.2%; P < 0.001 using Wilcoxon’s nonparametric test) and donor (0.73%; P < 0.0001) samples. At the genus level within pre-FMT samples, Cronobacter (17.9%) and an unclassified Enterobacteriaceae member (20.3%) were most abundant. Although these genera were observed in post-FMT samples (8.8%), they constituted only 0.12% of the sequences assigned in donor samples. Considerable genus-level diversity within the Firmicutes was detected in all samples, albeit at higher levels in post-FMT (51.6%) and donor (65.4%) samples compared to levels in pre-FMT samples (43.6%). In both post-FMT and donor samples, OTUs classified as Blautia (6.0%), Lachnospiraceae (incertae sedis, 5.6%), unclassified Lachnospiraceae (11.0%), and Faecalibacterium (4.3%) comprised the most abundant Firmicutes (Fig. 2). In contrast, Bacteroidetes members were severely depleted in pre-FMT samples (1.3%) compared to levels in post-FMT (32.3%; P < 0.0001) and donor (32.8%; P < 0.0001) samples. At the genus level within post-FMT and donor samples, OTUs classified as Bacteroides (24.0%) and Alistipes (3.7%) comprised the majority of the Bacteroidetes phylum members.
FIG 2 .

Relative abundances of most abundant OTUs in donor and pre- and post-FMT samples. Heat map of most abundant OTUs (>2% of all sequences), classified by phylum, family, and genus. Samples were clustered based on the Morisita-Horn distance matrix using the R package “vegan” and color coded by sample type (donor or pre- or post-FMT sample).
Interestingly, changes in less-abundant genera were observed following FMT procedure (see Fig. S1 and Table S2 in the supplemental material). Fusobacterium (phylum Fusobacteria), which was not detected in donor samples at >1 sequence total, was detected in 6/14 recipients prior to FMT and remained detectable in only two patients post-FMT. Conversely, Akkermansia (phylum Verrucomicrobia) was observed in 6/14 recipients post-FMT yet was absent in their pre-FMT samples. Of the three donor samples available for these recipients, all donor samples had high levels of Akkermansia. These results support the potential transfer of new organisms/communities from donor to recipient during FMT.
To evaluate overall compositional changes occurred following FMT, we calculated species richness by estimating the number of unique OTUs and utilized the Shannon diversity index to estimate diversity. Significance was calculated using the nonparametric Wilcoxon rank-sum test. The observed species richness was increased in post-FMT samples, nearing the level observed in donor samples (Fig. 3A). However, overall post-FMT community richness was significantly decreased compared to that for donor samples. Similarly, the Shannon diversity index was significantly increased following FMT but significantly decreased compared to that for donor samples (Fig. 3B). Taken together, these data support at least partial recovery of the community diversity following FMT.
FIG 3 .

Estimated diversity in donor and pre- and post-FMT samples. The observed species richness (estimated number of OTUs) (A) or Shannon diversity index (B) for donor and pre- and post-FMT samples is shown. Each recipient-donor pair is color coded as indicated by the legend. Box plots specify the median (second), third, and fourth quartiles. The nonparametric Wilcoxon test was used to calculate significance among the different sample groups (*, P < 0.01; **, P < 0.001; ***, P < 0.0001).
Comparison of structural similarity among donor and pre- and post-FMT samples.
To compare community structure and similarity among post-FMT, pre-FMT, and donor samples, we determined the number of OTUs shared between recipient-donor pairs. The shared species richness (number of shared OTUs) between recipient-donor pairs was significantly higher between post-FMT and donor samples than that for any comparison to pre-FMT samples (Fig. 4A), supporting the observation that donor organisms were transferred and maintained in the recipients.
FIG 4 .
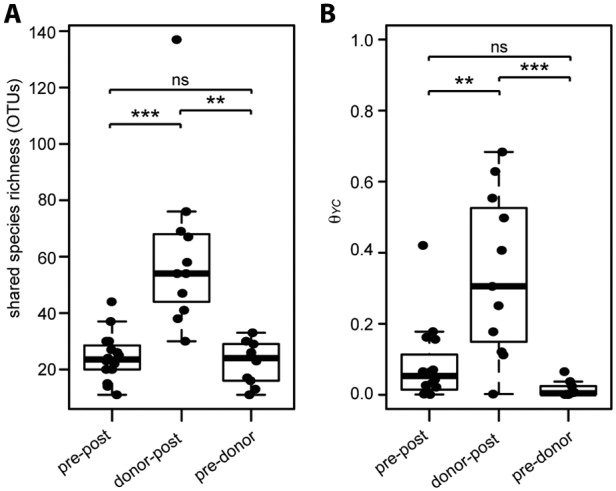
Similarity between donor, pre-, and post-FMT samples. Shared species richness (number of shared OTUs) (A) or the Yue-Clayton theta similarity (θYC) (B) between pre-FMT and post-FMT (pre-post), donor and post-FMT (donor-post), and pre-FMT and donor (pre-donor) samples within recipient-donor pairs is shown. The nonparametric Wilcoxon test was used to calculate significance among the different sample groups (*, P < 0.01; **, P < 0.001; ***, P < 0.0001; ns, not significant).
