

Abstract
The Blood Brain Barrier (BBB) is a specialized vascular structure tightly regulating central nervous system (CNS) homeostasis. Endothelial cells are the central component of the BBB and control of their barrier phenotype resides on astrocytes and pericytes. Interactions between these cells and the endothelium promote and maintain many of the physiological and metabolic characteristics that are unique to the BBB. In this review we describe recent findings related to the involvement of astroglial cells, including radial glial cells, in the induction of barrier properties during embryogenesis and adulthood. In addition, we describe changes that occur in astrocytes and endothelial cells during injury and inflammation with a particular emphasis on alterations of the BBB phenotype. GLIA 2013;61:1939–1958
Keywords: astrocyte, BBB, glial, astrogliosis, multiple sclerosis
Introduction
The circulatory system is made up of blood vessels that specialize in providing tissues with oxygen, nutrients, and hormones to stabilize body temperature, pH, and thus maintain homeostasis. The development of a mature vascular system is a multifaceted process divided into two separate stages. The first is vasculogenesis and it is defined as the de novo formation of endothelial tubes from newly differentiated endothelial precursor cells, or angioblasts. Aggregations of two or more angioblasts initiate the process of vascular tube formation by electrostatic cell surface repulsion of opposing apical cell surfaces in an endothelial cell (EC) cord. This is followed by EC elongation, flattening, and lumen widening in the developing vessel (Axnick and Lammert, 2012; Risau and Flamme, 1995). Establishment of apical-basal cell polarity and interactions between angioblasts and surrounding extracellular matrix (ECM) are also important steps in establishing early vasculature (Davis and Camarillo, 1996; Patan, 2000). The formation of vascular channels and capillary plexus are the latest stages of vasculogenesis, which are then remodeled into a circulatory network via angiogenesis (Kelly and Hirschi, 2009). In this second stage of vascular development, hypoxia leads the outgrowth of a sprout from a pre-existing vessel. Lateral inhibition is the mechanism driving the sprout into the hypoxic tissue. Sprouting can be intercellular or intracellular and it seems to be controlled by the mechanisms participating in lumen formation during vasculogenesis (Axnick and Lammert, 2012; Bicknell and Harris, 2004). Once the blood vessel is totally formed, the process of angiogenesis ceased and in the case of the central nervous system (CNS), the homeostatic control of the endothelial phenotype resides in cells in close proximity to the vessels such as astrocytes, pericytes, neurons, and microglia. Such interactions shape many of the unique characteristics of the brain endothelium known as the blood brain barrier (BBB).
The vasculature associated with the BBB is formed by highly specialized ECs characterized by the presence of specialized transport systems (Abbott et al., 2006), low pinocytic activity (Coomber and Stewart, 1985), higher mitochondrial volume fraction (Oldendorf et al., 1977), and a paracellular cleft between adjacent endothelial membranes (Alvarez et al., 2011a). This cleft is sealed by continuous strands of junctional complex proteins that include tight junctions (TJs) and adherens junctions (AJs) proteins (Dejana et al., 2008; Harhaj and Antonetti, 2004) which anastomose plasma membranes of one or several ECs, as shown in freeze-fracture electron-microscope (Nagy et al., 1984; Shivers et al., 1984); expression of AJs and TJs are known to induce a high transendothelial electric resistance (TEER) when compared to ECs in the periphery (Rubin et al., 1991). The BBB controls the movement of electrolytes, xenobiotics and circulating immune cells between the systemic circulation and the CNS parenchyma in order to maintain an optimal milieu for neuronal function (Alvarez et al., 2011a). Low transcellular passage results from the loss of cell fenestrations, and the tightly adhering junctional proteins between BBB-ECs that additionally promote low paracellular diffusion. In addition, paracellular permeability also depends on the balance between the contractile forces produced by the endothelial cytoskeleton and the adhering forces generated by the cell-cell junctional proteins and cell-ECM interactions (Garcia and Schaphorst, 1995). Thus, the opening and closing of the paracellular pathway depends on the dynamic interplay between structural components, which under pathological conditions are perturbed at different levels and result in significant alterations in the regulated exchange between the blood and the CNS. However, all these unique characteristics of the BBB are in great part due to the intricate cellular interactions between ECs and perivascular glial cells.
In this regard, the first clear demonstrations of astrocytes contributing to BBB formation and maintenance were provided independently by the groups of Raff and Brightman, in 1987. In seminal in vivo grafting and in vitro cell culture studies these two groups demonstrated that morphological and functional BBB characteristics are dependent on the CNS environment, and specifically on the contribution of perivascular astrocytes (Janzer and Raff, 1987; Tao-Cheng et al., 1987). In 1991, Rubin et al. demonstrated that isolated BBB-ECs cultured outside of the CNS environment lose their functional barrier capacity as measured by TEER recordings and permeability (Rubin et al., 1991). Nonetheless, electrical resistance and permeability to large and small molecular tracers can be reinduced on BBB-ECs when grown in the presence of astrocyte conditioned media (ACM) (Alvarez et al., 2011b; Neuhaus et al., 1991; Prat et al., 2001) or when astrocyte-EC contact is provided (Tao-Cheng et al., 1987). Conversely, when non-CNS ECs are cultured in the presence of astrocytes, or astrocyte-secreted factors, a series of BBB-specific properties, such as P-glycoprotein (p-gp) and TJ expression can be induced (Abbott et al., 2006; Prat et al., 2001; Wosik et al., 2007).
BBB Development
Barriergenesis and the unique EC properties in the CNS are not predetermined by brain-specific endothelial precursor cells but are induced by the local neural environment during the development of the vascular system. Blood vessel development within the CNS begins when vascular elements of the primitive capillary network located in the perineural vascular plexus expand by angiogenesis into the CNS parenchyma at embryonic day 11 in mouse (Risau, 1997; Risau and Wolburg, 1990) and around the 8th week of gestation in humans (Ballabh et al., 2005; Bar, 1980; Norman and O'Kusky, 1986). The vascular sprouts invade the embryonic parenchyma towards a Vascular Endothelial Growth Factor (VEGF) gradient generated by neuroectodermal cells that spans from the pial surface to the ependyma (Liebner et al., 2011). In addition to VEGF, multiple signaling pathways such as the Angiopoietin-Tie system, the Hedgehog pathway, the Ephrin receptor tyrosine kinases, the Dll4/Notch1 signaling and the Wnt signaling pathway are also involved in angiogenic sprouting and remodelling during early embryogenesis (Adams, 2002; Alvarez et al., 2011b; Daneman et al., 2009; Liebner et al., 2008; Shawber and Kitajewski, 2004; Suri et al., 1996). During this phase the vessels exhibit short and primitive TJs, lack fenestrations and restrict the movement of blood derived proteins into the CNS milieu (Liebner et al., 2011). The angiogenesis stage is followed by a differentiation phase in which interactions between newly formed vessels, pericytes and radial glial cells are established. The EC-pericyte interaction is driven by Platelet Derived Growth Factor (PDGF)-BB, who is secreted by ECs and binds the PDGFR-β on pericytes, by cross communication via the Angiopoietin-Tie system and by contact dependant mechanisms driven by sites enriched in N-cadherin expression (Daneman et al., 2009, 2010). In this phase (E14-E16 in mouse; around week 12 in humans), TJ strands become more complex, longer and the outer leaflets of adjacent membranes appear as a chain of fusion points forming sets of complicated networks that can be visualized by freeze fracture electron microscopy (Wolburg and Lippoldt, 2002). In contrast, meningeal blood vessels express TJ proteins at E14 but lack a competent barrier, as most endogenous and exo genous markers of vascular leakage can be detected in the meningeal space (Alvarez et al., 2011b; Norman and O'Kusky, 1986). This confirms the essential role of parenchymal signals, presumably glial, to establish a BBB phenotype and highlights the phenotypic and functional differences between vessels at distinct CNS locations. The BBB continues its development and maturation during late stages of embryogenesis and postnatally, as a highly complex network of strands and the increase in particle density reveal TJ maturation (Ballabh et al., 2005; Bar, 1980; Norman and O'Kusky, 1986; Wolburg and Lippoldt, 2002). The appearance of perivascular astrocytes further support the refinement of the BBB phenotype, which is also thought to be influenced by microglial- and neuronal-derived factors (Abbott et al., 2006). In addition, the higher pericyte coverage in the CNS as compared to the periphery highlights the functional importance of pericytes in CNS vascular biology in both embryogenesis and adulthood (Bell et al., 2010; Daneman et al., 2010). However, even in the absence of pericytes, embryonic CNS endothelial cells express many BBB-specific molecules, suggesting that in addition to pericytes, CNS derived signals also play an important role in promoting a BBB phenotype (Daneman et al., 2010).
In seminal histological studies using fetal human brain material, the groups of Praaven Ballabh and Jeanne E. Bell revealed the presence of astrocytes in the CNS as early as the 9th gestational week (Wilkinson et al., 1990), and their contact with CNS microvessels was detected at week 17th (El-Khoury et al., 2006). These astrocytes express glial fibrillary acidic protein (GFAP), S-100β, and Aquaporin-4 (AQP4)-immunoreactive material, which peaked and reached a plateau at week 23rd, especially in white matter and cortex (El-Khoury et al., 2006). In the developing mouse CNS, astrocytes are reported to appear in the second half of the gestational period (Molofsky et al., 2012), only after the formation of a competent BBB (Daneman et al., 2010). However, radial glial cells which are known to support neurogenesis and the migration of neural precursors are found throughout the developing CNS (Rakic, 1972). Radial glia cells are also known to closely interact with growing vessels of the cortex (Gerhardt et al., 2004) and during late embryonic corticogenesis they contact nascent blood vessels to regulate their stability via modulation of the canonical Wnt signaling (Ma et al., 2013). This suggests that glial-like cells modulate the phenotype of vessels during embryogenesis and that distinct cell types within the CNS might use similar mechanisms to induce BBB properties. At the end of neurogenesis and cortical development, most radial glial cells lose their ventricular attachment and migrate toward the cortical plate by a process known as somal translocation (Nadarajah, 2003). Astrocyte development is amplified in the later stages of embryogenesis as radial glial cells transform into multipolar astrocytes (Choi and Lapham, 1978; Molofsky et al., 2012; Schmechel and Rakic, 1979). During this process, radial glial cells convert from bipolar cells spanning the cortex to unipolar cells no longer establishing contact with the subventricular zone and then into multipolar cells with regressing radial process that progressively acquire astrocyte morphology. During postnatal development, astrocytes become the more abundant cells in the CNS and at this stage they participate in the induction and maintenance of BBB properties (Prat et al., 2001), while the role played by other constituents of the neurovascular unit (NVU) such as microglia and neurons still remains to be established.
Astrocyte Heterogeneity
The brain is an organ with high demand for oxygen and glucose. Such metabolic requirements are critical for optimal CNS function and both ECs and astrocytes play a crucial role in this process. Astrocyte endfeet ensheathe the CNS microvasculature and establish a close interaction known to control the CNS blood flow (neurovascular coupling), the energy supply to neurons and hence exert indirect control over CNS function. In the past, it was considered that neurons were responsible of the metabolic signals triggering changes in the blood flow (Attwell and Laughlin, 2001). However, the recent discovery of neurotransmitters being released by astrocytes and their ability to modulate the signaling involved in neurovascular coupling has bring strong support to the involvement of glial cells not only controlling BBB properties, but also in the regulation of blood flow. Glutamate-mediated signaling leads to the release of nitric oxide from neurons and of arachidonic acid derivatives from astrocytes (Attwell et al., 2010). These molecules can increase or decrease blood flow depending on the local oxygen concentration. Interestingly, the role of glutamate and its influence in the control of blood flow varies according to anatomical region analyzed (Northington et al., 1997). In this regard, multiple types of astrocytes with the potential to differentially modulate the microvascular phenotype include: protoplasmic astrocytes which are predominantly found in the cortex and hypothalamus, fibrous astrocytes that are found throughout the CNS and highly populate the white matter and the dorsal horn of the spinal cord, Bergmann glia which are found in the cerebellum extending processes from the Purkinje cells to the pial surface, Müller cells of the retina which have radial glia properties, velate astrocytes which are found in the cerebellum forming a sheath surrounding granule neurones, perivascular and marginal astrocytes that are localized close to the pia mater forming the pial and perivascular glia limitans, tanycytes which are found in the floor of the third ventricle supporting neuroendocrine functions and subependymal glia whose function remains to be determined (de Seranno et al., 2010; Reichenbach and Wolburg, 2005). In addition to that, there are substantial differences according to the brain region analyzed in terms of astrocyte density and their rate of proliferation. The level of complexity escalates as their electrophysiological characteristics differ. Astrocytes from different anatomical regions express distinct levels and types of ion channels which influence their electrophysiological properties, including their resting membrane potentials (Oberheim et al., 2012). Therefore, the high degree of heterogeneity within the astroglial population defines the unique cytoarchitecture of the CNS, and support the concept that distinct CNS microenvironments might reflect unique molecular and functional differences of the BBB.
Astrocyte-Endothelial Interactions
At the NVU the extracellular space consists of basement membranes in which the interplay between components of the ECM and matrix adhesion receptors is required for an appropriate interaction between cells and their microenvironment. At the level of the BBB there are two basement membranes (BMs) separating endothelium from astrocytes: the first is called the endothelial BM which is composed of fibronectin, collagen type IV, perlecan, and the laminins α4 and α5 chains, which associate with laminin β1 and γ1 to form laminin 411 and 511 (Engelhardt and Sorokin, 2009; Hamann et al., 1995; Sixt et al., 2001). The second membrane is called the parenchymal or astroglial BM (also called perivascular glia limitans) that is made of the ECM components fibronectin, agrin and the laminins α1 and α2 chains, which associate with laminin β1 and γ1 to form laminins 111 and 211 (Engelhardt and Sorokin, 2009; Sixt et al., 2001). All these ECM components have the potential to serve as scaffolding molecules, to bind to EC receptors and therefore to contribute to the BBB phenotype. In this regard, BBB-ECs are known to express the integrins α1β1, α3β1, α4β1, α5β1, α6β1, α6β4 and αvβ1/αvβ3/αvβ8 which bind to most proteins of the BM in the CNS (Dodelet-Devillers et al., 2009) as well as in the periphery (Milner and Campbell, 2002; Tagaya et al., 2001; Wagner et al., 1997). Astrocytes have also been reported to express dystroglycan and the integrins αvβ5, αvβ8, and α6β4 (Milner et al., 1999; Paulus et al., 1993; Sixt et al., 2001; Wagner et al., 1997). The BMs of cerebral microvessels are therefore seen as important scaffolding structures allowing both astrocyte-EC proximity and optimal astrocyte-EC communication.
Astrocytes also establish a close interaction with the brain vasculature through anchoring transmembrane proteins such as AQP4 and KIR4.1 (Higashi et al., 2001; Lien et al., 2012; Nielsen et al., 1997) and their adaptor molecules (syntrophin, dystrophin, and dystrobrevin). The water channel AQP4 is known to control water influx at the level of the BBB. AQP4 is anchored to the astrocyte cytoarchitecture via α-syntrophin, a component of the dystrophin-dystroglycan complex (Neely et al., 2001) that links dystrophin-associated proteins (DAP) such as dystroglycan, with various cytoskeletal components. DAP, dystroglycan, as well as other molecules preferentially expressed at the astrocyte endfeet are responsible for the polarized expression of AQP4 in CNS astrocytes (Amiry-Moghaddam and Ottersen 2003; Frigeri et al., 2001; Lien et al., 2012) and are associated with a high density of orthogonal arrays of particles (OAPs) that become less frequent in deeper areas of the neuropil (Wolburg et al., 2011). Both AQP4 knockout animals and mice specifically deficient for AQP4 in astrocytes have a significant reduction in brain water uptake and have reduced brain edema following stroke, as compared to WT animals (Haj-Yasein et al., 2011; Manley et al., 2000). However, mice specifically deficient for AQP4 in astrocytes had a fully competent BBB, suggesting that bidirectional alteration of water flow due to AQP4 deficiency does not compromise barrier function. In addition, AQP4 deletion affects the perivascular glial expression of α-syntrophin without affecting the ultrastructure of CNS ECs and their BBB properties (Eilert-Olsen et al., 2012). This indicates that expression of AQP4 is important either for the formation or for the stability of the cytoarchitecture of astrocytes.
