

Abstract
In vivo assembly of overlapping fragments by homologous recombination in Saccharomyces cerevisiae is a powerful method to engineer large DNA constructs. Whereas most in vivo assembly methods reported to date result in circular vectors, stable integrated constructs are often preferred for metabolic engineering as they are required for large-scale industrial application. The present study explores the potential of combining in vivo assembly of large, multigene expression constructs with their targeted chromosomal integration in S. cerevisiae. Combined assembly and targeted integration of a ten-fragment 22-kb construct to a single chromosomal locus was successfully achieved in a single transformation process, but with low efficiency (5% of the analyzed transformants contained the correctly assembled construct). The meganuclease I-SceI was therefore used to introduce a double-strand break at the targeted chromosomal locus, thus to facilitate integration of the assembled construct. I-SceI-assisted integration dramatically increased the efficiency of assembly and integration of the same construct to 95%. This study paves the way for the fast, efficient, and stable integration of large DNA constructs in S. cerevisiae chromosomes.
Keywords: synthetic biology, in vivo assembly, homologous recombination, pathway engineering
Introduction
The yeast Saccharomyces cerevisiae is intensively explored and applied as a platform for the industrial production of a wide range of endogenous and heterologous compounds. Its success in this role can be readily explained from its robustness, simple nutritional requirements, and easy genetic accessibility. This last feature has recently propelled the popularity of S. cerevisiae as a preferred platform in synthetic biology, especially for the assembly of large DNA constructs (Gibson et al., 2008). Improvement on the performance of industrial organisms (higher productivity and yield, increased robustness, expression of complex heterologous pathways, etc.) by metabolic engineering requires simultaneous expression of dozens of genes. Handling such numbers of genes by classical cloning methods is extremely time-consuming. Over the last decade, several methods have been developed for fast and efficient assembly of large DNA constructs. The most promising of these are recombination-based methods in which multiple linear DNA fragments with overlapping terminal sequences are recombined into a single vector (Ellis et al., 2011). While in vitro recombination-based methods such as SLIC (Li & Elledge, 2007), InFusion (Benoit et al., 2006) and Gibson's isothermal assembly (Gibson et al., 2009) are undeniably valuable, in vivo recombination using S. cerevisiae is proving to be the method of choice for large constructs assembled from many fragments (Gibson et al., 2008; Shao et al., 2009). This method can be used for efficient and accurate plasmid assembly (Kuijpers et al., 2013). However, plasmid-borne gene expression is not favored for industrial-scale production as plasmids are notoriously unstable, and maintaining the selection pressure necessary for the cells to retain the plasmid is typically difficult to achieve in industrial settings (Zhang et al., 1996). Conversely, chromosomal integration results in stable expression of genes, but methods capable of rapid and accurate assembly and integration of large constructs are currently not available. A recent pioneering study has demonstrated that S. cerevisiae can, in a single step, assemble multiple fragments in a 23.7-kb construct and integrate this construct into the yeast chromosome (Shao et al., 2009). To increase the probability of integration events, the abundant δ-sites were chosen as integration loci. Although reasonable efficiencies were obtained (up to 70% correct transformants), targeting to δ-sites randomizes the number and location of integration sites and therefore results in unpredictable copy numbers and integration loci. For strain engineering programs, where high integration efficiencies are required, control of the locus and the number of integration events is of paramount importance. Considering the relatively low chromosomal integration efficiencies of linear DNA fragments in S. cerevisiae [c. 10−6 transformants per viable cell using 50-bp flanks homologous to the integration site (Storici et al., 2003)], it is not surprising that targeting a single specific chromosomal site for combined assembly and integration of a multigene fragment results in low efficiencies. The aim of the present study was to evaluate the potential of Combined in vivo Assembly and Targeted chromosomal Integration (from now on referred to as CATI) of large DNA constructs in S. cerevisiae. Preliminary results identified integration as the bottleneck for the CATI approach. Use of the meganuclease I-SceI was therefore explored to create double-strand DNA breaks and thereby enhance the integration efficiency. I-SceI and its homolog I-SceII are native to S. cerevisiae, in which they are encoded by mitochondrial introns (Watabe et al., 1983; Shibata et al., 1984; Monteilhet et al., 1990). These meganucleases, also named homing endonucleases, are responsible for intron mobility in the mitochondria of yeast, in which they initiate a site-specific gene conversion (Plessis et al., 1992). Much like the well-studied HO meganuclease, I-SceI initiates a double-strand break at a specific recognition site. The recognition site of I-SceI extends over a 18-bp nonsymmetrical sequence and generates a cut with a 4-bp overhang within its recognition site (Monteilhet et al., 1990). In the early 90s, it was demonstrated that I-SceI, when expressed in the nucleus, was active on nuclear targets and, as predicted from the absence of I-SceI cutting sites in the genomic DNA, was not toxic upon expression in wild-type S. cerevisiae (Plessis et al., 1992). In the presented work, I-SceI was implemented and investigated to develop a robust system for combined assembly and targeted chromosomal integration of multigene constructs in S. cerevisiae.