As a further comparison of the paired samples, we calculated the Yue-Clayton similarity index (θYC), a multivariate metric that accounts for abundance of representative OTUs. Similar to the observed shared species richness, θYC similarity was significantly higher between donor and post-FMT samples than for either compared to pre-FMT samples (Fig. 4B). Additionally, ordination supported distinct clustering of pre-FMT, post-FMT, and donor samples (analysis of molecular variance [AMOVA], P < 0.001) (Fig. 5; see also Fig. S3 in the supplemental material). We calculated the correlation of each axis with OTU abundance and detected several OTUs likely to contribute significantly to the observed shifts within the axes (Fig. 5). Not surprisingly, many abundant OTUs were significantly correlated with sample type. Both Firmicutes and Bacteroidetes members, such as the unclassified OTU of the Lachnospiraceae family and Bacteroides, were significant drivers toward healthy and donor status. Conversely, Proteobacteria members, such as Cronobacter, were correlated toward the pre-FMT state direction.
FIG 5 .
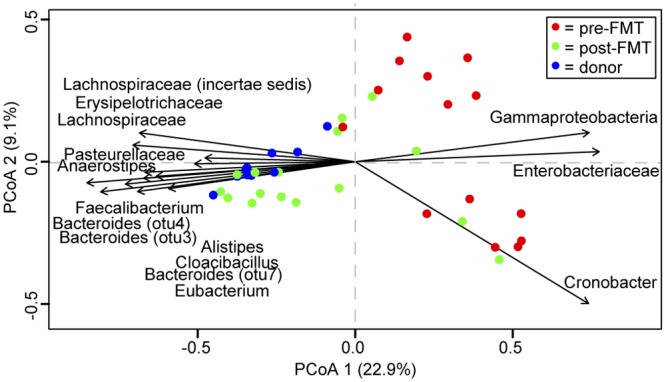
Compositional comparison of donor, pre-, and post-FMT samples. Principal coordinates analysis (PCoA) of the theta similarity (θYC) for all samples, color coded by sample type (donor and pre- and post-FMT). The direction of 15 most abundant significantly correlated operational taxonomic units (OTUs) for axes 1 and 2 are labeled, as calculated using the Spearman correlation (P < 0.001).
Predicted functional potential pre- and post-FMT.
In addition to the provision of a healthy community structure, FMTs may provide a functional component via the introduction of healthy members. We sought to analyze the relationship between the microbiota structure and function involved in maintaining colonization resistance against C. difficile by employing a computational method, PICRUSt (phylogenetic investigation of communities by reconstruction of unobserved states) to predict the metagenomic contribution of the observed microbial community within our FMT recipient and donor samples (20).
Similar to the OTU profile observed within our samples, comparison of the predicted Kyoto Encyclopedia of Genes and Genomes (KEGG) orthologies (KOs) demonstrated significant clustering of pre-FMT samples from post-FMT and donor samples (see Fig. S3 in the supplemental material). The relative abundance of KOs present in pre-FMT samples was predominantly decreased compared to that in post-FMT and donor samples. To encompass the major functional categories covered by individual KOs, we determined the overall gene pathway and module abundance using HUMAnN (HMP Unified Metabolic Analysis Network) (21). Using analysis of variance (ANOVA), we identified 75 gene modules (consisting of 5 to 20 genes per pathway) that were significantly differentially abundant among donor, pre-, and post-FMT samples (Fig. 6). Several modules related to basic metabolism and biosynthesis of amino acids, nucleotides, and carbohydrates were underrepresented in pre-FMT samples compared to donor and post-FMT samples. For instance, NAD (KEGG module M00115), histidine (M00026), and lysine (M00016) biosynthesis were all significantly decreased in pre-FMT samples. A similar pattern of underrepresentation was seen for genes involved in core metabolic pathways, such as glycolysis (M00001 and M00002) and the pentose phosphate pathway (M00004 and M00006), with these also being underrepresented in pre-FMT samples compared to findings for donor samples.
FIG 6 .

Metagenomic functional predictions for donor, pre-, and post-FMT samples. Mean relative gene pathway abundance of significantly differentially abundant modules (ANOVA; P < 0.01) for donor and pre- and post-FMT samples. Gene pathway abundances were calculated using the HUMAnN pipeline and grouped by major functional categories.