KIR4.1 is an inwardly rectifying potassium channel that colocalizes with AQP4, and it is important for maintaining potassium and water balance. KIR4.1 is also reported to anchor to the cytoskeleton via α-syntrophin (Connors et al., 2004), although studies using α-syntrophin knockout mice did not demonstrate a reduction or a redistribution of KIR4.1 in astrocyte endfeet (Puwarawuttipanit et al., 2006). These findings indicate that other intracellular proteins are involved in the polarized expression of KIR4.1.
α-Dystrobrevin is another cytoplasmic protein expressed by glial endfeet and known to be present in cells forming blood-tissue barriers (Lien et al., 2007). Dystrobrevins establish complex interactions with dystrophins, syntrophins and other proteins to provide anchorage and support to a multiple set of cytoskeleton components, receptors and channels located in the apical side of the cell membrane (Rees et al., 2007). Recent experiments using animals lacking α-dystrobrevin demonstrated a leaky BBB and progressive development of brain edema. These alterations coincided with perturbances in the interplay between astrocyte endfeet and BBB-ECs that were characterized by altered expression of TJ proteins and dysregulated expression of AQP4, KIR4.1 and dystroglycan (Lien et al., 2012). Thus, astrocyte endfeet are attached to the endothelial BM through dystroglycan which is connected through syntrophins and α-dystrobrevin to the intracytoplasmic/cytoskeletal machinery.
Astrocytes communicate with each other via gap junctions and establish a functional syncytium that is associated with effective and well-coordinated responses within large groups of cells (Theis et al., 2005). This functional interplay is also detected at the astroglial endfeet surrounding the brain vasculature as they express high levels of gap junction proteins mainly consisting of Connexin (Cx) 43 and, to a lesser extent, Cx30 (Giaume et al., 1991). These molecules also contribute to the extent of astrocyte heterogeneity as their expression levels vary during development and within distinct anatomical regions (Nagy and Rash, 2000). As the glial syncytium is highly prevalent in proximity to blood vessels, it has been suggested that astroglial signaling involved in vasodilation and vasoconstriction during neurovascular coupling is transmitted across inter-astroglial gap junctions (Rouach et al., 2008). In terms of BBB function, mice lacking Cx43 and Cx30 expression on astrocytes had a significant loss of AQP-4 and β-dystroglycan expression on their endfeet that was associated with edema and a disrupted BBB (Ezan et al., 2012). BBB-ECs also express Cxs that associate with junctional proteins and their inhibition affect barrier function (Nagasawa et al., 2006). In addition, astrocytes play an important role in supplying energy to neurons and their close interaction with the vasculature is fundamental in this process. In the classic “astrocyte-neuron lactate shuttle” astrocytes take glucose from ECs in response to glutamatergic neuronal activity. The energy needed for glutamate uptake is provided by glucose metabolism which in turn leads to production of lactate, an important source of energy taken by neurons (Giaume et al., 2010).
Control of BBB Phenotype by Astroglial Cues During Homeostasis
In addition to the support provided by astrocytes to post-natal BBB development, the persistence of a functional BBB throughout adulthood is maintained and regulated by numerous factors unique to the microniche of the NVU (Abbott et al., 2006; Haseloff et al., 2005). The multifaceted levels of cross-communication and signaling events occurring between BBB-ECs and astrocytes, pericytes, microglia and neurons, maintain an optimal BBB phenotype and promote junctional protein maintenance, expression of metabolic and specialized transporters, as well as immune quiescence of the CNS vasculature (Prat et al., 2001). The unique nature of such microenvironment also facilitates optimal regulation and remodeling of the BBB, a phenomenon crucial for maintaining CNS homeostasis in response to physiological and pathological stimuli. Astrocyte-BBB-EC interactions are also known to regulate vascular proliferation, angiogenesis, EC morphology, and ultimately influence the phenotype of the barrier under pathological conditions (Abbott et al., 2006; Prat et al., 2001; Wolburg and Lippoldt, 2002; Wosik et al., 2007).
Astrocytes are known to produce factors able to modulate EC function during development and adulthood. During embryogenesis there is a wide range of signaling pathways known to influence patterning and branching. One of these pathways is the Hedgehog (Hh) signaling cascade known to be involved in embryonic morphogenesis, neuronal guidance and angiogenesis (Nagase et al., 2008). In adulthood, it plays an important role supporting vascular proliferation, tissue repair and various homeostatic mechanisms throughout the body (Byrd and Grabel, 2004; Kanda et al., 2003; Robbins et al., 2012). Hh signaling is initiated by one of three secreted homologues of the Drosophila Hh: Indian Hh (Ihh), Desert Hh (Dhh), and Sonic Hh (Shh), the latter being widely associated with CNS morphogenic events (Briscoe et al., 1999; Marigo et al., 1995). We recently demonstrated that astrocytes secrete Shh and that BBB-ECs bear the Hh receptor Patched-1, the signal transducer Smoothened (Smo), as well as transcription factors of the Gli family (Fig. 1). TEER and permeability experiments using human primary cultures of BBB-ECs showed that activation of the Hh pathway stimulated expression of junctional proteins and promoted a BBB phenotype. In vivo permeability assays showed that blocking the Hh pathway increased barrier permeability and lack of Shh significantly compromised the BBB phenotype. In addition, mice specifically lacking the signal transducer Smo on ECs (Tie2-Cre; Smoc/c) showed a significant increase in BBB permeability that correlated with a decrease in junctional protein expression and disturbed BMs (Fig. 2) (Alvarez et al., 2011b). These findings correlated with a reduced association of astrocyte endfeet towards the vasculature of Tie2-Cre; Smoc/c mice at early postnatal stages, but after 8 wks there was a moderate increase in the expression of GFAP (Fig. 2), indicating that perturbances in the barrier function induce a reactive phenotype on perivascular astrocytes.
FIGURE 1.

Astrocyte-Endothelial cell crosstalk supporting BBB function. The BBB is formed by endothelial cells (ECs), surrounded by an endothelial and astroglial basement membranes with pericytes embedded in between. These are surrounded by astrocyte endfeet that provide support to endothelial function and establish communication with neurons. The right panel shows prototypic examples of factors secreted by astrocytes and ECs that are crucial in the establishment and maintenance of the BBB. The receptors for some of the molecules shown are included. Ang-1-2 (angiopoietin 1 and 2), ApoE3-E4 (apolipoprotein E3 and E4), AT1 (type 1 angiotensin receptor), CypA (cyclophylin A), ET (endothelin), FGF (fibroblast growth factor) GDNF (glial cell line-derived neurotrophic factor), MMP-9 (Matrix Metalloproteinase 9), Ptch1 (Patched 1), RA (retinoic acid), RARβ (RA-receptor β), Shh (Sonic Hedgehog), Smo (Smoothened), SSeCKS (Src suppressed C kinase substrate), TGFβ (transforming growth factor-β), Tie2 (receptor tyrosine kinase 2) and VEGF (Vascular endothelial growth factor). Arrow represent induction/activation; Blunted line represent inhibition/down-regulation.
FIGURE 2.
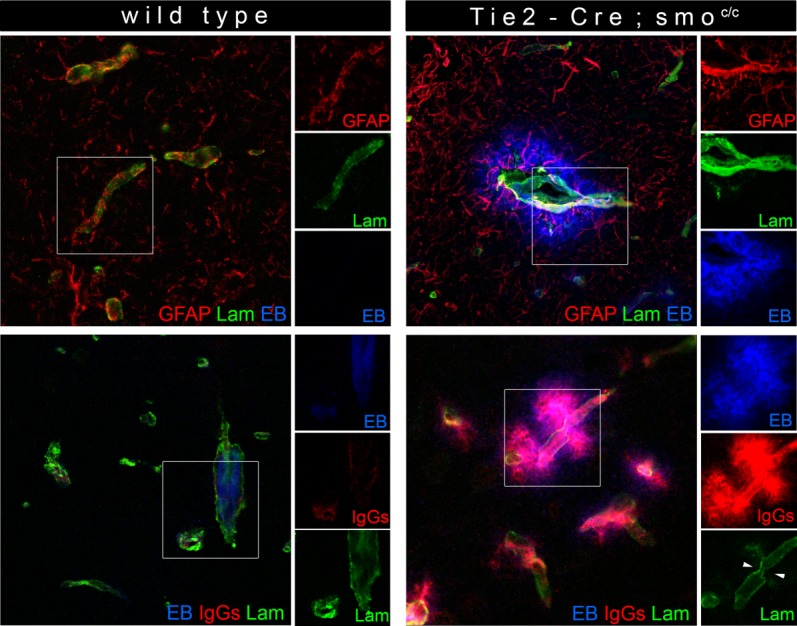
BBB permeability is compromised in mice lacking Hh signalling in CNS endothelium. Wild type (WT, left panels) and Tie2-Cre; Smoc/c (right panels) 8-week-old mice were intraperiteonally injected with Evans Blue (EB, blue). Brain cryosections from both groups were immunostained for GFAP (astrocytes, red) and laminin (Lam, basement membranes, green) (top panels), as well as for IgGs (indicators of BBB permeability, red (bottom panels)) and laminin (n = 3–5 animals). White rectangles indicate areas shown on the right of each panel. Arrowheads indicate areas showing basement membrane breakdown.
Astrocytes secrete angiogenic factors that promote vascular growth such as vascular endothelial growth factor (VEGF). VEGF binding to two tyrosine kinase receptors, VEGFR-1 (Flt-1) and VEGFR-2 (KDR/ Flk-1), elicits EC activation and mitosis (Robinson and Stringer, 2001; Shibuya, 2008) (Fig. 1). During development, VEGF is required for the formation, remodeling and survival of embryonic blood vessels as embryos lacking VEGF develop few or no angioblasts and die prematurely (Shalaby et al., 1995). As astrocytes are absent in early embryogenesis, radial glia cells seem to be a likely source of VEGF during BBB development (Virgintino et al., 2003). However, ECs have been described to promote cell-autonomous activation of the VEGF signaling pathway that during homeostasis is able to sustain a vascular phenotype (Lee et al., 2007a). In addition, neurons are also a source of VEGF (Shibuya, 2008), though their role in development or maintenance of the BBB phenotype via VEGF production remains to be determined. Thus, VEGF is a factor mostly known to promote angiogenesis and EC activation during development, while in adulthood it affects the phenotype and stability of the BBB upon inflammatory conditions (Argaw et al., 2009).
The angiopoietins (Ang-1, 2, 3, and 4) constitute a family of growth factors able to influence endothelial function by binding to the receptor tyrosine kinase Tie-2. Perivascular cells, including astrocytes, through the secretion of Ang-1 participate in the process of BBB differentiation by promoting angiogenesis, enhancing vascular stabilization and inducing a time- and dose-dependent decrease in endothelial permeability via the upregulation of junctional protein expression (Pfaff et al., 2006; Suri et al., 1996) (Fig. 1). In contrast, Ang-2 which is an autocrine multifunctional factor produced by ECs is known to function as a driver of postnatal vascular remodeling in the eye (Gale et al., 2002), to promote angiogenesis, to participate in the early phases of BBB breakdown (Nourhaghighi et al., 2003) (Fig. 1) and to act as protective factor that reduces barrier permeability in stressed ECs. Interestingly, when factors known to compromise BBB function such as VEGF are co-expressed with Ang-1, the barrier integrity is enhanced and neuroprotective properties are induced (Shen et al., 2011).
Src suppressed C kinase substrate (SSeCKS) is a factor produce by astrocytes, that increases gradually during BBB maturation. SSeCKS regulates angiogenesis and influences the BBB phenotype by decreasing the expression of VEGF, by stimulating astrocyte-Ang-1 expression, by augmenting the expression of ZO-1 and claudin-1 and by decreasing the permeability of cerebral EC monolayers (Lee et al., 2003) (Fig. 1). Surprisingly, no further evaluation or understanding of SSeCKS's role in controlling BBB under normal or pathological conditions has emerged since the original publication more than 10 years ago. This leaves the subject of the exact role of SSeCKS in disease conditions opened for discussion. Astrocytes also produce the angiotensin converting enzyme-1 (ACE-1), which converts angiotensin I into angiotensin II and acts on type 1 angiotensin receptors (AT1) expressed by BBB-ECs. Angiotensin II induces tightening of vessels increasing blood pressure (Lavoie and Sigmund, 2003) and at the CNS, activation of AT1 restricts BBB permeability and this correlates with stabilization of junctional protein function (Fig. 1). AGT deficient mice showed an aberrant expression of occludin at the BBB level and this correlated with a leaky BBB, suggesting that astrocytes via Angiotensin II promote TJ formation at BBB-ECs (Wosik et al., 2007).
A common denominator of the astroglial factors influencing BBB function is their well-documented involvement in CNS neural cell development. The family of fibroblast growth factors (FGFs) is recognized to support multiple mechanisms during CNS embryogenesis and adulthood. Astrocytes express FGF receptors and FGF-2 has been described as a major candidate in the autocrine and/or paracrine regulation of astrocyte differentiation (Reuss et al., 2000) (Fig. 1). Studies using FGF-2−/− and FGF5−/− single and FGF-2−/−/FGF-5−/− double mutant mice demonstrated that astroglial differentiation is compromised in the absence of FGF-2 and FGP5 and this results in reduced expression of junctional proteins, leakage of serum derived factors and in general a defective BBB (Reuss et al., 2003). More recent studies have strengthened the role of FGFs in supporting vascular integrity: suppression of FGF signaling was shown to promote the dissociation of junctional proteins in both arterial and venous vasculature (Fig. 1). This inhibition induced p120-catenin and VE-cadherin uncoupling that led to VE-cadherin internalization, to the loss of endothelial cell-cell contact and compromised the endothelial barrier integrity (Murakami et al., 2008). However, the exact role of FGF factors in controlling astrocyte function influencing endothelial physiology remains to be established.
Glia-derived neurotropic factor (GDNF) is secreted by astrocytes and the GDNF receptor alpha 1 (GDNFR-α1) is preferentially expressed by BBB-ECs at the level of the cortex. BBB-ECs showed an increase in TEER and decreased permeability when treated with GDNF, suggesting the influence of this neurotrophic factor in modulating junctional protein expression at the BBB (Igarashi et al., 1999) (Fig. 1). In addition to astrocytes, pericytes of the BBB and of the Blood Nerve Barrier (BNB) are able to secrete GNDF and modulate BBB and BNB phenotype by increasing the expression of claudin-5 and the TEER of these barriers (Shimizu et al., 2012). As it occurs with the FGF family, it remains to be established how astrocytes induce expression of GDNF during development and homeostasis, how neurotrophic factors influence distinct and unique barrier properties of CNS ECs under normal and pathological conditions and what differences, if any, exist between the GDNF activation induced by astrocytes and pericytes.
TGF-β is a multifunctional cytokine involved in cell growth, embryogenesis, differentiation, morphogenesis, wound healing, apoptosis and immunomodulation (Blobe et al., 2000). In the CNS, TGF-β is neuroprotective and in vitro studies have shown its capacity to induce p-gp activity and to reduce BBB permeability (Dohgu et al., 2004). TGF-β is secreted by astrocytes and CNS-ECs and TGF-β is known to downregulate the extent of leukocyte transmigration across the endothelium (Fabry et al., 1995). However, in the last decade the immunoregulatory and neuroprotective role of TGF-β has been challenged as this molecule has been found to support astroglial responses during CNS injury and to be upregulated in numerous CNS disorders including MS, Alzheimer's disease, stroke, tumors and neuro-AIDS (Heinemann et al., 2012). In addition, TGF-β seems to be a crucial factor in the development of epileptic activity as it was shown to support aberrant transformation of astrocytes during the latent period of epileptogenesis (Heinemann et al., 2012). TGF-β is highly pleiotropic most probably due to the numerous signaling pathways resulting from TGF receptor activation, TGF receptors in different cells and under any given condition. For instance, TGF-β binding to the TGF-β type II receptor activates the EC-restricted ALK1 and the broadly expressed ALK-5. ALK1 via Smad1/5 transcription factors stimulates EC proliferation and migration, whereas ALK5 via Smad2/3 has the opposite effect (Lebrin et al., 2005) demonstrating that the response to TGF-β highly depends on the signaling mechanisms at play in given circumstances within individual ECs (Fig. 1). Therefore, the overwhelming pleomorphic roles of TGF-β do not currently allow to conclude on the exact role of astrocyte derived TGF-β in BBB physiology.