Materials and methods
Strains and media
The S. cerevisiae strains used in this study are derived from the CEN.PK family (Table 1; Entian & Kotter, 2007; Nijkamp et al., 2012). Cultures for transformation were grown in complex medium containing 10 g L−1 Bacto yeast extract, 20 g L−1 Bacto peptone, and 20 g L−1 glucose as carbon source. When galactose induction of SCEI was required, cultures were transferred to synthetic medium (SM) containing galactose as the sole carbon source and grown for 4 h on that medium prior to transformation. SM contained, per liter of demineralized water, 5 g (NH4)2SO4, 3 g KH2PO4, 0.5 g MgSO4.7·H2O, and trace elements (Verduyn et al., 1992). Vitamins (Verduyn et al., 1992) were added after heat sterilization of the medium at 120 °C for 20 min. Glucose or galactose was separately sterilized at 110 °C and added to a final concentration of 20 g L−1. Where required, the medium was supplemented with appropriate amounts of auxotrophic requirements (Pronk, 2002). Solid medium was prepared by adding 2% (w/v) agar to the media prior to heat sterilization. Selective medium for the amdS marker was prepared as previously described (Solis-Escalante et al., 2013).
Table 1.
Strains used in this study
| Strain | Relevant genotype | Source |
|---|---|---|
| CEN.PK113-7D | MATa MAL2-8c SUC2 | Nijkamp et al. (2012) and van Dijken et al. (2000) |
| CEN.PK113-5D | MATa ura3-52 MAL2-8c SUC2 | van Dijken et al. (2000) |
| CEN.PK102-3A | MATa ura3-52 leu2-3 MAL2-8c SUC2 | van Dijken et al. (2000) |
| IMX212 | MATa ura3-52 leu2-3 MAL2-8c SUC2 spr3::(PGAL1-SCEI-Tcyc1; KlURA3) | This study |
| IMX221 | MATa ura3-52 MAL2-8c SUC2 spr3::(TagG-KlURA3- PGAL1-SCEI-Tcyc1-TagF) | This study |
| IMX222 | MATa ura3-52 leu2-3 MAL2-8c SUC2 spr3::(TagG-KlURA3- PGAL1-SCEI-Tcyc1-TagF) | This study |
| IMX224 | MATa ura3-52 MAL2-8c SUC2 spr3::(TagG-amdSYM-TagF) | This study |
Molecular biology techniques
PCR amplification was performed using Phusion® Hot Start II High-Fidelity DNA Polymerase (Thermo Fisher Scientific, Waltham, MA). To improve PCR efficiency, the conditions in the PCR as recommended by the supplier were modified by decreasing the primer concentration from 500 to 200 nM and increasing the Phusion™ Hot Start High-Fidelity polymerase concentration from 0.02 to 0.03 U μL−1. All other conditions followed the manufacturer's instructions. Genomic template DNA was isolated from S. cerevisiae CEN.PK113-7D using the Qiagen 100/G kit (Qiagen, Hilden, Germany). Plasmids maintained in E. coli DH5α were isolated with the GenElute™ Plasmid Miniprep Kit (Sigma, St. Louis, MI). DNA fragments were separated on 1% (w/v) agarose (Sigma) gels in 1× TAE (40 mM Tris-acetate pH 8.0 and 1 mM EDTA) and in 2% (w/v) agarose in 0.5× TBE (45 mM Tris-borate pH 8.0 1 mM EDTA) when fragments were smaller than 500 bp. Fragments were isolated from gel using the Zymoclean Gel DNA Recovery kit (Zymo Research, Irvine, CA). The glycolytic gene fragments for assembly were not gel-purified, but concentrated directly after PCR amplification by Vivacon® 500 spin columns (Sartorius Stedim, Aubagne, France). DNA concentrations were measured in a NanoDrop 2000 spectrophotometer (wavelength 260 nm; Thermo Fisher Scientific). Genomic DNA of transformants was isolated using the YeaStar™ Genomic DNA kit (Zymo Research). Multiplex PCR was performed with primers (Table 2) at a concentration of 150 nM. Cycling parameters were 94 °C for 3 min, then 35 cycles of 94 °C for 30 s, 60 °C for 90 s, and 72 °C for 60 s, followed by a 10-min incubation at 72 °C. Prior to transformation, fragments were pooled, maintaining equimolar concentrations with the marker fragment. Transformation to yeast was performed with the LiAc/ssDNA method (Gietz & Woods, 2002).