Conversely, multiple gene pathways associated with stress response and peptide transport were overrepresented in pre-FMT samples compared to findings for both post-FMT and donor samples. The presence of pathways involved in both glutamate metabolism (KEGG modules M00045 and M00121) and gamma-aminobutyric acid (GABA) biosynthesis (M00136) were increased in pre-FMT samples (Fig. 6). In many prokaryotes, GABA production is a stress response (22). Interestingly, some pathways that utilize glutamate as a substrate, such as the biosynthesis of UMP (M00051) or ornithine (M00028), were increased in donor and post-FMT samples, suggesting an imbalance of pathways involved in controlling glutamate metabolism within pre- and post-FMT samples. Transport systems for amino acids, such as for histidine (M00226), glutathione (M00348), and arginine (M00229), were also overrepresented in pre-FMT samples. These data suggest that in addition to the observed structural differences between recipients and donors, the potential metabolic contribution by the community is also greatly different between pre-FMT and donor/post-FMT samples.
DISCUSSION
Although clinical recovery of CDI is ultimately diagnosed by the resolution of clinical CDI symptoms, study protocol required clinical testing for C. difficile during the patient’s follow-up visit. Three out of five recipients who tested positive for C. difficile using the Illumigene C. difficile assay (Meridian Bioscience, Inc., Cincinnati, OH) during their post-FMT appointment (Fig. 1, recipients 002, 003, and 012) were asymptomatic and considered to be clinically recovered. For one symptomatic patient who tested positive for C. difficile (recipient 005), a 7-day vancomycin course resulted in successful clinical recovery and an eventual negative test. Patient 006, who also tested positive, became symptomatic after the initial post-FMT visit and was treated with vancomycin for an indefinite time. Other than a brief follow-up phone contact to assess the status of the patient’s CDI symptoms up to 6 months after the procedure, study protocol did not dictate that further data be collected upon a positive C. difficile test. Since 12 participants remained asymptomatic throughout study contact, we consider this FMT study to have an 86% success rate.
We observed significant differences in the microbiome following FMT that support restoration of community diversity and structure at an individual level. Our data suggest that the fecal microbiota in post-FMT samples resembles the community of each respective donor rather than the community observed prior to FMT, supporting previous observations about the fecal microbiota community following FMT for the treatment of C. difficile (7, 11, 14, 17, 23). By comparing community composition and structure per FMT pair, we observed differences in functional potential of the microbiome based on the type of community present within individual recipient-donor pairs. The results presented here suggest that an imbalance of the microbiome during recurrent CDI is at least partially returned to healthy status following FMT despite interindividual differences in community structure observed prior to FMT. Interestingly, the resulting post-FMT microbial community within the recipient did not necessarily predict C. difficile test results or clinical recovery status of the recipient patients. For instance, the post-FMT sample from patients 006 (symptomatic) and 012 (asymptomatic) both clustered within the donor samples despite a positive C. difficile test at their follow-up visit (Fig. 2; see also Fig. S2 in the supplemental material). Similarly, the post-FMT sample for patient 007 clustered among the pre-FMT samples despite testing negative during their follow-up. However, the post-FMT samples from patients 005 and 012, which harbored a high abundance of Proteobacteria, clustered within the pre-FMT samples (Fig. 2; see also Fig. S1).
Phylogenetically, a decreased abundance of Proteobacteria members and an increased abundance of various Firmicutes and Bacteroidetes were observed following FMT (Fig. 2; see also Fig. S1 in the supplemental material). However, we observed that the most abundant OTUs within post-FMT samples (collected up to 1 month following the procedure) were not necessarily the most abundant OTUs of the donor (see Table S2). It is hypothesized that microbes from the donor directly colonize the recipient, thus restoring colonization resistance. A longitudinal study by Hamilton et al. investigated community dynamics up to 4 months following FMT in three patients (14). While the community post-FMT was structurally more similar to the donor sample than the pre-FMT sample, the most abundant OTUs >3 months post-FMT were not those of the donor. Similar observations of transient colonization by the donor community were described by Angelberger et al., who analyzed the temporal dynamics following FMT in ulcerative colitis patients (24). While the donor’s most abundant OTUs were considered the most abundant within a week post-FMT, the authors concluded that colonization by the donor community was transient in most patients, followed by colonization patterns that were not directly from the donor source. This suggests a less direct contribution of FMTs in restoring colonization resistance, such as functional provision of an environment conducive for growth of existing Bacteroidetes and/or Firmicutes that can now repopulate the microbiota and/or direct inhibition of C. difficile growth via niche exclusion.