Retinoic acid (RA) derived from radial glia cells plays a crucial role in supporting neurogenesis during embryogenesis (Kornyei et al., 2007). Recent findings suggest that RA is secreted by astrocytes and its receptor, RA-receptor β (RARβ) is expressed by developing vasculature. In vitro RARβ-dependent activation induces barrier properties, including increase in TEER, expression of BBB related molecules such as VE-cadherin, P-gp, and ZO-1, while the expression of the permeability factor VEGF was downregulated (Fig. 1). In vivo pharmacologic inhibition of RARβ affected the differentiation of the murine vasculature that resulted in a perturbed BBB (Mizee et al., 2013). In addition, RA is known to regulate the Hh, Wnt and FGF pathways during CNS development (Halilagic et al., 2007; Paschaki et al., 2012) and as these pathways play crucial roles in BBB induction and maintenance is plausible that RA secreted by radial glial cells is a master upstream regulator of BBB development.
Thrombospondins (TSPs) are matricellular proteins well regarded by their role in supporting synapse formation (Eroglu, 2009). Astrocyte-secreted (TSP)-1 and TSP2 have anti-angiogenic properties, inhibit EC proliferation and have been suggested to promote vascular maturation (Park et al., 2008). TSP-2 influences ECM assembly via modulation of MMP levels and animals deficient in TSP-2 show deficiencies in ECM remodelling (Kyriakides and Maclauchlan, 2009). The expression and secretion of TSP-1/-2 is regulated by Meteorin (Park et al., 2008), a protein highly expressed by astrocyte endfeet during embryonic and post-natal development and known to be a potent neurotrophic factor able to affect glial cell differentiation (Nishino et al., 2004).
Apolipoprotein E (ApoE) is a glycoprotein with three isoforms, ApoE2, ApoE3, and ApoE4, with the latter being a major risk factor for Alzheimer disease (Mahley, 1988). Lipoprotein particles containing ApoE are mainly produced by astrocytes to deliver cholesterol and other essential lipids to neurons (Herz and Bock 2002). Mice deficient in ApoE spontaneously develop BBB and BNB breakdown that increases with age and equally depended from blood- and tissue-derived ApoE (Fullerton et al., 2001; Hafezi-Moghadam et al., 2007). Using a triple co-culture model of primary ECs, astrocyte and pericytes, Nishitsuji et al. demonstrated that astrocyte-derived ApoE4 regulates PKCη activity and the phosphorylation of occludin which reflected lower TEER readings in an ApoE4−/− BBB model. Likewise, BBB permeability was enhanced in ApoE4−/− mice (Nishitsuji et al., 2011). However, divergent data provided by a more recent study showed that astrocyte-secreted ApoE4 is able to disrupt BBB integrity by regulating the activation of the cyclophilin A–NF-kB–MMP-9 pathway in pericytes (Bell et al., 2012) (Fig. 1). This clearly indicates the presence of cross-communication pathways between distinct elements of the NVU, in this case the ECs, the pericytes and the astrocytes.
Most of the influence provided by contact or secreted factors is endothelial-centered and little attention has been given to the effect of BBB-EC derived factors to the phenotype of astrocytes and other cells in the neurovascular niche. In this regard, in vitro experiments have shown that BBB-ECs can modulate multiple aspects of astrocyte physiology especially via endothelins (ETs). ETs are a family of endothelium-derived peptides that signal through the receptors ETA and ETB, which are preferentially found on smooth muscle cells mediating vasoconstriction (Adner et al., 1994). ETB is also expressed by astrocytes and by means of in vitro and in vivo experiments it has been shown that exogenous activation of the ETB receptor increased the production of neurotrophic factors, especially those known to promote BBB properties such as BDNF, GDNF, Nerve Growth Factor (NGF) as well as neurotrophin-3 (NT-3) (Koyama et al., 2003, 2005; Wall, 2003) (Fig. 1). However, in these experiments a direct correlation between the production of these factors and the induction of barrier properties on BBB-ECs was not determined. In addition, endothelial ET1 has been reported to regulate the production of astrocytic VEGF-A and Ang-1, and in this setting the increased production of VEGF-A potentiates ET-induced astrocytic proliferation by an autocrine mechanism (Koyama et al., 2012) (Fig. 1).
All the above-mentioned factors provided by astrocytes have been reported to impact on some aspects of the physiology and the biology of the BBB. This highlights the extreme diversity of the CNS inputs needed to generate this physiological barrier and emphasizes the wide redundancy of molecular signals affecting junctional molecules, BBB formation, and stability. Despite such redundancy, these numerous signals can be integrated into a more general concept using knowledge available from the field of cell signaling (pathways). Although little of the information provided below (or in Fig. 3) come specifically from CNS-ECs, a large part of these signaling pathways are likely conserved among distinct cell types. Our review of the literature identified “central” signaling pathways and transcription factors with either barrier promoting properties (Wnt, Hh, Sox-18, nrf-2, ERG, Nkx2-1, and SP3/YY1) or with barrier disrupting effects (NF-κb, Snail, FoxO1, PKC, and eNOS (Fig. 3)). Within the signaling pathways promoting BBB functioning, Wnt and Hh seem to be dominant and cooperate in promoting a BBB phenotype. Wnt ligand binding to Frizzled/LRP5/6 receptor complexes activates β-Catenin which leads to transcription, expression and proper targeting of the junctional proteins claudin-3 and p120 to the cell membrane (Hong et al., 2010; Liebner et al., 2008). β-Catenin is also known to regulate the activity of Snail, which has a negative effect on the stability of p120/VE-cadherin complexes and on the expression of TJ molecules occludin and claudin-5. On the other hand, loss of the Wnt co-receptor Lrp5 has been associated with downregulation of claudin-5 expression (Chen et al., 2011). The Hh signaling pathway appears to drive the transcription and expression of junctional proteins, but also dampens inflammatory responses on CNS-ECs. Activation of Gli-1 by the Hh ligands is reported to activate Sox-18 (Alvarez et al., 2011b), a transcription factor known to control claudin-5 expression and barrier function (Fontijn et al., 2008) and wnt signaling also influences Sox-18 expression. Likewise, activation of the Hh and Wnt pathways also promote the expression of Nkx2-1 through Gli independent mechanisms and via β-catenin (Gilbert-Sirieix et al., 2011; Gulacsi and Anderson 2006). Nkx2-1 (also known as TTF-1) transcriptionally regulates occludin expression (Runkle et al., 2012). In addition, β-catenin can influence the Hh signaling pathway by enhancing the transcriptional activity of Gli (Maeda et al., 2006). Wnt and Hh activation also induce the expression of NR2F2, a transcription factor known to promote Ang-1 expression, which promotes junctional protein expression and dampens inflammatory responses supported by the Nf-kB pathway (Lee et al., 2007b; Okamura et al., 2009). In contrast, NR2F2 is known to downregulate the expression of Ang-2, a factor known to negatively impact on junctional protein expression on BBB-ECs. Collectively, these findings suggest that Wnt/β-Catenin and Gli pathways could converge or synergize. Surprisingly, the additional signaling pathways/transcription factors promoting AJ and TJ protein expression or stability do not seem to cooperate or influence each other. For example, activation of the nrf-2 pathway by oxidative stress activates antioxidant response elements (ARE) known to promote the accumulation of NADPH-dehydrogenase Quinone-1 (NQO-1) and Glutathione which are known to stabilize ZO-1, occludin and claudin-5 expression (Fan et al., 2013). Nrf2 translocation to the nucleus occurs in response to activation by the protein kinase C and the Map kinase pathways (McCubrey et al., 2006; Nguyen et al., 2009) and independently of the Hh and Wnt signaling pathways. In addition, nrf-2 protects ECs during injury by supressing the expression of inflammatory genes (Chen et al., 2006). The transcription factors SP3 and YY1 have been reported to sustain transcription and stabilise the expression of claudin-5 (SP3) and occludin (YY1) (Sade et al., 2009), though the factors and signals controlling their expression at the CNS endothelium remain to be identified. The ETS related gene (ERG) is a transcription factor constitutively expressed by ECs that controls their permeability and the expression of the junctional proteins VE-cadherin and claudin-5 (Birdsey et al., 2008; Yuan et al., 2012). Interestingly, the expression of ERG is under the control of VE-cadherin and ERG can act as a transcriptional repressor of the Nf-KB pathway and other inflammatory genes (Sperone et al., 2011). Thus, signaling pathways and transcription factors supporting barrier function also tend to promote anti-inflammatory responses. Conversely, inflammation activates the NF-κB pathway through Akt, PKC(isomers) Fyn/Lyn and Rip which have a direct negative impact on the expression, transcription and stability of occludin, ZO-1 and claudin-5 (Aveleira et al., 2010; Stamatovic et al., 2008), as well as an indirect effect via activation of Snail (Wu et al., 2009b), which disrupts p120/VE-Cadherin complexes and decreases expression of occludin and claudin-5 (Ikenouchi et al., 2003; Ohkubo and Ozawa 2004). In addition Iκκ-α negatively regulates β-catenin and Iκκ-β associates with β-catenin to induce cell proliferation via cyclin D1, therefore reducing cell-cell interactions (Albanese et al., 2003; Lamberti et al., 2001). VEGFR signaling can also impact on barrier function by activating the PKC and eNOs pathways and thus affecting occludin and claudin-5 expression (Argaw et al., 2009, 2012). Interestingly, NF-κB activation has been reported to induce expression of Shh, which can promote barrier function via Smo and Gli-1 (Kasperczyk et al., 2009). Finally, Foxo1 is a transcription factor that downregulates claudin-5 expression, however such mechanism is subject to a remarkable level of transcriptional regulation as cytoplasmic VE-cadherin and β-catenin can independently repress Foxo1 activity (Jang et al., 2011; Kamo et al., 2013; Taddei et al., 2008). Taken together, available information suggest that NF-κB and Snail might be dominant negative regulators of BBB functioning, while the Wnt and Hh signaling pathways as well as Nrf-2 and ERG appear to be dominant promoters, or at least facilitators, of an optimal BBB phenotype (Fig. 3).
FIGURE 3.
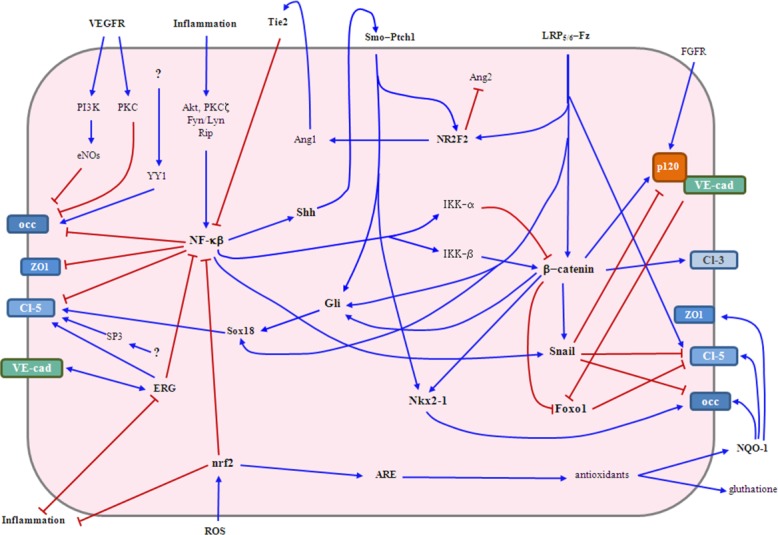
Signaling pathways and transcription factors supporting or affecting the barrier phenotype of CNS-ECs. The Wnt and Hh signaling pathways as well as the nrf-2 and ERG transcription factors appear to be playing a dominant role in promoting barrier properties at the junctional complex level. The Nf-κβ signaling pathway and the transcription factor snail seem to act as dominant negative regulators. Additional factors promoting or affecting the expression of junctional molecules and the BBB phenotype are included. Blue arrows indicate activation/stimulation and red lines denote downregulation/blockage. Akt (protein kinase B), Ang-1-2 (angiopoietin 1 and 2), ARE (antioxidant response elements), Cl-3 (claudin-3), Cl-5 (claudin-5), eNOs (endothelial nitric oxide synthase), ERG (ETS-related gene), Foxo1 (Forkhead box protein O1), Fz (frizzled), Gli (glioma-associated oncogene family zinc finger), IKK-α and -β (inhibitor of kappa B kinase α and β), LRP5/6 (low-density lipoprotein receptor-related protein 5), NF-κβ (nuclear factor kappa B), Nkx2-1 (NK2 homeobox 1), NQO-1 (NADPH-dehydrogenase quinone-1), NR2F2 (nuclear receptor subfamily 2, group F, member 2), nrf-2 (nuclear factor (erythroid-derived 2)-like 2), occ (occludin), PI3K (Phosphatidylinositide 3-kinase), PKC (Protein kinase C), Ptch1 (Patched1), ROS (reactive oxygen species), Shh (Sonic Hegdehog), Smo (smoothened), Tie2 (receptor tyrosine kinase 2), VE-cad (VE-cadherin), VEGFR (vascular endothelial growth factor receptor), YY1 (Ying and Yang 1 protein) and ZO1 (zonula occludens 1).
Therefore, the current view, which is based on an overwhelming number of scientific articles, is that astrocytes are key regulators of BBB development, maintenance, and regulation. The natural extension of this concept would be that understanding the array of astrocyte-BBB-EC interactions may lead to the development of novel therapeutic strategies for CNS disorders, especially those in which BBB function is compromised. However, given the multiple cells involved in controlling barrier function at the NVU, more data is needed to decipher the distinct (and maybe specific) roles of astrocytes, pericytes, neurons and microglia, as well as to further document the interactions of their cellular machineries in supporting BBB function.
Astrocyte and BBB Responses During Inflammatory CNS Diseases
The CNS microenvironment is very tightly controlled and even any minor perturbances in such delicate balance induce rapid phenotypical changes in astrocytes known as reactive astrogliosis. Due to their extraordinary capacity to quickly respond to adverse signals, many efforts have been made in understating how astrocytes respond to injury, inflammation, or any adverse condition in their vicinity. As there is a wide range of changes occurring under pathophysiological conditions, reactive astrogliosis has been defined as a series of inter-independent features in which (i) a spectrum of morphologic and molecular changes occur in response to CNS injury and disease, (ii) correlates with the nature and severity of the insult, (iii) it is regulated in a context-specific manner by particular signaling events that have the potential to modify both the nature and degree of those changes and (iv) has the potential to induce either gain or loss of function of astrocytes and therefore affect the surrounding neural and non-neural cells (Sofroniew, 2009). In this regard, the influence of distinct CNS microenvironments to the reactive phenotype seen in astrocytes remains to be established, as well as the variability in terms of reactivity according to the astrocyte type studied. In general terms, astrogliosis is characterized by changes associated with hypertrophy of cell bodies and glial processes as well as upregulation of the intermediate filaments vimentin and GFAP and the ECM components tenascin-C and chondroitin sulfate proteoglycans (CSPGs) (Ridet et al., 1997; Robel et al., 2011). Of interest, reactive astrocytes express nestin, CD15, DSD1 proteoglycan, and brain-lipid lipid-binding protein (BLBP), resembling the phenotype of radial glial cells and neural stem cells (Robel et al., 2011). Some of these cells also display long-term self renewal and multipotency when compared to astrocytes from a non-injury site (Buffo et al., 2008) and recently it has been shown that Shh is necessary and sufficient to induce such stem cell responses in astrocytes. Depending on the extent of injury or inflammation, astrocytes can proliferate and this is reflected by the high number of cells found in the proximity of a lesion, injury or glial scar (Sofroniew, 2009). However, recent findings using in vivo two-photon laser scanning microscopy showed that upon injury not all astrocytes proliferate and migrate. Astroglial proliferation was found to be predominant in proximity to the injury and selectively on astrocytes opposing the CNS vasculature (Bardehle et al., 2013). The close interaction of astrocytes with ECs maintains their endfeet polarized towards the vasculature serving as injury sensors (Robel et al., 2011), which in part explains their preferential proliferation upon injury. The loss of signaling between astrocytes and CNS vasculature can trigger most of the signaling programme associated with reactive astrogliosis. In particular, post-natal deletion of β1 integrin in astrocytes is sufficient to depolarize the endfeet and induce massive changes that include hypertrophy, upregulation of intermediate filaments, alterations in the localization of DAP members and the water channel AQP4, as well as secretion of chondroitin sulphate proteoglycans (Robel et al., 2009). Again, these changes directly correlated with vascular perturbances as fibrinogen, a soluble plasma protein entering the CNS upon BBB disruption, modulates the expression of both TGF-β and molecules associated with the phenotype of reactive astrocytes (Schachtrup et al., 2010). However, this activation pathway and pattern are not unique for astrocytes as fibrinogen induces rapid microglial responses toward the vasculature (Davalos et al., 2012).