To gain insight into the functional differences among recipients with CDI and what a donor community might provide via FMT, we used PICRUSt to predict the metagenomic contribution of the communities observed. PICRUSt predicts metagenomic potential by imputing the available annotated genes within a known sequenced database, such as the KEGG catalogue, based on the presence/absence of OTUs of a 16S rRNA survey. PICRUSt has been utilized previously to describe differences in potential function within human samples and positively corresponds to actual metagenomic and metabolic comparisons, despite flaws used in imputation (25, 26). Similar to the structural observations based on 16S profiles, our results suggest an imbalanced representation of gene pathways between pre- and post-FMT samples. PICRUSt analysis suggested that core metabolic pathways, such as glycolysis or the pentose phosphate pathway, were underrepresented, while an increase in the types of transport pathways were overrepresented, within pre-FMT samples. Furthermore, this analysis suggested that differential representation of amino acid biosynthesis pathways was present in pre-FMT compared to donor and post-FMT samples, such as an increased coverage of pathways involved in biosynthesis of histidine, an amino acid observed to promote C. difficile growth (27). Additionally, several peptide transport systems found in the Proteobacteria, such as those for histidine, glutathione, and arginine, were overrepresented in pre-FMT samples. Interestingly, Morgan et al. observed similar results in samples from individuals with inflammatory bowel disease (IBD) (25). For instance, glutathione is involved in inflammatory cascades during oxidative stress, which may be relevant in the development of gastrointestinal inflammation. Although experimental validation of the human metabolic environment that is able to promote C. difficile is necessary, metagenomic prediction tools, such as PICRUSt, provide an important first glance at the functional contribution of a healthy community within the gut, leading to testable hypotheses.
Our results support previous observations that in addition to changing the structural landscape of the gut (removal of specific organisms), antibiotic-induced loss of colonization resistance affects the metabolic potential of the environment as a whole (10, 28). In some individuals, this may develop into a more favorable environment for C. difficile to either colonize or grow. A study by Theriot et al. identified significant shifts in the intestinal metabolome of antibiotic-treated mice (29). In particular, levels of sugars that C. difficile can utilize were greatly increased in susceptible mice, reflective of a less diverse community structure. Additionally, the abundance of taurocholic acid, a known germinant for C. difficile, was increased following antibiotic use. Few studies have described the metabolic profile of CDI patients in addition to the structural changes. Antharam et al. observed a depleted butyrate-producing bacterial profile in CDI and diarrheal control patients, suggesting that butyrate production may be important in limiting C. difficile growth (8). A recent study by Weingarden et al. employed targeted and untargeted metabolomics to describe changes in the metabolome before and after FMT and observed similar shifts in the full metabolome, as with microbial composition (17). Specifically, they observed increased amounts of primary bile acids in recurrent CDI patients, consistent with in vitro findings of C. difficile growth and in vivo mouse models. However, it is unclear whether these observations are a result or cause of CDI. The availability of prospective samples prior to CDI infection in human studies is limited, making functional comparisons prior to initial CDI difficult. Additionally, the interindividual variation of the microbiome in humans and differences in immunity complicate the identification of specific members that may provide functional or structural colonization resistance. However, identifying the specific metabolites, as well as the microbes that can potentially provide these, that contribute to C. difficile growth will hopefully lead to new hypotheses of why certain individuals become susceptible to CDI.
While current success rates are high for treatment of CDI, a clear understanding of how FMT works has the potential to aid the development of alternative biotherapeutics. Future methods may include a defined bacterial community that is effective in all patients without the need for donor screening or prebiotics that improve the restoration of colonization resistance. Reports of short-term risks, such as bacteremia, acute norovirus infection, and IBD relapse, have been recently reported following FMT (30–32), and a clear understanding of these potential dangers is necessary to ensure the continued safety of FMT. Meanwhile, the long-term effects of exposing the recipient to nonself microbiota are undetermined. Because of the potential for negative downstream effects, complete understanding of how FMT promotes microbiota recovery is necessary for the advancement of biotherapeutics in the treatment of recurrent CDI.
MATERIALS AND METHODS
Patient enrollment and sample collection.
Patients were enrolled at the Essentia Health Duluth Clinic, Duluth, MN. Informed consent was received from all participants under an approved Institutional Review Board (IRB) protocol at Essentia Health Duluth Clinic (IRB no. SMDC-09068; principal investigator, Timothy Rubin, FDA Investigational New Drug [IND] no. 15460). To become a candidate for stool transplantation, patients must have had a laboratory-confirmed diagnosis of CDI followed by at least two laboratory-confirmed recurrences of CDI after antibiotic treatment with metronidazole and/or vancomycin (Fig. 1; see also Fig. S1 in the supplemental material). Samples pre- and post-FMT were collected at designated appointments before and after transplantation procedure. Patients had brief scheduled telephone contacts at 2 weeks, 4 weeks, 3 months, and 6 months to discuss recovery and presence of recurrence or adverse effects. Laboratory testing for C. difficile toxin was conducted during the patient’s post-FMT appointment at the time of stool collection. If a patient reported recurrence of symptoms of CDI (diarrhea or abdominal pain) or if the follow-up C. difficile toxin testing was positive, the patient was referred to one of the study members (T. A. Rubin) for clinical care outside of the study protocol.
Donor screening and preparation of donor stool.
A healthy donor was defined as someone lacking acute gastrointestinal conditions (diarrhea) or chronic gastrointestinal conditions (inflammatory bowel disease, microcolitis, celiac disease, sprue, or a history of colon cancer). Donors must not have been treated with antibiotics in the 3 months preceding the event and were screened for the following not more than 1 month before the procedure: C. difficile (toxin), hepatitis A, B, and C, syphilis, ova and parasites, enteric bacterial pathogens, and HIV-1/HIV-2. Donor stool (approximately 30 g or 3 cm3 in volume) was collected <6 h prior to the procedure and then brought to the clinic for preparation of the stool suspension by laboratory staff. The stool was then combined with 90 ml sterile saline and processed in a blender until a smooth consistency was reached. The suspension was then filtered using a coffee filter twice, yielding 40 to 60 ml of stool suspension to be used for transplantation.
Fecal transplantation protocol.
All protocols were approved by and conducted at the Essentia Health Duluth Clinic, Duluth, MN, and conducted as described previously (33). Prior to transplantation, patients were treated with vancomycin (125 mg four times per day for at least 4 days) and a proton pump inhibitor the day before and the day of transplantation. Transplantation was conducted nasogastrically, confirming the placement of the tube tip in the patient’s gastric antrum verified via radiography. Approximately 25 to 50 ml of the stool suspension was aspirated into a syringe and administered through the nasogastric tube, followed by flushing with normal saline before removing the tube.
Sample preparation and sequencing.
Total DNA was isolated using the PowerMag soil DNA isolation kit optimized for epMotion (catalog no. 27100-4-EP; MoBio, Carlsbad, CA) according to the manufacturer’s directions and adapted for a Biomek FXp lab automation workstation (Beckman Coulter, Brea, CA). The bacterial V3-V5 16S rRNA region was amplified using the bar-coded 357F/926R primer set as used by the Human Microbiome Project (HMP) Consortium 454 sequencing protocol as described previously (34). Samples were sequenced using 454 FLX Titanium chemistry according to the manufacturer’s directions (Roche Diagnostics, Brandford, CT). Raw sequences were placed in the NCBI Sequence Read Archive (SRA) under the associated BioProject ID, PRJNA238042.
Data analysis.
Data were analyzed using a combination of the software programs R, mothur, and JMP v.10.0. Sequences were processed with mothur v.1.29.0 using the described 454 standard operating procedure (SOP) (18). All OTUs were clustered at a cutoff of 0.02 (98% similarity) and classified using the Ribosomal DNA Project (RDP) database (v9) (19). Heat maps, principal coordinates analysis (PCoA), and bar plots were created in R using the packages vegan and gplots (35, 36). Statistics using the Wilcoxon test, analysis of molecular variation (AMOVA), and analysis of variance (ANOVA) were calculated using JMP v.10.0 and R. For PICRUSt, we normalized the OTU results by subsampling to the lowest sequence count (3,500 OTUs/sample). This table was input into the QIIME pipeline, which was used to reference the Greengenes 16S rRNA database (v9), the required input for PICRUSt (20, 37). Within PICRUSt, OTUs were normalized by 16S rRNA copy number, and metagenomes were predicted from the Kyoto Encyclopedia of Genes and Genomes (KEGG) catalogue (38). HUMAnN was used to predict downstream pathway coverage from the KEGG Ortholog results (21). ANOVA in R was used to calculate the significantly differentially abundant pathways (P < 0.001).
SUPPLEMENTAL MATERIAL
Abundant genera in fecal microbiota transplant (FMT) recipients. Relative abundances of most abundant genera (>2%) in fecal samples from donor and pre- and post-FMT samples, by recipient-donor pairs, are shown. Genera are color coded as indicated and ordered by abundance within the main phyla. Download
Structural similarities in fecal microbiota transplant (FMT) recipients. Principal coordinates analysis (PCoA) of donor and pre- and post-FMT samples, based on the Yue-Clayton theta similarity (θYC). Axis 1, 2, and 3 comparisons are shown in panels A, B, and C. Post-FMT samples that tested positive (+) for C. difficile toxin are labeled as indicated. Download
KEGG orthologs (KOs) in fecal microbiota transplant (FMT) recipients. The heat map shows the relative abundances of individual KEGG orthologs (KOs) calculated for donor and pre- and post-FMT samples using PICRUSt. Samples were clustered using the Euclidean distance measure. Download
FMT recipient information and C. difficile detection. Recipient metadata (sex, age, antibiotics use, and sample numbers collected) for each FMT recipient.