As a result of the reactive phenotype, astrocytes influence the pathophysiological response of the CNS to injury by producing a wide range of molecules that include neurotrophic factors, growth factors, cytokines, chemokines, neurotransmitters, reactive oxygen species, and proteases (Sofroniew, 2009). These factors can influence the remodeling of CNS vasculature, the state of activation of migrating leukocytes and depending on the context they can have beneficial and/or detrimental effects on neurons and oligodendrocytes (Allman et al., 1994; Koyama and Michinaga, 2012). However, most studies characterizing these factors have been carried out using in vitro methodologies designed to understand reactive astrogliosis in the context of injury and have not taken into account the extent of astrocyte heterogeneity within distinct anatomical areas of the CNS.
In the following section the pathophysiological role of astrocytes within the NVU will be discussed in terms of the neuroinflammatory responses observed during neuromyelitis optica (NMO) as well as multiple sclerosis (MS) and its animal model experimental autoimmune encephalomyelitis (EAE).
Neuromyelitis optica, multiple sclerosis, and EAE
In most CNS pathologies the NVU/BBB microniche is affected as a result of the inflammation, injury or degeneration processes specific to the pathology. However, in only a few diseases the NVU/BBB is specifically targeted by the pathogenic process or by the disease determinants. NMO and MS are amongst these diseases.
In NMO, the astrocyte water channel AQP4 is targeted by the immune system, resulting in the production of anti-AQP4 IgG antibodies (Lennon et al., 2004). The disease directly affects BBB function as AQP4 OAPs are present on the astrocyte endfeet surrounding the CNS vasculature. Binding of anti-AQP4 antibodies to their target result in the activation of complement-dependent cytotoxic cell damage that lead to the loss of AQP4, GFAP and the excitatory amino acid transporter2 (EAAT2) (Hinson et al., 2007, 2008). In addition, the BBB damage is associated with focal areas of perivascular immune cell infiltration and demyelination. In addition, the further deposition of APQ4-IgG complexes enhances immune cell infiltration, particularly granulocytes and eosinophils that degranulate in the perivascular space causing local damage that includes astrocyte injury. Oligodendrocytes appeared to be affected as a result of the pathophysiological changes in the NVU and this leads to myelin loss and axonal damage (Ratelade and Verkman, 2012). However, the exact mechanism(s) driving oligodendrocyte and neuronal damage due to the targeting of an astrocyte-related antigen remain to be determined.
In contrast to NMO, astrocytes are not classically regarded as a target of the immune system in MS, although BBB disruption and alterations in astrocyte physiology are hallmarks of MS pathogenesis. The etiology of MS remains elusive, but it is clear that multiple factors are involved disease development. Studies showing strong concordance between both dizygotic and monozygotic twins demonstrate that hereditary factors play an important role (Willer et al., 2003). MS susceptibility is strongly associated with the gene encoding the major histocompatibility complex (MHC) class II DRB1 gene (Hafler et al., 2007) and genome-wide association studies (GWAS) have identified additional risk alleles that indicate a strong involvement of the immune system and suggest a polygenic influence on the disease pathogenesis (Bush et al., 2010). Environmental factors such as Vitamin D and sunlight exposure are thought to be associated with MS prevalence as Vitamin D promotes a microenvironment that suppresses the activation of encephalitogenic T cells, downregulating autoimmune responses in the CNS (Farias et al., 2013; Simon et al., 2012). Various viruses have been suggested as triggers of MS with Epstein-Barr virus (EBV) showing the strongest association (Compston and Coles, 2008). MS and mononucleosis distribute similarly and it has been shown that MS risk increases tenfold in patients infected with EBV during childhood/teenage years, when compared to populations of EBV-seronegatives (Ascherio and Munger, 2007). Interestingly, the risk of developing MS increases if low levels of vitamin D and EBV exposure occur concomitantly (Ramagopalan et al., 2011).
MS is considered a T-cell–mediated disease in which CD4 T helper (Th) cells of the Th17 and Th1 phenotype play a fundamental role in its pathogenesis (Kebir et al., 2009). B cells are also essential in MS immunopathogenesis, as antibodies produced intrathecally are a fundamental feature of the disease (i.e. oligoclonal bands) and as B-cell–directed therapies provide strong protection against lesion formation (Cross and Waubant, 2011). Professional antigen presenting cells (APCs) are also recognized as important drivers of the immune activation process within the CNS and their phenotype is known to be shape by the NVU (Chastain et al., 2011; Ifergan et al., 2008). These and other immune cells need to migrate from the periphery to the CNS and although the mechanism used by distinct leukocytes to cross the NVU/BBB are not well understood, it is clear that during this process the barrier function becomes compromised (Larochelle et al., 2011). Such phenotype is characterized by BBB leakage which is associated with alterations of junctional proteins, as well BBB activation, which relates to the capacity of ECs, astrocytes, and possibly pericytes to express and secrete immune factors able to influence the recruitment, the effector functions and the survival of leukocytes entering the CNS (Alvarez et al., 2011a). Analysis of MS tissues demonstrates that abnormalities in the expression of junctional proteins coincide with perivascular astrogliosis and such changes are detected in very early stages of lesion formation (Alvarez et al., 2011a). This has been in part explored by Luo et al. when inducing active EAE in mice expressing luciferase under the control of GFAP. Despite showing clinical signs only at day 11, increases in bioluminescence associated with GFAP expression could be detected in the brain of these animals as early as 3 days post-induction, (Luo et al., 2008) suggesting that astrocytes are activated in the very early stages of EAE and in the absence of clinical signs of the disease. In this regard, we have analyzed the state of astrocyte activation in a spontaneous relapsing remitting EAE (sRR-EAE) model that closely resembles MS. These animals develop EAE symptoms around day 60 (Pollinger et al., 2009) and analysis of presymptomatic mice showed that at day 35 perivascular astrocytes show a reactive phenotype as GFAP expression is higher than in wild type littermates and in the absence of immune cell infiltration. The level of astrogliosis increases by day 45 and is associated with low levels of macrophage and T cell infiltration. By day 55, the immune cell infiltration increased with preferential recruitment in perivascular cuffs and this correlated with prominent astrogliosis on perivascular endfeet and parenchymal astrocytes (Fig. 4).
FIGURE 4.
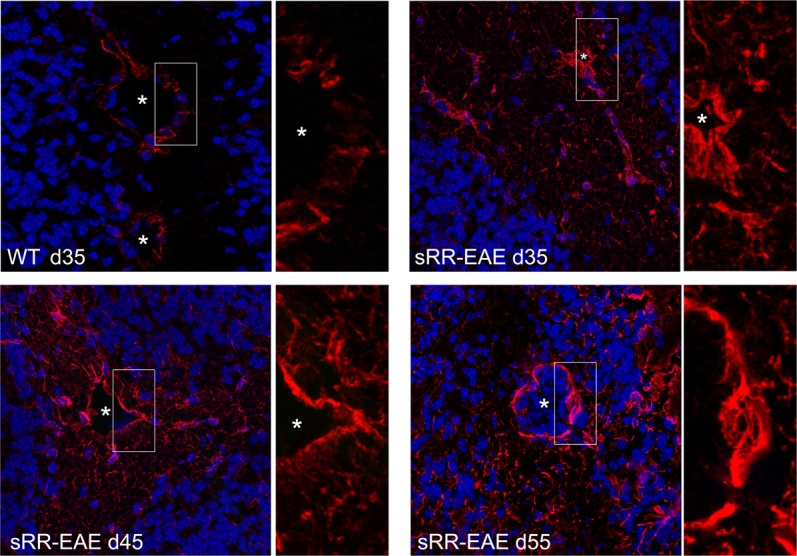
GFAP expression in asymptomatic spontaneous relapsing remitting EAE mice. Astrocytes were labeled with GFAP (red) and images were acquired by confocal microscopy and using the same settings. Representative images of GFAP in the cerebellar white matter of asymptomatic sRR-EAE mice at day (d)35, d45, and d55 are shown. Asterisks denote blood vessels and white rectangles indicate the high magnification areas shown on the right of each panel. Nuclear staining in blue. Scale bar: 50 μm.
Early disturbances of the BBB are associated with EC activation that enhances the process of leukocyte infiltration and leads to multifocal perivascular infiltrates. These infiltrates are characterized by the presence of important contributors of the inflammatory and demyelinating process, including APCs, cytotoxic CD8 T cells, and Th1 and Th17 CD4 T cells (Friese and Fugger, 2009; Ifergan et al., 2008; Kebir et al., 2009). While essentially protective for the CNS, the BBB/NVU can also promote inflammatory reactions by producing, secreting, expressing or transporting proinflammatory cytokines, chemokines, and adhesion molecules (Fig. 5). Therefore, besides its primary neuroprotective function, the BBB has also been shown to actively promote neuroinflammation by orchestrating immune responses during CNS-targeted autoimmune aggression. In fact, BBB-ECs can favor migration of immune cells to the CNS perivascular space by expressing E- and P-selectin (Engelhardt and Ransohoff, 2005), although their exact role in supporting the process of lymphocyte recruitment to the CNS remains a matter of debate. Furthermore, BBB-ECs are an important source of pro-inflammatory chemokines CCL2/MCP-1, CXCL8/IL-8 (Biernacki et al., 2001; Kebir et al., 2007), CCL5/RANTES and CXCL10/IP-10 (Ifergan et al., 2006) (Fig. 5), that are required for lymphocyte and monocyte recruitment to the CNS (Ransohoff et al., 2003). Immune cell infiltration into the CNS correlates with production of proinflammatory mediators such as IL-17, IL-22, GM-CSF, IFN-γ, and TNF-α (Alvarez et al., 2011a). These cytokines have been implicated in the modulation of EC function by upregulating the expression of proinflammatory mediators and by affecting the expression of junctional proteins and thus compromising BBB permeability (Alvarez et al., 2011a; Huppert et al., 2010; Kebir et al., 2007) (Fig. 5). Lastly and most importantly, upon activation with proinflammatory cytokines, BBB-ECs express intercellular adhesion molecule (ICAM)-1, ICAM-2, vascular CAM (VCAM)-1, activated leukocyte CAM (ALCAM), melanoma CAM (MCAM), and ninjurin-1 which mediate, at least in part, the adhesion process and transmigration of leukocytes and leukocyte subtypes to the CNS (Cayrol et al., 2008; Ifergan et al., 2011; Larochelle et al., 2012; Prat et al., 2000; Steiner et al., 2010; Wong and Dorovini-Zis, 1992) (Fig. 5). Additional effectors of leukocyte diapedesis include platelet endothelial cell adhesion molecule (PECAM) (Williams et al., 1996), JAM-A (Ostermann et al., 2002), CD73 and vascular associated protein-1 (VAP-1) (Jalkanen and Salmi, 2008) (Fig. 5) but their role in immune cell recruitment to the CNS is not clear. Thus, although the BBB protects against CNS-directed inflammation by restricting immune cell access to the brain, it can also regulate the local inflammatory response by expressing proinflammatory molecules that promote the recruitment of peripheral immune cells into the CNS.
FIGURE 5.

BBB changes during MS/EAE. During neuroinflammation, early BBB disturbances are associated with EC activation that is characterized by increased expression of E- and P-selectins, cell adhesion molecules (CAMs) including ALCAM, ICAM-1, ICAM-2, VCAM-1, MCAM, Ninjurin-1, VAP-1 and CD73 [1], chemokines (CCLs-CXCLs) [2] and cytokine receptors such as IFNR, TNFR, IL-17R, IL-22R, and GM-CSFR [3]. Such activation compromises the BBB phenotype by affecting junctional proteins [4] and enhances the expression of additional factors supporting the infiltration of T helper (Th) 1, Th17, and CD8 T cells as well as antigen presenting cells (APCs) [5] that accumulate in the perivascular space in a multifocal pattern. To gain access into the CNS, the infiltrating leukocytes secrete Matrix Metalloproteinases (MMPs) which target components of the astroglial basement membrane and dystroglycan (DG) at the astrocyte endfeet [6], during this process the polarity of the water channel AQP4 is perturbed leading to edema [7]. The endfeet depolarization activates the reactive gliotic program which results in increase expression of the intermediate filament GFAP [8]. Astrocytes also increase the expression of Shh [9] and ECs upregulate the Hh receptors Patched 1 (Ptch1) and Smoothened (Smo) [10] to repair the BBB and downregulate EC activation and leukocyte migration. Shh also modulates the phenotype of pathogenic Th cells by downregulating their expression of inflammatory cytokines and CAMs [11]. In the parenchymal milieu, microglia secrete IL-1β and activate the production of VEGF on astrocytes [12], which induces endothelial nitric oxidase synthase (eNOs) production and promotes junctional protein damage [13]. Finally, 50% of MS patients have autoantibodies against KIR4.1 that fix complement and induce the reactive gliosis program [14].
Upon migration across ECs, leukocytes cross the endothelial BM and subsequently the parenchymal BM to get access into the CNS (Sixt et al., 2001) (Fig. 5). The composition of the endothelial BM can regulate the extent of perivascular infiltration as large amounts of leukocytes are detected in vessels expressing laminin 411 and low levels of 511, while in the absence of 411, laminin 511 is ubiquitously expressed and associates with low T cell infiltration and milder disease (Wu et al., 2009a). In EAE and MS, immune cell infiltrates are in great part contained to the perivascular space and the process of leukocyte migration across the parenchymal BM and astrocyte endfeet appears to be more tightly controlled than the diapedesis across ECs (Engelhardt and Sorokin, 2009). In EAE, CD4+ T cell infiltration across the parenchymal BM is not laminin dependent, but rather requires focal MMP-2 and MMP-9 to selectively cleave dystroglycan, affecting the BM stability and integrity (Agrawal et al., 2006) (Fig. 5). Interestingly, parenchymal BM components and other ECM-binding receptors on the astrocyte endfeet remain unaffected, indicating the existence of specific and specialized protective mechanisms under the control of astrocytes and possibly other cells within the NVU (Engelhardt and Sorokin, 2009). Thus, further understanding is needed in terms of astrocyte involvement in supporting or inhibiting the activation and migration of immune cells, as well as the repair of the affected BBB/NVU during MS/EAE and other CNS disorders.
Astrocytes have been traditionally seen as promoters of CNS inflammation in MS and little is known about their anti-inflammatory role. Recently, both the Khoury group in Boston (Wang et al., 2008) and our group in Montreal (Alvarez et al., 2011b) have demonstrated that reactive perivascular astrocytes found in MS lesions up-regulate Shh (Fig. 5). Surprisingly, Shh was found to promote immune quiescence of human BBB-ECs by down-regulating secretion of proinflammatory chemokines CCL2, the expression of cell adhesion molecules ICAM-1 and VCAM-1 and the migration of immune cells, a phenomenon that is dysregulated during neuroinflammation (Fig. 5). In addition, Shh was found to impact on cytokine production of local infiltrating Th1, Th2 and Th17 lymphocytes (Fig. 5). Finally, blockade of the Hh pathway during the course of EAE, increased disease morbidity and this detrimental outcome was associated with an exacerbated neuroinflammatory response, suggesting that an active Hh pathway is needed to mitigate auto-inflammatory responses in the CNS. Thus, these data indicate that Hh activation at the level of the NVU provides barrier-promoting effects and an endogenous anti-inflammatory balance to CNS-directed immune attack (Alvarez et al., 2011b).
Conversely, reactive astrocytes can be the source of factors that will negatively affect barrier function at the NVU. In MS and EAE, VEGF-A is expressed by reactive astrocytes and in vitro/in vivo studies demonstrate its capacity to induce BBB breakdown by disrupting claudin-5 and occludin expression and thus promoting immune cell infiltration to the CNS (Argaw et al., 2009). Additional studies propose that IL-1β production by microglia induces VEGF-A upregulation (Fig. 5). VEGF-A is released from the astrocytes and binding to its receptor VEGFR2 on BBB-ECs activates eNOS-dependant downregulation of the junctional proteins claudin-5 and occludin that leads to BBB breakdown (Argaw et al., 2012) (Fig. 5). The authors also examined the role of Hypoxia-inducible factor (HIF)-1α, a molecule that induces VEGF expression and is upregulated in reactive astrocytes in vitro and in MS lesions (Argaw et al., 2006). However, mice specifically lacking HIF-1α on astrocytes displayed little effect on VEGF-A expression or BBB disruption, indicating that mechanisms regulating VEGF-A production by astrocytes in acute conditions like EAE differ to those seen in MS.