16S rRNA sequence information. Raw OTU counts, with taxonomic classification, for all samples. Sequences were classified using the Ribosomal Database Project (RDP-II, v9) reference.
ACKNOWLEDGMENTS
We thank Jeanette A. Palcher, program analyst at the Essentia Institute of Rural Health, for database management, Jennifer L. Gunderson, clinical research nurse at the Essentia Institute of Rural Health, for serving as the clinical assistant on this study, and Luanne L. Borowicz, clinical assistant in gastroenterology, for assisting in patient management.
This research was funded in part by NIH grant U19AI090871 (to V.B.Y.) and with support from the Michigan Gastrointestinal Peptide Research Center (P30DK034933). Additional support came from the Essentia Health Foundation in Duluth, MN.
Footnotes
Citation Seekatz AM, Aas J, Gessert CE, Rubin TA, Saman DM, Bakken JS, Young VB. 2014. Recovery of the gut microbiome following fecal microbiota transplantation. mBio 5(3):e00893-14. doi:10.1128/mBio.00893-14.
REFERENCES
- 1. Ananthakrishnan AN. 2011. Clostridium difficile infection: epidemiology, risk factors and management. Nat. Rev. Gastroenterol. Hepatol. 8:17–26. 10.1038/nrgastro.2010.190 [DOI] [PubMed] [Google Scholar]
- 2. Dubberke ER, Olsen MA. 2012. Burden of Clostridium difficile on the healthcare system. Clin. Infect. Dis. 55(Suppl 2):S88–S92. 10.1093/cid/cis335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cohen SH, Gerding DN, Johnson S, Kelly CP, Loo VG, McDonald LC, Pepin J, Wilcox MH, Society for Healthcare Epidemiology of America. Infectious Diseases Society of America 2010. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the Society for Healthcare Epidemiology of America (SHEA) and the Infectious Diseases Society of America (IDSA). Infect. Control Hosp. Epidemiol. 31:431–455. 10.1086/651706 [DOI] [PubMed] [Google Scholar]
- 4. Musher DM, Aslam S, Logan N, Nallacheru S, Bhaila I, Borchert F, Hamill RJ. 2005. Relatively poor outcome after treatment of Clostridium difficile colitis with metronidazole. Clin. Infect. Dis. 40:1586–1590. 10.1086/430311 [DOI] [PubMed] [Google Scholar]
- 5. Kelly CP. 2012. Current strategies for management of initial Clostridium difficile infection. J. Hosp. Med. 7(Suppl 3):S5–S10. 10.1002/jhm.1909 [DOI] [PubMed] [Google Scholar]
- 6. van der Waaij D. 1989. The ecology of the human intestine and its consequences for overgrowth by pathogens such as Clostridium difficile. Annu. Rev. Microbiol. 43:69–87. 10.1146/annurev.micro.43.1.69 [DOI] [PubMed] [Google Scholar]
- 7. Khoruts A, Dicksved J, Jansson JK, Sadowsky MJ. 2010. Changes in the composition of the human fecal microbiome after bacteriotherapy for recurrent Clostridium difficile-associated diarrhea. J. Clin. Gastroenterol. 44:354–360. 10.1097/MCG.0b013e3181c87e02 [DOI] [PubMed] [Google Scholar]
- 8. Antharam VC, Li EC, Ishmael A, Sharma A, Mai V, Rand KH, Wang GP. 2013. Intestinal dysbiosis and depletion of butyrogenic bacteria in Clostridium difficile infection and nosocomial diarrhea. J. Clin. Microbiol. 51:2884–2892. 10.1128/JCM.00845-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chang JY, Antonopoulos DA, Kalra A, Tonelli A, Khalife WT, Schmidt TM, Young VB. 2008. Decreased diversity of the fecal microbiome in recurrent Clostridium difficile-associated diarrhea. J. Infect. Dis. 197:435–438. 10.1086/525047 [DOI] [PubMed] [Google Scholar]
- 10. Theriot CM, Young VB. 2014. Microbial and metabolic interactions between the gastrointestinal tract and Clostridium difficile infection. Gut Microbes 5:86–95. 10.4161/gmic.27131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. van Nood E, Vrieze A, Nieuwdorp M, Fuentes S, Zoetendal EG, de Vos WM, Visser CE, Kuijper EJ, Bartelsman JF, Tijssen JG, Speelman P, Dijkgraaf MG, Keller JJ. 2013. Duodenal infusion of donor feces for recurrent Clostridium difficile. N. Engl. J. Med. 368:407–415. 10.1056/NEJMoa1205037 [DOI] [PubMed] [Google Scholar]
- 12. Brandt LJ, Aroniadis OC. 2013. An overview of fecal microbiota transplantation: techniques, indications, and outcomes. Gastrointest. Endosc. 78:240–249. 10.1016/j.gie.2013.03.1329 [DOI] [PubMed] [Google Scholar]