While reactive astrocytes can produce BBB-promoting (i.e. Hh) or BBB-disrupting (i.e. VEGF) factors, they can also loose or down-regulate factors which have the capacity to promote barrier function. In this regard, astrocytes produce AGT (which is cleaved into Angiotensin II) and analysis of MS tissues showed that expression of AGT in astrocytes and occludin in ECs is decreased in MS lesions when compared to normal appearing white matter. This pattern correlates with the downregulated expression of AGT detected in astrocytes stimulated in vitro with IFN-γ and TNF-α. Interestingly, nonimmunized (non-EAE) AGT-deficient mice have compromised BBB function which correlates with decreased and disrupted expression of occludin. Therefore local inflammatory mediators present in perivascular cuffs can also negatively impact on the capacity of reactive astrocytes to promote BBB function, by down-regulating their production of BBB-promoting factors (Wosik et al., 2007).
Non-cytokine immune factors can also have a direct negative influence on astrocyte- EC interactions. The study of serum derived from MS patients led Srivastava et al. to find anti-KIR4.1 specific IgGs in about 50% of the patients (Srivastava et al., 2012). These IgGs were demonstrated to bind to astrocyte endfeet and can activate the complement system (Fig. 5). In vivo validation of the pathogenic role of anti-KIR4.1 antibodies was provided by injecting anti-KIR4.1 IgGs in mice; this led to loss of KIR4.1 expression, activation of the complement cascade and alterations in GFAP expression on astrocytes. Thus, the study suggests that autoantibodies present in the serum of a yet-to-be-confirmed proportion of human MS patients are directed against KIR4.1. This autoimmune and antibody-dependent cell-mediated cytotoxicity interferes with the local K+ buffering at the NVU and could be the cause of lesion initiation or tissue damage. Further studies will be needed to determine if KIR4.1 autoimmunity is specific to MS, to a subtype of the disease or if generation of anti-KIR4.1 antibodies is the result (i.e. is secondary) of the immune response occurring at the neurovascular niche.
Nevertheless, important divergences in the anti-KIR4.1 and anti-AQP4 responses are very informative of the biology of astrocytes and the NVU. Interestingly, KIR4.1 and AQP4 are co-localized on astrocyte endfeet and are two molecules associated with both BBB/NVU physiology and pathology. Anti-KIR4.1 antibodies seem to be associated with MS, while anti-AQP4 antibodies are clearly associated with NMO. However, loss of polarized expression of AQP4 is seen in CNS vessels displaying perivascular inflammation as well as dystroglycan disruption and these pathological changes contribute to edema development (Fukuda and Badaut, 2012; Wolburg et al., 2009) (Fig. 5). These two inflammatory diseases have distinct clinical courses, treatment responses, and pathological features. Therefore, targeting different determinants on astrocytes endfeet can lead to distinct pathological and clinical diseases, even if these antigens are expressed by the exact same cells. These important biological, pathological, and clinical discrepancies remain unexplained.
In conclusion, accumulating evidence reported over the last hundred years point to the critical influence of perivascular astrocytes on the anatomical and functional integrity of the BBB. Identification of the molecular pathways involved in the communication between astrocytes and BBB-ECs is however slowly progressing. This is in part due to important biological differences between species (artiodactyla vs. rodentia vs. human and non-human primates), differences between the in vitro and in vivo models used by different groups, as well as the in vivo relevance of some in vitro models, the various levels of redundancy in the function of the molecular pathways, the different astrocyte populations studied, the absence of a single, unique or specific phenotypic/molecular determinant for BBB-ECs and the immense complexity of new genetic tools used to knock-down or knock in specific molecules in distinct cell populations. To identify the array of factors that are involved in astrocyte-BBB-EC communication in the human brain, additional research efforts and novel technologies will be needed.
Acknowledgments
Grant sponsor: MS Society of Canada (MSSC); Grant sponsor: CIHR; Grant numbers: MOP 89885 and MOP14828; Grant sponsors: MSSC (to J.I.A.), CIHR Strategic Training Initiative in Health Research Neuroinflammation Training Program (to T.K.), and Fonds de la recherche en santé du Québec (FRSQ) (to A.P.).
The authors thank Casper Briels for helpful discussions and Alejandro Alvarez Espitia for his collaboration during the preparation of the figures for the manuscript.
References
- Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- Adams RH. Vascular patterning by Eph receptor tyrosine kinases and ephrins. Semin Cell Dev Biol. 2002;13:55–60. doi: 10.1006/scdb.2001.0289. [DOI] [PubMed] [Google Scholar]
- Adner M, Jansen I, Edvinsson L. Endothelin-A receptors mediate contraction in human cerebral, meningeal and temporal arteries. J Auton Nerv Syst. 1994;49(Suppl):S117–S121. doi: 10.1016/0165-1838(94)90098-1. [DOI] [PubMed] [Google Scholar]
- Agrawal S, Anderson P, Durbeej M, van Rooijen N, Ivars F, Opdenakker G, Sorokin LM. Dystroglycan is selectively cleaved at the parenchymal basement membrane at sites of leukocyte extravasation in experimental autoimmune encephalomyelitis. J Exp Med. 2006;203:1007–1019. doi: 10.1084/jem.20051342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albanese C, Wu K, D'Amico M, Jarrett C, Joyce D, Hughes J, Hulit J, Sakamaki T, Fu M, Ben-Ze'ev A, Bromberg JF, Lamberti C, Verma U, Gaynor RB, Byers SW, Pestell RG. IKKalpha regulates mitogenic signaling through transcriptional induction of cyclin D1 via Tcf. Mol Biol Cell. 2003;14:585–599. doi: 10.1091/mbc.02-06-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allman BE, Klein AG, Nugent KA, Opat GI. Refractive-index-profile determinations by using Lloyd's mirage. Appl Opt. 1994;33:1806–1811. doi: 10.1364/AO.33.001806. [DOI] [PubMed] [Google Scholar]
- Alvarez JI, Cayrol R, Prat A. Disruption of central nervous system barriers in multiple sclerosis. Biochim Biophys Acta. 2011a;1812:252–264. doi: 10.1016/j.bbadis.2010.06.017. [DOI] [PubMed] [Google Scholar]
- Alvarez JI, Dodelet-Devillers A, Kebir H, Ifergan I, Fabre PJ, Terouz S, Sabbagh M, Wosik K, Bourbonniere L, Bernard M, van Horssen J, de Vries HE, Charron F, Prat A. The Hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science. 2011b;334:1727–1731. doi: 10.1126/science.1206936. [DOI] [PubMed] [Google Scholar]
- Amiry-Moghaddam M, Ottersen OP. The molecular basis of water transport in the brain. Nat Rev Neurosci. 2003;4:991–1001. doi: 10.1038/nrn1252. [DOI] [PubMed] [Google Scholar]
- Argaw AT, Asp L, Zhang J, Navrazhina K, Pham T, Mariani JN, Mahase S, Dutta DJ, Seto J, Kramer EG, Ferrara N, Sofroniew MV, John GR. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J Clin Invest. 2012;122:2454–2468. doi: 10.1172/JCI60842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argaw AT, Gurfein BT, Zhang Y, Zameer A, John GR. VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc Natl Acad Sci USA. 2009;106:1977–1982. doi: 10.1073/pnas.0808698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argaw AT, Zhang Y, Snyder BJ, Zhao ML, Kopp N, Lee SC, Raine CS, Brosnan CF, John GR. IL-1beta regulates blood-brain barrier permeability via reactivation of the hypoxia-angiogenesis program. J Immunol. 2006;177:5574–5584. doi: 10.4049/jimmunol.177.8.5574. [DOI] [PubMed] [Google Scholar]
- Ascherio A, Munger KL. Environmental risk factors for multiple sclerosis. Part II: Noninfectious factors. Ann Neurol. 2007;61:504–513. doi: 10.1002/ana.21141. [DOI] [PubMed] [Google Scholar]
- Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA. Glial and neuronal control of brain blood flow. Nature. 2010;468:232–243. doi: 10.1038/nature09613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwell D, Laughlin SB. An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab. 2001;21:1133–1145. doi: 10.1097/00004647-200110000-00001. [DOI] [PubMed] [Google Scholar]
- Aveleira CA, Lin CM, Abcouwer SF, Ambrosio AF, Antonetti DA. TNF-alpha signals through PKCzeta/NF-kappaB to alter the tight junction complex and increase retinal endothelial cell permeability. Diabetes. 2010;59:2872–2882. doi: 10.2337/db09-1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axnick J, Lammert E. Vascular lumen formation. Curr Opin Hematol. 2012;19:192–198. doi: 10.1097/MOH.0b013e3283523ebc. [DOI] [PubMed] [Google Scholar]
- Ballabh P, Hu F, Kumarasiri M, Braun A, Nedergaard M. Development of tight junction molecules in blood vessels of germinal matrix, cerebral cortex, and white matter. Pediatr Res. 2005;58:791–798. doi: 10.1203/01.PDR.0000180535.14093.FB. [DOI] [PubMed] [Google Scholar]
- Bar T. The vascular system of the cerebral cortex. Adv Anat Embryol Cell Biol. 1980;59:1–62. doi: 10.1007/978-3-642-67432-7. I-VI. [DOI] [PubMed] [Google Scholar]
- Bardehle S, Kruger M, Buggenthin F, Schwausch J, Ninkovic J, Clevers H, Snippert HJ, Theis FJ, Meyer-Luehmann M, Bechmann I, Dimou L, Gotz M. Live imaging of astrocyte responses to acute injury reveals selective juxtavascular proliferation. Nat Neurosci. 2013;16:580–586. doi: 10.1038/nn.3371. [DOI] [PubMed] [Google Scholar]
- Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R, Zlokovic BV. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron. 2010;68:409–427. doi: 10.1016/j.neuron.2010.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, Holtzman DM, Betsholtz C, Armulik A, Sallstrom J, Berk BC, Zlokovic BV. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485:512–516. doi: 10.1038/nature11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bicknell R, Harris AL. Novel angiogenic signaling pathways and vascular targets. Annu Rev Pharmacol Toxicol. 2004;44:219–238. doi: 10.1146/annurev.pharmtox.44.101802.121650. [DOI] [PubMed] [Google Scholar]
- Biernacki K, Prat A, Blain M, Antel JP. Regulation of Th1 and Th2 lymphocyte migration by human adult brain endothelial cells. J Neuropathol Exp Neurol. 2001;60:1127–1136. doi: 10.1093/jnen/60.12.1127. [DOI] [PubMed] [Google Scholar]
- Birdsey GM, Dryden NH, Amsellem V, Gebhardt F, Sahnan K, Haskard DO, Dejana E, Mason JC, Randi AM. Transcription factor Erg regulates angiogenesis and endothelial apoptosis through VE-cadherin. Blood. 2008;111:3498–3506. doi: 10.1182/blood-2007-08-105346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. N Engl J Med. 2000;342:1350–1358. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- Briscoe J, Sussel L, Serup P, Hartigan-O'Connor D, Jessell TM, Rubenstein JL, Ericson J. Homeobox gene Nkx2.2 and specification of neuronal identity by graded Sonic hedgehog signalling. Nature. 1999;398:622–627. doi: 10.1038/19315. [DOI] [PubMed] [Google Scholar]
- Buffo A, Rite I, Tripathi P, Lepier A, Colak D, Horn AP, Mori T, Gotz M. Origin and progeny of reactive gliosis: A source of multipotent cells in the injured brain. Proc Natl Acad Sci USA. 2008;105:3581–3586. doi: 10.1073/pnas.0709002105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush WS, Sawcer SJ, de Jager PL, Oksenberg JR, McCauley JL, Pericak-Vance MA, Haines JL. Evidence for polygenic susceptibility to multiple sclerosis--The shape of things to come. Am J Hum Genet. 2010;86:621–625. doi: 10.1016/j.ajhg.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd N, Grabel L. Hedgehog signaling in murine vasculogenesis and angiogenesis. Trends Cardiovasc Med. 2004;14:308–313. doi: 10.1016/j.tcm.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Cayrol R, Wosik K, Berard JL, Dodelet-Devillers A, Ifergan I, Kebir H, Haqqani AS, Kreymborg K, Krug S, Moumdjian R, Bouthillier A, Becher B, Arbour N, David S, Stanimirovic D, Prat A. Activated leukocyte cell adhesion molecule promotes leukocyte trafficking into the central nervous system. Nat Immunol. 2008;9:137–145. doi: 10.1038/ni1551. [DOI] [PubMed] [Google Scholar]
- Chastain EM, Duncan DS, Rodgers JM, Miller SD. The role of antigen presenting cells in multiple sclerosis. Biochim Biophys Acta. 2011;1812:265–274. doi: 10.1016/j.bbadis.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Stahl A, Krah NM, Seaward MR, Dennison RJ, Sapieha P, Hua J, Hatton CJ, Juan AM, Aderman CM, Willett KL, Guerin KI, Mammoto A, Campbell M, Smith LE. Wnt signaling mediates pathological vascular growth in proliferative retinopathy. Circulation. 2011;124:1871–1881. doi: 10.1161/CIRCULATIONAHA.111.040337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XL, Dodd G, Thomas S, Zhang X, Wasserman MA, Rovin BH, Kunsch C. Activation of Nrf2/ARE pathway protects endothelial cells from oxidant injury and inhibits inflammatory gene expression. Am J Physiol Heart Circ Physiol. 2006;290:H1862–H1870. doi: 10.1152/ajpheart.00651.2005. [DOI] [PubMed] [Google Scholar]
- Choi BH, Lapham LW. Radial glia in the human fetal cerebrum: A combined Golgi, immunofluorescent and electron microscopic study. Brain Res. 1978;148:295–311. doi: 10.1016/0006-8993(78)90721-7. [DOI] [PubMed] [Google Scholar]
- Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372:1502–1517. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- Connors NC, Adams ME, Froehner SC, Kofuji P. The potassium channel Kir4.1 associates with the dystrophin-glycoprotein complex via alpha-syntrophin in glia. J Biol Chem. 2004;279:28387–28392. doi: 10.1074/jbc.M402604200. [DOI] [PubMed] [Google Scholar]
- Coomber BL, Stewart PA. Morphometric analysis of CNS microvascular endothelium. Microvasc Res. 1985;30:99–115. doi: 10.1016/0026-2862(85)90042-1. [DOI] [PubMed] [Google Scholar]
- Cross AH, Waubant E. MS and the B cell controversy. Biochim Biophys Acta. 2011;1812:231–238. doi: 10.1016/j.bbadis.2010.07.020. [DOI] [PubMed] [Google Scholar]
- Daneman R, Agalliu D, Zhou L, Kuhnert F, Kuo CJ, Barres BA. Wnt/beta-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc Natl Acad Sci USA. 2009;106:641–646. doi: 10.1073/pnas.0805165106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature. 2010;468:562–566. doi: 10.1038/nature09513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos D, Ryu JK, Merlini M, Baeten KM, Le Moan N, Petersen MA, Deerinck TJ, Smirnoff DS, Bedard C, Hakozaki H, Gonias Murray S, Ling JB, Lassmann H, Degen JL, Ellisman MH, Akassoglou K. Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat Commun. 2012;3:1227. doi: 10.1038/ncomms2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GE, Camarillo CW. An alpha 2 beta 1 integrin-dependent pinocytic mechanism involving intracellular vacuole formation and coalescence regulates capillary lumen and tube formation in three-dimensional collagen matrix. Exp Cell Res. 1996;224:39–51. doi: 10.1006/excr.1996.0109. [DOI] [PubMed] [Google Scholar]
- de Seranno S, d'Anglemont de Tassigny X, Estrella C, Loyens A, Kasparov S, Leroy D, Ojeda SR, Beauvillain JC, Prevot V. Role of estradiol in the dynamic control of tanycyte plasticity mediated by vascular endothelial cells in the median eminence. Endocrinology. 2010;151:1760–1772. doi: 10.1210/en.2009-0870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejana E, Orsenigo F, Lampugnani MG. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J Cell Sci. 2008;121:2115–2122. doi: 10.1242/jcs.017897. [DOI] [PubMed] [Google Scholar]
- Dodelet-Devillers A, Cayrol R, van Horssen J, Haqqani AS, de Vries HE, Engelhardt B, Greenwood J, Prat A. Functions of lipid raft membrane microdomains at the blood-brain barrier. J Mol Med (Berl) 2009;87:765–774. doi: 10.1007/s00109-009-0488-6. [DOI] [PubMed] [Google Scholar]
- Dohgu S, Yamauchi A, Takata F, Naito M, Tsuruo T, Higuchi S, Sawada Y, Kataoka Y. Transforming growth factor-beta1 upregulates the tight junction and P-glycoprotein of brain microvascular endothelial cells. Cell Mol Neurobiol. 2004;24:491–497. doi: 10.1023/B:CEMN.0000022776.47302.ce. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilert-Olsen M, Haj-Yasein NN, Vindedal GF, Enger R, Gundersen GA, Hoddevik EH, Petersen PH, Haug FM, Skare O, Adams ME, Froehner SC, Burkhardt JM, Thoren AE, Nagelhus EA. Deletion of aquaporin-4 changes the perivascular glial protein scaffold without disrupting the brain endothelial barrier. Glia. 2012;60:432–440. doi: 10.1002/glia.22277. [DOI] [PubMed] [Google Scholar]
- El-Khoury N, Braun A, Hu F, Pandey M, Nedergaard M, Lagamma EF, Ballabh P. Astrocyte end-feet in germinal matrix, cerebral cortex, and white matter in developing infants. Pediatr Res. 2006;59:673–679. doi: 10.1203/01.pdr.0000214975.85311.9c. [DOI] [PubMed] [Google Scholar]
- Engelhardt B, Ransohoff RM. The ins and outs of T-lymphocyte trafficking to the CNS: Anatomical sites and molecular mechanisms. Trends Immunol. 2005;26:485–495. doi: 10.1016/j.it.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Engelhardt B, Sorokin L. The blood-brain and the blood-cerebrospinal fluid barriers: Function and dysfunction. Semin Immunopathol. 2009;31:497–511. doi: 10.1007/s00281-009-0177-0. [DOI] [PubMed] [Google Scholar]
- Eroglu C. The role of astrocyte-secreted matricellular proteins in central nervous system development and function. J Cell Commun Signal. 2009;3:167–176. doi: 10.1007/s12079-009-0078-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezan P, Andre P, Cisternino S, Saubamea B, Boulay AC, Doutremer S, Thomas MA, Quenech'du N, Giaume C, Cohen-Salmon M. Deletion of astroglial connexins weakens the blood-brain barrier. J Cereb Blood Flow Metab. 2012;32:1457–1467. doi: 10.1038/jcbfm.2012.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabry Z, Topham DJ, Fee D, Herlein J, Carlino JA, Hart MN, Sriram S. TGF-beta 2 decreases migration of lymphocytes in vitro and homing of cells into the central nervous system in vivo. J Immunol. 1995;155:325–332. [PubMed] [Google Scholar]
- Fan X, Staitieh BS, Jensen JS, Mould KJ, Greenberg JA, Joshi PC, Koval M, Guidot DM. Activating the Nrf2-mediated antioxidant response element restores barrier function in the alveolar epithelium of HIV-1 transgenic rats. Am J Physiol Lung Cell Mol Physiol. 2013;305:L267–L277. doi: 10.1152/ajplung.00288.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farias AS, Spagnol GS, Bordeaux-Rego P, Oliveira CO, Fontana AG, de Paula RF, Santos MP, Pradella F, Moraes AS, Oliveira EC, Longhini AL, Rezende AC, Vaisberg MW, Santos LM. Vitamin D3 induces IDO(+) tolerogenic DCs and enhances Treg, reducing the severity of EAE. CNS Neurosci Ther. 2013;19:269–277. doi: 10.1111/cns.12071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontijn RD, Volger OL, Fledderus JO, Reijerkerk A, de Vries HE, Horrevoets AJ. SOX-18 controls endothelial-specific claudin-5 gene expression and barrier function. Am J Physiol Heart Circ Physiol. 2008;294:H891–900. doi: 10.1152/ajpheart.01248.2007. [DOI] [PubMed] [Google Scholar]
- Friese MA, Fugger L. Pathogenic CD8(+) T cells in multiple sclerosis. Ann Neurol. 2009;66:132–141. doi: 10.1002/ana.21744. [DOI] [PubMed] [Google Scholar]
- Frigeri A, Nicchia GP, Nico B, Quondamatteo F, Herken R, Roncali L, Svelto M. Aquaporin-4 deficiency in skeletal muscle and brain of dystrophic mdx mice. FASEB J. 2001;15:90–98. doi: 10.1096/fj.00-0260com. [DOI] [PubMed] [Google Scholar]
- Fukuda AM, Badaut J. Aquaporin 4: A player in cerebral edema and neuroinflammation. J Neuroinflammation. 2012;9:279. doi: 10.1186/1742-2094-9-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullerton SM, Shirman GA, Strittmatter WJ, Matthew WD. Impairment of the blood-nerve and blood-brain barriers in apolipoprotein e knockout mice. Exp Neurol. 2001;169:13–22. doi: 10.1006/exnr.2001.7631. [DOI] [PubMed] [Google Scholar]
- Gale NW, Thurston G, Hackett SF, Renard R, Wang Q, McClain J, Martin C, Witte C, Witte MH, Jackson D, Suri C, Campochiaro PA, Wiegand SJ, Yancopoulos GD. Angiopoietin-2 is required for postnatal angiogenesis and lymphatic patterning, and only the latter role is rescued by Angiopoietin-1. Dev Cell. 2002;3:411–423. doi: 10.1016/s1534-5807(02)00217-4. [DOI] [PubMed] [Google Scholar]
- Garcia JG, Schaphorst KL. Regulation of endothelial cell gap formation and paracellular permeability. J Investig Med. 1995;43:117–126. [PubMed] [Google Scholar]
- Gerhardt H, Ruhrberg C, Abramsson A, Fujisawa H, Shima D, Betsholtz C. Neuropilin-1 is required for endothelial tip cell guidance in the developing central nervous system. Dev Dyn. 2004;231:503–509. doi: 10.1002/dvdy.20148. [DOI] [PubMed] [Google Scholar]
- Giaume C, Fromaget C, el Aoumari A, Cordier J, Glowinski J, Gros D. Gap junctions in cultured astrocytes: Single-channel currents and characterization of channel-forming protein. Neuron. 1991;6:133–143. doi: 10.1016/0896-6273(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Giaume C, Koulakoff A, Roux L, Holcman D, Rouach N. Astroglial networks: A step further in neuroglial and gliovascular interactions. Nat Rev Neurosci. 2010;11:87–99. doi: 10.1038/nrn2757. [DOI] [PubMed] [Google Scholar]
- Gilbert-Sirieix M, Makoukji J, Kimura S, Talbot M, Caillou B, Massaad C, Massaad-Massade L. Wnt/beta-catenin signaling pathway is a direct enhancer of thyroid transcription factor-1 in human papillary thyroid carcinoma cells. PLoS One. 2011;6:e22280. doi: 10.1371/journal.pone.0022280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulacsi A, Anderson SA. Shh maintains Nkx2.1 in the MGE by a Gli3-independent mechanism. Cereb Cortex. 2006;16(Suppl 1):i89–i95. doi: 10.1093/cercor/bhk018. [DOI] [PubMed] [Google Scholar]
- Hafezi-Moghadam A, Thomas KL, Wagner DD. ApoE deficiency leads to a progressive age-dependent blood-brain barrier leakage. Am J Physiol Cell Physiol. 2007;292:C1256–C1262. doi: 10.1152/ajpcell.00563.2005. [DOI] [PubMed] [Google Scholar]
- Hafler DA, Compston A, Sawcer S, Lander ES, Daly MJ, De Jager PL, de Bakker PI, Gabriel SB, Mirel DB, Ivinson AJ, Pericak-Vance MA, Gregory SG, Rioux JD, McCauley JL, Haines JL, Barcellos LF, Cree B, Oksenberg JR, Hauser SL. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med. 2007;357:851–862. doi: 10.1056/NEJMoa073493. [DOI] [PubMed] [Google Scholar]
- Haj-Yasein NN, Vindedal GF, Eilert-Olsen M, Gundersen GA, Skare O, Laake P, Klungland A, Thoren AE, Burkhardt JM, Ottersen OP, Nagelhus EA. Glial-conditional deletion of aquaporin-4 (Aqp4) reduces blood-brain water uptake and confers barrier function on perivascular astrocyte endfeet. Proc Natl Acad Sci USA. 2011;108:17815–17820. doi: 10.1073/pnas.1110655108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halilagic A, Ribes V, Ghyselinck NB, Zile MH, Dolle P, Studer M. Retinoids control anterior and dorsal properties in the developing forebrain. Dev Biol. 2007;303:362–375. doi: 10.1016/j.ydbio.2006.11.021. [DOI] [PubMed] [Google Scholar]
- Hamann GF, Okada Y, Fitridge R, del Zoppo GJ. Microvascular basal lamina antigens disappear during cerebral ischemia and reperfusion. Stroke. 1995;26:2120–2126. doi: 10.1161/01.str.26.11.2120. [DOI] [PubMed] [Google Scholar]
- Harhaj NS, Antonetti DA. Regulation of tight junctions and loss of barrier function in pathophysiology. Int J Biochem Cell Biol. 2004;36:1206–1237. doi: 10.1016/j.biocel.2003.08.007. [DOI] [PubMed] [Google Scholar]
- Haseloff RF, Blasig IE, Bauer HC, Bauer H. In search of the astrocytic factor(s) modulating blood-brain barrier functions in brain capillary endothelial cells in vitro. Cell Mol Neurobiol. 2005;25:25–39. doi: 10.1007/s10571-004-1375-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann U, Kaufer D, Friedman A. Blood-brain barrier dysfunction, TGFbeta signaling, and astrocyte dysfunction in epilepsy. Glia. 2012;60:1251–1257. doi: 10.1002/glia.22311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz J, Bock HH. Lipoprotein receptors in the nervous system. Annu Rev Biochem. 2002;71:405–434. doi: 10.1146/annurev.biochem.71.110601.135342. [DOI] [PubMed] [Google Scholar]
- Higashi K, Fujita A, Inanobe A, Tanemoto M, Doi K, Kubo T, Kurachi Y. An inwardly rectifying K(+) channel, Kir4.1, expressed in astrocytes surrounds synapses and blood vessels in brain. Am J Physiol Cell Physiol. 2001;281:C922–C931. doi: 10.1152/ajpcell.2001.281.3.C922. [DOI] [PubMed] [Google Scholar]
- Hinson SR, Pittock SJ, Lucchinetti CF, Roemer SF, Fryer JP, Kryzer TJ, Lennon VA. Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology. 2007;69:2221–2231. doi: 10.1212/01.WNL.0000289761.64862.ce. [DOI] [PubMed] [Google Scholar]
- Hinson SR, Roemer SF, Lucchinetti CF, Fryer JP, Kryzer TJ, Chamberlain JL, Howe CL, Pittock SJ, Lennon VA. Aquaporin-4-binding autoantibodies in patients with neuromyelitis optica impair glutamate transport by down-regulating EAAT2. J Exp Med. 2008;205:2473–2481. doi: 10.1084/jem.20081241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong JY, Park JI, Cho K, Gu D, Ji H, Artandi SE, McCrea PD. Shared molecular mechanisms regulate multiple catenin proteins: Canonical Wnt signals and components modulate p120-catenin isoform-1 and additional p120 subfamily members. J Cell Sci. 2010;123:4351–4365. doi: 10.1242/jcs.067199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppert J, Closhen D, Croxford A, White R, Kulig P, Pietrowski E, Bechmann I, Becher B, Luhmann HJ, Waisman A, Kuhlmann CR. Cellular mechanisms of IL-17-induced blood-brain barrier disruption. FASEB J. 2010;24:1023–1034. doi: 10.1096/fj.09-141978. [DOI] [PubMed] [Google Scholar]
- Ifergan I, Kebir H, Bernard M, Wosik K, Dodelet-Devillers A, Cayrol R, Arbour N, Prat A. The blood-brain barrier induces differentiation of migrating monocytes into Th17-polarizing dendritic cells. Brain. 2008;131:785–799. doi: 10.1093/brain/awm295. [DOI] [PubMed] [Google Scholar]
- Ifergan I, Kebir H, Terouz S, Alvarez JI, Lecuyer MA, Gendron S, Bourbonniere L, Dunay IR, Bouthillier A, Moumdjian R, Fontana A, Haqqani A, Klopstein A, Prinz M, Lopez-Vales R, Birchler T, Prat A. Role of Ninjurin-1 in the migration of myeloid cells to central nervous system inflammatory lesions. Ann Neurol. 2011;70:751–763. doi: 10.1002/ana.22519. [DOI] [PubMed] [Google Scholar]
- Ifergan I, Wosik K, Cayrol R, Kebir H, Auger C, Bernard M, Bouthillier A, Moumdjian R, Duquette P, Prat A. Statins reduce human blood-brain barrier permeability and restrict leukocyte migration: Relevance to multiple sclerosis. Ann Neurol. 2006;60:45–55. doi: 10.1002/ana.20875. [DOI] [PubMed] [Google Scholar]
- Igarashi Y, Utsumi H, Chiba H, Yamada-Sasamori Y, Tobioka H, Kamimura Y, Furuuchi K, Kokai Y, Nakagawa T, Mori M, Sawada N. Glial cell line-derived neurotrophic factor induces barrier function of endothelial cells forming the blood-brain barrier. Biochem Biophys Res Commun. 1999;261:108–112. doi: 10.1006/bbrc.1999.0992. [DOI] [PubMed] [Google Scholar]
- Ikenouchi J, Matsuda M, Furuse M, Tsukita S. Regulation of tight junctions during the epithelium-mesenchyme transition: Direct repression of the gene expression of claudins/occludin by Snail. J Cell Sci. 2003;116:1959–1967. doi: 10.1242/jcs.00389. [DOI] [PubMed] [Google Scholar]
- Jalkanen S, Salmi M. VAP-1 and CD73, endothelial cell surface enzymes in leukocyte extravasation. Arterioscler Thromb Vasc Biol. 2008;28:18–26. doi: 10.1161/ATVBAHA.107.153130. [DOI] [PubMed] [Google Scholar]
- Jang AS, Concel VJ, Bein K, Brant KA, Liu S, Pope-Varsalona H, Dopico RA, Jr, Di YP, Knoell DL, Barchowsky A, Leikauf GD. Endothelial dysfunction and claudin 5 regulation during acrolein-induced lung injury. Am J Respir Cell Mol Biol. 2011;44:483–490. doi: 10.1165/rcmb.2009-0391OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janzer RC, Raff MC. Astrocytes induce blood-brain barrier properties in endothelial cells. Nature. 1987;325:253–257. doi: 10.1038/325253a0. [DOI] [PubMed] [Google Scholar]
- Kamo N, Ke B, Busuttil RW, Kupiec-Weglinski JW. PTEN-mediated Akt/beta-catenin/Foxo1 signaling regulates innate immune responses in mouse liver ischemia/reperfusion injury. Hepatology. 2013;57:289–298. doi: 10.1002/hep.25958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda S, Mochizuki Y, Suematsu T, Miyata Y, Nomata K, Kanetake H. Sonic hedgehog induces capillary morphogenesis by endothelial cells through phosphoinositide 3-kinase. J Biol Chem. 2003;278:8244–8249. doi: 10.1074/jbc.M210635200. [DOI] [PubMed] [Google Scholar]
- Kasperczyk H, Baumann B, Debatin KM, Fulda S. Characterization of sonic hedgehog as a novel NF-kappaB target gene that promotes NF-kappaB-mediated apoptosis resistance and tumor growth in vivo. FASEB J. 2009;23:21–33. doi: 10.1096/fj.08-111096. [DOI] [PubMed] [Google Scholar]
- Kebir H, Ifergan I, Alvarez JI, Bernard M, Poirier J, Arbour N, Duquette P, Prat A. Preferential recruitment of interferon-gamma-expressing TH17 cells in multiple sclerosis. Ann Neurol. 2009;66:390–402. doi: 10.1002/ana.21748. [DOI] [PubMed] [Google Scholar]