- 13. Yoon SS, Brandt LJ. 2010. Treatment of refractory/recurrent C. difficile-associated disease by donated stool transplanted via colonoscopy: a case series of 12 patients. J. Clin. Gastroenterol. 44:562–566. 10.1097/MCG.0b013e3181dac035 [DOI] [PubMed] [Google Scholar]
- 14. Hamilton MJ, Weingarden AR, Unno T, Khoruts A, Sadowsky MJ. 2013. High-throughput DNA sequence analysis reveals stable engraftment of gut microbiota following transplantation of previously frozen fecal bacteria. Gut Microbes 4:125–135. 10.4161/gmic.23571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lawley TD, Clare S, Walker AW, Stares MD, Connor TR, Raisen C, Goulding D, Rad R, Schreiber F, Brandt C, Deakin LJ, Pickard DJ, Duncan SH, Flint HJ, Clark TG, Parkhill J, Dougan G. 2012. Targeted restoration of the intestinal microbiota with a simple, defined bacteriotherapy resolves relapsing Clostridium difficile disease in mice. PLoS Pathog. 8:e1002995. 10.1371/journal.ppat.1002995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Reeves AE, Koenigsknecht MJ, Bergin IL, Young VB. 2012. Suppression of Clostridium difficile in the gastrointestinal tracts of germfree mice inoculated with a murine isolate from the family Lachnospiraceae. Infect. Immun. 80:3786–3794. 10.1128/IAI.00647-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Weingarden AR, Chen C, Bobr A, Yao D, Lu Y, Nelson VM, Sadowsky MJ, Khoruts A. 2014. Microbiota transplantation restores normal fecal bile acid composition in recurrent Clostridium difficile infection. Am. J. Physiol. Gastrointest. Liver Physiol. 306:G310–G319. 10.1152/ajpgi.00282.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75:7537–7541. 10.1128/AEM.01541-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang Q, Garrity GM, Tiedje JM, Cole JR. 2007. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73:5261–5267. 10.1128/AEM.00062-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, Beiko RG, Huttenhower C. 2013. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31:814–821. 10.1038/nbt.2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Abubucker S, Segata N, Goll J, Schubert AM, Izard J, Cantarel BL, Rodriguez-Mueller B, Zucker J, Thiagarajan M, Henrissat B, White O, Kelley ST, Methé B, Schloss PD, Gevers D, Mitreva M, Huttenhower C. 2012. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput. Biol. 8:e1002358. 10.1371/journal.pcbi.1002358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Feehily C, Karatzas KA. 2013. Role of glutamate metabolism in bacterial responses towards acid and other stresses. J. Appl. Microbiol. 114:11–24. 10.1111/j.1365-2672.2012.05434.x [DOI] [PubMed] [Google Scholar]
- 23. Song Y, Garg S, Girotra M, Maddox C, von Rosenvinge EC, Dutta A, Dutta S, Fricke FW. 2013. Microbiota dynamics in patients treated with fecal microbiota transplantation for recurrent Clostridium difficile infection. PLoS One 8:e81330. 10.1371/journal.pone.0081330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Angelberger S, Reinisch W, Makristathis A, Lichtenberger C, Dejaco C, Papay P, Novacek G, Trauner M, Loy A, Berry D. 2013. Temporal bacterial community dynamics vary among ulcerative colitis patients after fecal microbiota transplantation. Am. J. Gastroenterol. 108:1620–1630. 10.1038/ajg.2013.257 [DOI] [PubMed] [Google Scholar]
- 25. Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, Reyes JA, Shah SA, LeLeiko N, Snapper SB, Bousvaros A, Korzenik J, Sands BE, Xavier RJ, Huttenhower C. 2012. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 13:R79. 10.1186/gb-2012-13-9-r79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McHardy IH, Goudarzi M, Tong M, Ruegger PM, Schwager E, Weger JR, Graeber TG, Sonnenburg JL, Horvath S, Huttenhower C, McGovern DPB, Fornace AJ, Borneman J, Braun J. 2013. Integrative analysis of the microbiome and metabolome of the human intestinal mucosal surface reveals exquisite inter-relationships. Microbiome 1:17. 10.1186/2049-2618-1-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Karasawa T, Ikoma S, Yamakawa K, Nakamura S. 1995. A defined growth medium for Clostridium difficile. Microbiology 141:371–375. 10.1099/13500872-141-2-371 [DOI] [PubMed] [Google Scholar]