- Kebir H, Kreymborg K, Ifergan I, Dodelet-Devillers A, Cayrol R, Bernard M, Giuliani F, Arbour N, Becher B, Prat A. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med. 2007;13:1173–1175. doi: 10.1038/nm1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly MA, Hirschi KK. Signaling hierarchy regulating human endothelial cell development. Arterioscler Thromb Vasc Biol. 2009;29:718–724. doi: 10.1161/ATVBAHA.109.184200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornyei Z, Gocza E, Ruhl R, Orsolits B, Voros E, Szabo B, Vagovits B, Madarasz E. Astroglia-derived retinoic acid is a key factor in glia-induced neurogenesis. FASEB J. 2007;21:2496–2509. doi: 10.1096/fj.06-7756com. [DOI] [PubMed] [Google Scholar]
- Koyama Y, Baba A, Matsuda T. Endothelins stimulate the expression of neurotrophin-3 in rat brain and rat cultured astrocytes. Neuroscience. 2005;136:425–433. doi: 10.1016/j.neuroscience.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Koyama Y, Maebara Y, Hayashi M, Nagae R, Tokuyama S, Michinaga S. Endothelins reciprocally regulate VEGF-A and angiopoietin-1 production in cultured rat astrocytes: Implications on astrocytic proliferation. Glia. 2012;60:1954–1963. doi: 10.1002/glia.22411. [DOI] [PubMed] [Google Scholar]
- Koyama Y, Michinaga S. Regulations of astrocytic functions by endothelins: Roles in the pathophysiological responses of damaged brains. J Pharmacol Sci. 2012;118:401–407. doi: 10.1254/jphs.11r13cp. [DOI] [PubMed] [Google Scholar]
- Koyama Y, Tsujikawa K, Matsuda T, Baba A. Endothelin-1 stimulates glial cell line-derived neurotrophic factor expression in cultured rat astrocytes. Biochem Biophys Res Commun. 2003;303:1101–1105. doi: 10.1016/s0006-291x(03)00491-1. [DOI] [PubMed] [Google Scholar]
- Kyriakides TR, Maclauchlan S. The role of thrombospondins in wound healing, ischemia, and the foreign body reaction. J Cell Commun Signal. 2009;3:215–225. doi: 10.1007/s12079-009-0077-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamberti C, Lin KM, Yamamoto Y, Verma U, Verma IM, Byers S, Gaynor RB. Regulation of beta-catenin function by the IkappaB kinases. J Biol Chem. 2001;276:42276–42286. doi: 10.1074/jbc.M104227200. [DOI] [PubMed] [Google Scholar]
- Larochelle C, Alvarez JI, Prat A. How do immune cells overcome the blood-brain barrier in multiple sclerosis? FEBS Lett. 2011;585:3770–3780. doi: 10.1016/j.febslet.2011.04.066. [DOI] [PubMed] [Google Scholar]
- Larochelle C, Cayrol R, Kebir H, Alvarez JI, Lecuyer MA, Ifergan I, Viel E, Bourbonniere L, Beauseigle D, Terouz S, Hachehouche L, Gendron S, Poirier J, Jobin C, Duquette P, Flanagan K, Yednock T, Arbour N, Prat A. Melanoma cell adhesion molecule identifies encephalitogenic T lymphocytes and promotes their recruitment to the central nervous system. Brain. 2012;135:2906–2924. doi: 10.1093/brain/aws212. [DOI] [PubMed] [Google Scholar]
- Lavoie JL, Sigmund CD. Minireview: Overview of the renin-angiotensin system--An endocrine and paracrine system. Endocrinology. 2003;144:2179–2183. doi: 10.1210/en.2003-0150. [DOI] [PubMed] [Google Scholar]
- Lebrin F, Deckers M, Bertolino P, Ten Dijke P. TGF-beta receptor function in the endothelium. Cardiovasc Res. 2005;65:599–608. doi: 10.1016/j.cardiores.2004.10.036. [DOI] [PubMed] [Google Scholar]
- Lee S, Chen TT, Barber CL, Jordan MC, Murdock J, Desai S, Ferrara N, Nagy A, Roos KP, Iruela-Arispe ML. Autocrine VEGF signaling is required for vascular homeostasis. Cell. 2007a;130:691–703. doi: 10.1016/j.cell.2007.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SW, Kim WJ, Choi YK, Song HS, Son MJ, Gelman IH, Kim YJ, Kim KW. SSeCKS regulates angiogenesis and tight junction formation in blood-brain barrier. Nat Med. 2003;9:900–906. doi: 10.1038/nm889. [DOI] [PubMed] [Google Scholar]
- Lee SW, Moskowitz MA, Sims JR. Sonic hedgehog inversely regulates the expression of angiopoietin-1 and angiopoietin-2 in fibroblasts. Int J Mol Med. 2007b;19:445–451. [PubMed] [Google Scholar]
- Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K, Nakashima I, Weinshenker BG. A serum autoantibody marker of neuromyelitis optica: Distinction from multiple sclerosis. Lancet. 2004;364:2106–2112. doi: 10.1016/S0140-6736(04)17551-X. [DOI] [PubMed] [Google Scholar]
- Liebner S, Corada M, Bangsow T, Babbage J, Taddei A, Czupalla CJ, Reis M, Felici A, Wolburg H, Fruttiger M, Taketo MM, von Melchner H, Plate KH, Gerhardt H, Dejana E. Wnt/beta-catenin signaling controls development of the blood-brain barrier. J Cell Biol. 2008;183:409–417. doi: 10.1083/jcb.200806024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebner S, Czupalla CJ, Wolburg H. Current concepts of blood-brain barrier development. Int J Dev Biol. 2011;55:467–476. doi: 10.1387/ijdb.103224sl. [DOI] [PubMed] [Google Scholar]
- Lien CF, Hazai D, Yeung D, Tan J, Fuchtbauer EM, Jancsik V, Gorecki DC. Expression of alpha-dystrobrevin in blood-tissue barriers: Sub-cellular localisation and molecular characterisation in normal and dystrophic mice. Cell Tissue Res. 2007;327:67–82. doi: 10.1007/s00441-006-0241-1. [DOI] [PubMed] [Google Scholar]
- Lien CF, Mohanta SK, Frontczak-Baniewicz M, Swinny JD, Zablocka B, Gorecki DC. Absence of glial alpha-dystrobrevin causes abnormalities of the blood-brain barrier and progressive brain edema. J Biol Chem. 2012;287:41374–41385. doi: 10.1074/jbc.M112.400044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Ho P, Steinman L, Wyss-Coray T. Bioluminescence in vivo imaging of autoimmune encephalomyelitis predicts disease. J Neuroinflammation. 2008;5:6. doi: 10.1186/1742-2094-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S, Kwon HJ, Johng H, Zang K, Huang Z. Radial glial neural progenitors regulate nascent brain vascular network stabilization via inhibition of Wnt signaling. PLoS Biol. 2013;11:e1001469. doi: 10.1371/journal.pbio.1001469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda O, Kondo M, Fujita T, Usami N, Fukui T, Shimokata K, Ando T, Goto H, Sekido Y. Enhancement of GLI1-transcriptional activity by beta-catenin in human cancer cells. Oncol Rep. 2006;16:91–96. [PubMed] [Google Scholar]
- Mahley RW. Apolipoprotein E: Cholesterol transport protein with expanding role in cell biology. Science. 1988;240:622–630. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- Manley GT, Fujimura M, Ma T, Noshita N, Filiz F, Bollen AW, Chan P, Verkman AS. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med. 2000;6:159–163. doi: 10.1038/72256. [DOI] [PubMed] [Google Scholar]
- Marigo V, Roberts DJ, Lee SM, Tsukurov O, Levi T, Gastier JM, Epstein DJ, Gilbert DJ, Copeland NG, Seidman CE, et al. Cloning, expression, and chromosomal location of SHH and IHH: Two human homologues of the Drosophila segment polarity gene hedgehog. Genomics. 1995;28:44–51. doi: 10.1006/geno.1995.1104. [DOI] [PubMed] [Google Scholar]
- McCubrey JA, Lahair MM, Franklin RA. Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxid Redox Signal. 2006;8:1775–1789. doi: 10.1089/ars.2006.8.1775. [DOI] [PubMed] [Google Scholar]
- Milner R, Campbell IL. The integrin family of cell adhesion molecules has multiple functions within the CNS. J Neurosci Res. 2002;69:286–291. doi: 10.1002/jnr.10321. [DOI] [PubMed] [Google Scholar]
- Milner R, Huang X, Wu J, Nishimura S, Pytela R, Sheppard D, ffrench-Constant C. Distinct roles for astrocyte alphavbeta5 and alphavbeta8 integrins in adhesion and migration. J Cell Sci. 1999;112:4271–4279. doi: 10.1242/jcs.112.23.4271. [DOI] [PubMed] [Google Scholar]
- Mizee MR, Wooldrik D, Lakeman KA, van het Hof B, Drexhage JA, Geerts D, Bugiani M, Aronica E, Mebius RE, Prat A, de Vries HE, Reijerkerk A. Retinoic acid induces blood-brain barrier development. J Neurosci. 2013;33:1660–1671. doi: 10.1523/JNEUROSCI.1338-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molofsky AV, Krencik R, Ullian EM, Tsai HH, Deneen B, Richardson WD, Barres BA, Rowitch DH. Astrocytes and disease: A neurodevelopmental perspective. Genes Dev. 2012;26:891–907. doi: 10.1101/gad.188326.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami M, Nguyen LT, Zhuang ZW, Moodie KL, Carmeliet P, Stan RV, Simons M. The FGF system has a key role in regulating vascular integrity. J Clin Invest. 2008;118:3355–3366. doi: 10.1172/JCI35298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadarajah B. Radial glia and somal translocation of radial neurons in the developing cerebral cortex. Glia. 2003;43:33–36. doi: 10.1002/glia.10245. [DOI] [PubMed] [Google Scholar]
- Nagasawa K, Chiba H, Fujita H, Kojima T, Saito T, Endo T, Sawada N. Possible involvement of gap junctions in the barrier function of tight junctions of brain and lung endothelial cells. J Cell Physiol. 2006;208:123–132. doi: 10.1002/jcp.20647. [DOI] [PubMed] [Google Scholar]
- Nagase T, Nagase M, Machida M, Fujita T. Hedgehog signalling in vascular development. Angiogenesis. 2008;11:71–77. doi: 10.1007/s10456-008-9105-5. [DOI] [PubMed] [Google Scholar]
- Nagy JI, Rash JE. Connexins and gap junctions of astrocytes and oligodendrocytes in the CNS. Brain Res Brain Res Rev. 2000;32:29–44. doi: 10.1016/s0165-0173(99)00066-1. [DOI] [PubMed] [Google Scholar]
- Nagy Z, Peters H, Huttner I. Fracture faces of cell junctions in cerebral endothelium during normal and hyperosmotic conditions. Lab Invest. 1984;50:313–322. [PubMed] [Google Scholar]
- Neely JD, Amiry-Moghaddam M, Ottersen OP, Froehner SC, Agre P, Adams ME. Syntrophin-dependent expression and localization of Aquaporin-4 water channel protein. Proc Natl Acad Sci USA. 2001;98:14108–14113. doi: 10.1073/pnas.241508198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuhaus J, Risau W, Wolburg H. Induction of blood-brain barrier characteristics in bovine brain endothelial cells by rat astroglial cells in transfilter coculture. Ann NY Acad Sci. 1991;633:578–580. doi: 10.1111/j.1749-6632.1991.tb15667.x. [DOI] [PubMed] [Google Scholar]
- Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem. 2009;284:13291–13295. doi: 10.1074/jbc.R900010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen S, Nagelhus EA, Amiry-Moghaddam M, Bourque C, Agre P, Ottersen OP. Specialized membrane domains for water transport in glial cells: High-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J Neurosci. 1997;17:171–180. doi: 10.1523/JNEUROSCI.17-01-00171.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino J, Yamashita K, Hashiguchi H, Fujii H, Shimazaki T, Hamada H. Meteorin: A secreted protein that regulates glial cell differentiation and promotes axonal extension. EMBO J. 2004;23:1998–2008. doi: 10.1038/sj.emboj.7600202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishitsuji K, Hosono T, Nakamura T, Bu G, Michikawa M. Apolipoprotein E regulates the integrity of tight junctions in an isoform-dependent manner in an in vitro blood-brain barrier model. J Biol Chem. 2011;286:17536–17542. doi: 10.1074/jbc.M111.225532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman MG, O'Kusky JR. The growth and development of microvasculature in human cerebral cortex. J Neuropathol Exp Neurol. 1986;45:222–232. [PubMed] [Google Scholar]
- Northington FJ, Tobin JR, Harris AP, Traystman RJ, Koehler RC. Developmental and regional differences in nitric oxide synthase activity and blood flow in the sheep brain. J Cereb Blood Flow Metab. 1997;17:109–115. doi: 10.1097/00004647-199701000-00014. [DOI] [PubMed] [Google Scholar]
- Nourhaghighi N, Teichert-Kuliszewska K, Davis J, Stewart DJ, Nag S. Altered expression of angiopoietins during blood-brain barrier breakdown and angiogenesis. Lab Invest. 2003;83:1211–1222. doi: 10.1097/01.lab.0000082383.40635.fe. [DOI] [PubMed] [Google Scholar]
- Oberheim NA, Goldman SA, Nedergaard M. Heterogeneity of astrocytic form and function. Methods Mol Biol. 2012;814:23–45. doi: 10.1007/978-1-61779-452-0_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkubo T, Ozawa M. The transcription factor Snail downregulates the tight junction components independently of E-cadherin downregulation. J Cell Sci. 2004;117:1675–1685. doi: 10.1242/jcs.01004. [DOI] [PubMed] [Google Scholar]
- Okamura M, Kudo H, Wakabayashi K, Tanaka T, Nonaka A, Uchida A, Tsutsumi S, Sakakibara I, Naito M, Osborne TF, Hamakubo T, Ito S, Aburatani H, Yanagisawa M, Kodama T, Sakai J. COUP-TFII acts downstream of Wnt/beta-catenin signal to silence PPARgamma gene expression and repress adipogenesis. Proc Natl Acad Sci U S A. 2009;106:5819–5824. doi: 10.1073/pnas.0901676106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldendorf WH, Cornford ME, Brown WJ. The large apparent work capability of the blood-brain barrier: A study of the mitochondrial content of capillary endothelial cells in brain and other tissues of the rat. Ann Neurol. 1977;1:409–417. doi: 10.1002/ana.410010502. [DOI] [PubMed] [Google Scholar]
- Ostermann G, Weber KS, Zernecke A, Schroder A, Weber C. JAM-1 is a ligand of the beta(2) integrin LFA-1 involved in transendothelial migration of leukocytes. Nat Immunol. 2002;3:151–158. doi: 10.1038/ni755. [DOI] [PubMed] [Google Scholar]
- Park JA, Lee HS, Ko KJ, Park SY, Kim JH, Choe G, Kweon HS, Song HS, Ahn JC, Yu YS, Kim KW. Meteorin regulates angiogenesis at the gliovascular interface. Glia. 2008;56:247–258. doi: 10.1002/glia.20600. [DOI] [PubMed] [Google Scholar]
- Paschaki M, Lin SC, Wong RL, Finnell RH, Dolle P, Niederreither K. Retinoic acid-dependent signaling pathways and lineage events in the developing mouse spinal cord. PLoS One. 2012;7:e32447. doi: 10.1371/journal.pone.0032447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patan S. Vasculogenesis and angiogenesis as mechanisms of vascular network formation, growth and remodeling. J Neurooncol. 2000;50:1–15. doi: 10.1023/a:1006493130855. [DOI] [PubMed] [Google Scholar]
- Paulus W, Baur I, Schuppan D, Roggendorf W. Characterization of integrin receptors in normal and neoplastic human brain. Am J Pathol. 1993;143:154–163. [PMC free article] [PubMed] [Google Scholar]
- Pfaff D, Fiedler U, Augustin HG. Emerging roles of the Angiopoietin-Tie and the ephrin-Eph systems as regulators of cell trafficking. J Leukoc Biol. 2006;80:719–726. doi: 10.1189/jlb.1105652. [DOI] [PubMed] [Google Scholar]
- Pollinger B, Krishnamoorthy G, Berer K, Lassmann H, Bosl MR, Dunn R, Domingues HS, Holz A, Kurschus FC, Wekerle H. Spontaneous relapsing-remitting EAE in the SJL/J mouse: MOG-reactive transgenic T cells recruit endogenous MOG-specific B cells. J Exp Med. 2009;206:1303–1316. doi: 10.1084/jem.20090299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prat A, Biernacki K, Becher B, Antel JP. B7 expression and antigen presentation by human brain endothelial cells: Requirement for proinflammatory cytokines. J Neuropathol Exp Neurol. 2000;59:129–136. doi: 10.1093/jnen/59.2.129. [DOI] [PubMed] [Google Scholar]
- Prat A, Biernacki K, Wosik K, Antel JP. Glial cell influence on the human blood-brain barrier. Glia. 2001;36:145–155. doi: 10.1002/glia.1104. [DOI] [PubMed] [Google Scholar]
- Puwarawuttipanit W, Bragg AD, Frydenlund DS, Mylonakou MN, Nagelhus EA, Peters MF, Kotchabhakdi N, Adams ME, Froehner SC, Haug FM, Ottersen OP, Amiry-Moghaddam M. Differential effect of alpha-syntrophin knockout on aquaporin-4 and Kir4.1 expression in retinal macroglial cells in mice. Neuroscience. 2006;137:165–175. doi: 10.1016/j.neuroscience.2005.08.051. [DOI] [PubMed] [Google Scholar]
- Rakic P. Mode of cell migration to the superficial layers of fetal monkey neocortex. J Comp Neurol. 1972;145:61–83. doi: 10.1002/cne.901450105. [DOI] [PubMed] [Google Scholar]
- Ramagopalan SV, Handel AE, Giovannoni G, Rutherford Siegel S, Ebers GC, Chaplin G. Relationship of UV exposure to prevalence of multiple sclerosis in England. Neurology. 2011;76:1410–1414. doi: 10.1212/WNL.0b013e318216715e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM, Kivisakk P, Kidd G. Three or more routes for leukocyte migration into the central nervous system. Nat Rev Immunol. 2003;3:569–581. doi: 10.1038/nri1130. [DOI] [PubMed] [Google Scholar]
- Ratelade J, Verkman AS. Neuromyelitis optica: Aquaporin-4 based pathogenesis mechanisms and new therapies. Int J Biochem Cell Biol. 2012;44:1519–1530. doi: 10.1016/j.biocel.2012.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees ML, Lien CF, Gorecki DC. Dystrobrevins in muscle and non-muscle tissues. Neuromuscul Disord. 2007;17:123–134. doi: 10.1016/j.nmd.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Reichenbach A, Wolburg H. Astrocytes and ependymal glia. In: Kettenmann H, Ransom BR, editors. Neuroglia. Oxford: Oxford University Press; 2005. pp. 19–35. [Google Scholar]
- Reuss B, Dono R, Unsicker K. Functions of fibroblast growth factor (FGF)-2 and FGF-5 in astroglial differentiation and blood-brain barrier permeability: Evidence from mouse mutants. J Neurosci. 2003;23:6404–6412. doi: 10.1523/JNEUROSCI.23-16-06404.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuss B, Hertel M, Werner S, Unsicker K. Fibroblast growth factors-5 and [minus]9 distinctly regulate expression and function of the gap junction protein connexin43 in cultured astroglial cells from different brain regions. Glia. 2000;30:231–241. [PubMed] [Google Scholar]
- Ridet JL, Malhotra SK, Privat A, Gage FH. Reactive astrocytes: Cellular and molecular cues to biological function. Trends Neurosci. 1997;20:570–577. doi: 10.1016/s0166-2236(97)01139-9. [DOI] [PubMed] [Google Scholar]
- Risau W. Mechanisms of angiogenesis. Nature. 1997;386:671–674. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- Risau W, Flamme I. Vasculogenesis. Annu Rev Cell Dev Biol. 1995;11:73–91. doi: 10.1146/annurev.cb.11.110195.000445. [DOI] [PubMed] [Google Scholar]
- Risau W, Wolburg H. Development of the blood-brain barrier. Trends Neurosci. 1990;13:174–178. doi: 10.1016/0166-2236(90)90043-a. [DOI] [PubMed] [Google Scholar]
- Robbins DJ, Fei DL, Riobo NA. The Hedgehog signal transduction network. Sci Signal. 2012;5:re6. doi: 10.1126/scisignal.2002906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robel S, Berninger B, Gotz M. The stem cell potential of glia: Lessons from reactive gliosis. Nat Rev Neurosci. 2011;12:88–104. doi: 10.1038/nrn2978. [DOI] [PubMed] [Google Scholar]
- Robel S, Mori T, Zoubaa S, Schlegel J, Sirko S, Faissner A, Goebbels S, Dimou L, Gotz M. Conditional deletion of beta1-integrin in astroglia causes partial reactive gliosis. Glia. 2009;57:1630–1647. doi: 10.1002/glia.20876. [DOI] [PubMed] [Google Scholar]
- Robinson CJ, Stringer SE. The splice variants of vascular endothelial growth factor (VEGF) and their receptors. J Cell Sci. 2001;114:853–865. doi: 10.1242/jcs.114.5.853. [DOI] [PubMed] [Google Scholar]
- Rouach N, Koulakoff A, Abudara V, Willecke K, Giaume C. Astroglial metabolic networks sustain hippocampal synaptic transmission. Science. 2008;322:1551–1555. doi: 10.1126/science.1164022. [DOI] [PubMed] [Google Scholar]
- Rubin LL, Hall DE, Porter S, Barbu K, Cannon C, Horner HC, Janatpour M, Liaw CW, Manning K, Morales J, Tanner LI, Tomaselli KJ, Bard F. A cell culture model of the blood-brain barrier. J Cell Biol. 1991;115:1725–1735. doi: 10.1083/jcb.115.6.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runkle EA, Rice SJ, Qi J, Masser D, Antonetti DA, Winslow MM, Mu D. Occludin is a direct target of thyroid transcription factor-1 (TTF-1/NKX2-1) J Biol Chem. 2012;287:28790–28801. doi: 10.1074/jbc.M112.367987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sade H, Holloway K, Romero IA, Male D. Transcriptional control of occludin expression in vascular endothelia: Regulation by Sp3 and YY1. Biochim Biophys Acta. 2009;1789:175–184. doi: 10.1016/j.bbagrm.2009.01.006. [DOI] [PubMed] [Google Scholar]
- Schachtrup C, Ryu JK, Helmrick MJ, Vagena E, Galanakis DK, Degen JL, Margolis RU, Akassoglou K. Fibrinogen triggers astrocyte scar formation by promoting the availability of active TGF-beta after vascular damage. J Neurosci. 2010;30:5843–5854. doi: 10.1523/JNEUROSCI.0137-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmechel DE, Rakic P. Arrested proliferation of radial glial cells during midgestation in rhesus monkey. Nature. 1979;277:303–305. doi: 10.1038/277303a0. [DOI] [PubMed] [Google Scholar]
- Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 1995;376:62–66. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- Shawber CJ, Kitajewski J. Notch function in the vasculature: Insights from zebrafish, mouse and man. Bioessays. 2004;26:225–234. doi: 10.1002/bies.20004. [DOI] [PubMed] [Google Scholar]
- Shen F, Walker EJ, Jiang L, Degos V, Li J, Sun B, Heriyanto F, Young WL, Su H. Coexpression of angiopoietin-1 with VEGF increases the structural integrity of the blood-brain barrier and reduces atrophy volume. J Cereb Blood Flow Metab. 2011;31:2343–2351. doi: 10.1038/jcbfm.2011.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya M. Vascular endothelial growth factor-dependent and -independent regulation of angiogenesis. BMB Rep. 2008;41:278–286. doi: 10.5483/bmbrep.2008.41.4.278. [DOI] [PubMed] [Google Scholar]
- Shimizu F, Sano Y, Saito K, Abe MA, Maeda T, Haruki H, Kanda T. Pericyte-derived glial cell line-derived neurotrophic factor increase the expression of claudin-5 in the blood-brain barrier and the blood-nerve barrier. Neurochem Res. 2012;37:401–409. doi: 10.1007/s11064-011-0626-8. [DOI] [PubMed] [Google Scholar]
- Shivers RR, Betz AL, Goldstein GW. Isolated rat brain capillaries possess intact, structurally complex, interendothelial tight junctions; freeze-fracture verification of tight junction integrity. Brain Res. 1984;324:313–322. doi: 10.1016/0006-8993(84)90042-8. [DOI] [PubMed] [Google Scholar]
- Simon KC, Munger KL, Ascherio A. Vitamin D and multiple sclerosis: Epidemiology, immunology, and genetics. Curr Opin Neurol. 2012;25:246–251. doi: 10.1097/WCO.0b013e3283533a7e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sixt M, Engelhardt B, Pausch F, Hallmann R, Wendler O, Sorokin LM. Endothelial cell laminin isoforms, laminins 8 and 10, play decisive roles in T cell recruitment across the blood-brain barrier in experimental autoimmune encephalomyelitis. J Cell Biol. 2001;153:933–946. doi: 10.1083/jcb.153.5.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperone A, Dryden NH, Birdsey GM, Madden L, Johns M, Evans PC, Mason JC, Haskard DO, Boyle JJ, Paleolog EM, Randi AM. The transcription factor Erg inhibits vascular inflammation by repressing NF-kappaB activation and proinflammatory gene expression in endothelial cells. Arterioscler Thromb Vasc Biol. 2011;31:142–150. doi: 10.1161/ATVBAHA.110.216473. [DOI] [PubMed] [Google Scholar]
- Srivastava R, Aslam M, Kalluri SR, Schirmer L, Buck D, Tackenberg B, Rothhammer V, Chan A, Gold R, Berthele A, Bennett JL, Korn T, Hemmer B. Potassium channel KIR4.1 as an immune target in multiple sclerosis. N Engl J Med. 2012;367:115–123. doi: 10.1056/NEJMoa1110740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatovic SM, Keep RF, Andjelkovic AV. Brain endothelial cell-cell junctions: How to "open" the blood brain barrier. Curr Neuropharmacol. 2008;6:179–192. doi: 10.2174/157015908785777210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner O, Coisne C, Cecchelli R, Boscacci R, Deutsch U, Engelhardt B, Lyck R. Differential roles for endothelial ICAM-1, ICAM-2, and VCAM-1 in shear-resistant T cell arrest, polarization, and directed crawling on blood-brain barrier endothelium. J Immunol. 2010;185:4846–4855. doi: 10.4049/jimmunol.0903732. [DOI] [PubMed] [Google Scholar]
- Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, Davis S, Sato TN, Yancopoulos GD. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell. 1996;87:1171–1180. doi: 10.1016/s0092-8674(00)81813-9. [DOI] [PubMed] [Google Scholar]
- Taddei A, Giampietro C, Conti A, Orsenigo F, Breviario F, Pirazzoli V, Potente M, Daly C, Dimmeler S, Dejana E. Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat Cell Biol. 2008;10:923–934. doi: 10.1038/ncb1752. [DOI] [PubMed] [Google Scholar]
- Tagaya M, Haring HP, Stuiver I, Wagner S, Abumiya T, Lucero J, Lee P, Copeland BR, Seiffert D, del Zoppo GJ. Rapid loss of microvascular integrin expression during focal brain ischemia reflects neuron injury. J Cereb Blood Flow Metab. 2001;21:835–846. doi: 10.1097/00004647-200107000-00009. [DOI] [PubMed] [Google Scholar]
- Tao-Cheng JH, Nagy Z, Brightman MW. Tight junctions of brain endothelium in vitro are enhanced by astroglia. J Neurosci. 1987;7:3293–3299. doi: 10.1523/JNEUROSCI.07-10-03293.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theis M, Sohl G, Eiberger J, Willecke K. Emerging complexities in identity and function of glial connexins. Trends Neurosci. 2005;28:188–195. doi: 10.1016/j.tins.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Virgintino D, Errede M, Robertson D, Girolamo F, Masciandaro A, Bertossi M. VEGF expression is developmentally regulated during human brain angiogenesis. Histochem Cell Biol. 2003;119:227–232. doi: 10.1007/s00418-003-0510-y. [DOI] [PubMed] [Google Scholar]
- Wagner S, Tagaya M, Koziol JA, Quaranta V, del Zoppo GJ. Rapid disruption of an astrocyte interaction with the extracellular matrix mediated by integrin alpha 6 beta 4 during focal cerebral ischemia/reperfusion. Stroke. 1997;28:858–865. doi: 10.1161/01.str.28.4.858. [DOI] [PubMed] [Google Scholar]
- Wall MJ. Endogenous nitric oxide modulates GABAergic transmission to granule cells in adult rat cerebellum. Eur J Neurosci. 2003;18:869–878. doi: 10.1046/j.1460-9568.2003.02822.x. [DOI] [PubMed] [Google Scholar]
- Wang Y, Imitola J, Rasmussen S, O'Connor KC, Khoury SJ. Paradoxical dysregulation of the neural stem cell pathway sonic hedgehog-Gli1 in autoimmune encephalomyelitis and multiple sclerosis. Ann Neurol. 2008;64:417–427. doi: 10.1002/ana.21457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson M, Hume R, Strange R, Bell JE. Glial and neuronal differentiation in the human fetal brain 9-23 weeks of gestation. Neuropathol Appl Neurobiol. 1990;16:193–204. doi: 10.1111/j.1365-2990.1990.tb01156.x. [DOI] [PubMed] [Google Scholar]
- Willer CJ, Dyment DA, Risch NJ, Sadovnick AD, Ebers GC. Twin concordance and sibling recurrence rates in multiple sclerosis. Proc Natl Acad Sci USA. 2003;100:12877–12882. doi: 10.1073/pnas.1932604100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KC, Zhao RW, Ueno K, Hickey WF. PECAM-1 (CD31) expression in the central nervous system and its role in experimental allergic encephalomyelitis in the rat. J Neurosci Res. 1996;45:747–757. doi: 10.1002/(SICI)1097-4547(19960915)45:6<747::AID-JNR11>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Wolburg H, Lippoldt A. Tight junctions of the blood-brain barrier: Development, composition and regulation. Vascul Pharmacol. 2002;38:323–337. doi: 10.1016/s1537-1891(02)00200-8. [DOI] [PubMed] [Google Scholar]
- Wolburg H, Noell S, Wolburg-Buchholz K, Mack A, Fallier-Becker P. Agrin, aquaporin-4, and astrocyte polarity as an important feature of the blood-brain barrier. Neuroscientist. 2009;15:180–193. doi: 10.1177/1073858408329509. [DOI] [PubMed] [Google Scholar]
- Wolburg H, Wolburg-Buchholz K, Fallier-Becker P, Noell S, Mack AF. Structure and functions of aquaporin-4-based orthogonal arrays of particles. Int Rev Cell Mol Biol. 2011;287:1–41. doi: 10.1016/B978-0-12-386043-9.00001-3. [DOI] [PubMed] [Google Scholar]
- Wong D, Dorovini-Zis K. Upregulation of intercellular adhesion molecule-1 (ICAM-1) expression in primary cultures of human brain microvessel endothelial cells by cytokines and lipopolysaccharide. J Neuroimmunol. 1992;39:11–21. doi: 10.1016/0165-5728(92)90170-p. [DOI] [PubMed] [Google Scholar]
- Wosik K, Cayrol R, Dodelet-Devillers A, Berthelet F, Bernard M, Moumdjian R, Bouthillier A, Reudelhuber TL, Prat A. Angiotensin II controls occludin function and is required for blood brain barrier maintenance: Relevance to multiple sclerosis. J Neurosci. 2007;27:9032–9042. doi: 10.1523/JNEUROSCI.2088-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Ivars F, Anderson P, Hallmann R, Vestweber D, Nilsson P, Robenek H, Tryggvason K, Song J, Korpos E, Loser K, Beissert S, Georges-Labouesse E, Sorokin LM. Endothelial basement membrane laminin alpha5 selectively inhibits T lymphocyte extravasation into the brain. Nat Med. 2009a;15:519–527. doi: 10.1038/nm.1957. [DOI] [PubMed] [Google Scholar]
- Wu Y, Deng J, Rychahou PG, Qiu S, Evers BM, Zhou BP. Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer Cell. 2009b;15:416–428. doi: 10.1016/j.ccr.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan L, Le Bras A, Sacharidou A, Itagaki K, Zhan Y, Kondo M, Carman CV, Davis GE, Aird WC, Oettgen P. ETS-related gene (ERG) controls endothelial cell permeability via transcriptional regulation of the claudin 5 (CLDN5) gene. J Biol Chem. 2012;287:6582–6591. doi: 10.1074/jbc.M111.300236. [DOI] [PMC free article] [PubMed] [Google Scholar]