- 28. Perez-Cobas AE, Artacho A, Knecht H, Ferrus ML, Friedrichs A, Ott SJ, Moya A, Latorre A, Gosalbes MJ. 2013. Differential effects of antibiotic therapy on the structure and function of human gut microbiota. PLoS One 8:e80201. 10.1371/journal.pone.0080201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Theriot CM, Koenigsknecht MJ, Carlson PE, Hatton GE, Nelson AM, Li B, Huffnagle GB, Li JZ, Young VB. 2014. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increases susceptibility to Clostridium difficile infection. Nat. Commun 5:3114. 10.1038/ncomms4114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Quera R, Espinoza R, Estay C, Rivera D. 2014. Bacteremia as an adverse event of fecal microbiota transplantation in a patient with Crohn’s disease and recurrent Clostridium difficile infection. J. Crohns Colitis 8:252–253. 10.1016/j.crohns.2013.10.002 [DOI] [PubMed] [Google Scholar]
- 31. Schwartz M, Gluck M, Koon S. 2013. Norovirus gastroenteritis after fecal microbiota transplantation for treatment of Clostridium difficile infection despite asymptomatic donors and lack of sick contacts. Am. J. Gastroenterol. 108:1367. 10.1038/ajg.2013.164 [DOI] [PubMed] [Google Scholar]
- 32. De Leon LM, Watson JB, Kelly CR. 2013. Transient flare of ulcerative colitis after fecal microbiota transplantation for recurrent Clostridium difficile infection. Clin. Gastroenterol. Hepatol. 11:1036–1038. 10.1016/j.cgh.2013.04.045 [DOI] [PubMed] [Google Scholar]
- 33. Aas J, Gessert CE, Bakken JS. 2003. Recurrent Clostridium difficile colitis: case series involving 18 patients treated with donor stool administered via a nasogastric tube. Clin. Infect. Dis. 36:580–585. 10.1086/367657 [DOI] [PubMed] [Google Scholar]
- 34. Hashway SA, Bergin IL, Bassis CM, Uchihashi M, Schmidt KC, Young VB, Aronoff DM, Patton DL, Bell JD. 2014. Impact of a hormone-releasing intrauterine system on the vaginal microbiome: a prospective baboon model. J. Med. Primatol. 43:89–99. 10.1111/jmp.12090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Stevens MHH, Wagner H. 2013. Vegan: Community Ecology R Package, v2.0-10. http://vegan.r-forge.r-project.org/
- 36. Warnes GR, Bolker B, Bonebakker L, Gentleman R, Liaw WHA, Lumley T, Maechler M, Magnusson A, Moeller S, Schwartz M, Venables B. 2013. Gplots: various R programming tools for plotting data, v2.12.1. http://cran.r-project.org/web/packages/gplots/index.html
- 37. Kuczynski J, Stombaugh J, Walters WA, Gonzalez A, Caporaso JG, Knight R. 2012. Using QIIME to analyze 16S rRNA gene sequences from microbial communities. Curr. Protoc. Microinform. 36:10.7.1–10.7.20. 10.1002/0471250953.bi1007s36 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kanehisa M, Goto S. 2000. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30. 10.1093/nar/28.7.e27 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Abundant genera in fecal microbiota transplant (FMT) recipients. Relative abundances of most abundant genera (>2%) in fecal samples from donor and pre- and post-FMT samples, by recipient-donor pairs, are shown. Genera are color coded as indicated and ordered by abundance within the main phyla. Download
Structural similarities in fecal microbiota transplant (FMT) recipients. Principal coordinates analysis (PCoA) of donor and pre- and post-FMT samples, based on the Yue-Clayton theta similarity (θYC). Axis 1, 2, and 3 comparisons are shown in panels A, B, and C. Post-FMT samples that tested positive (+) for C. difficile toxin are labeled as indicated. Download
KEGG orthologs (KOs) in fecal microbiota transplant (FMT) recipients. The heat map shows the relative abundances of individual KEGG orthologs (KOs) calculated for donor and pre- and post-FMT samples using PICRUSt. Samples were clustered using the Euclidean distance measure. Download
FMT recipient information and C. difficile detection. Recipient metadata (sex, age, antibiotics use, and sample numbers collected) for each FMT recipient.
16S rRNA sequence information. Raw OTU counts, with taxonomic classification, for all samples. Sequences were classified using the Ribosomal Database Project (RDP-II, v9) reference.