Table 2.
Oligonucleotide primers used in this study
| Primers | Sequence 5′ → 3′ |
|---|---|
| To add SHR-sequences | |
| TPI1 Rv+H | AGATTACTCTAACGCCTCAGCCATCATCGGTAATAGCTCGAATTGCTGAGAACCCGTGACTAGTGTGAGCGGGATTTAAACTGTG |
| TPI1 Fw+I | GCCTACGGTTCCCGAAGTATGCTGCTGATGTCTGGCTATACCTATCCGTCTACGTGAATAGCGAAAATGACGCTTGCAGTG |
| FBA1 Rv+H | GTCACGGGTTCTCAGCAATTCGAGCTATTACCGATGATGGCTGAGGCGTTAGAGTAATCTAAAATCTCAAAAATGTGTGGGTCATTACG |
| FBA1 Fw+G | GCCAGAGGTATAGACATAGCCAGACCTACCTAATTGGTGCATCAGGTGGTCATGGCCCTTAGTGCATGACAAAAGATGAGCTAGG |
| Amds-GPD Fw+A | ACTATATGTGAAGGCATGGCTATGGCACGGCAGACATTCCGCCAGATCATCAATAGGCACGCTGGAGCTCTTCGA |
| Amds-GPD Rv+B | GTTGAACATTCTTAGGCTGGTCGAATCATTTAGACACGGGCATCGTCCTCTCGAAAGGTGGGCCGCAAATTAAAGCCTTCGAG |
| Amds-TEF Fw+A | ACTATATGTGAAGGCATGGCTATGGCACGGCAGACATTCCGCCAGATCATCAATAGGCACGCGACATGGAGGCCCAGAATACC |
| Amds-TEF RV+B | GTTGAACATTCTTAGGCTGGTCGAATCATTTAGACACGGGCATCGTCCTCTCGAAAGGTGAGTATAGCGACCAGCATTCACATACG |
| KlLEU2-Fw+A | ACTATATGTGAAGGCATGGCTATGGCACGGCAGACATTCCGCCAGATCATCAATAGGCACAGAGATCCGCAGGCTAACCG |
| KlLEU2-Rv+B | GTTGAACATTCTTAGGCTGGTCGAATCATTTAGACACGGGCATCGTCCTCTCGAAAGGTGGCTGTGAAGATCCCAGCAAAGG |
| PFK2 Rv+F | TGCCGAACTTTCCCTGTATGAAGCGATCTGACCAATCCTTTGCCGTAGTTTCAACGTATGATAGCCATTCTCTGCTGCTTTGTTG |
| PFK2 Fw+J | GGCCGTCATATACGCGAAGATGTCCAAGCAGGTAGAACACATAGTCTGAGCATCTCGTCGGAGATCCGAGGGACGTTTATTGG |
| PFK1 Rv+D | ACGCATCTACGACTGTGGGTCCCGTGGAGAAATGTATGAAACCCTGTATGGAGAGTGATTTCGAGATTCCTCAATCCATACACCATTATAG |
| PFK1 Fw+J | CGACGAGATGCTCAGACTATGTGTTCTACCTGCTTGGACATCTTCGCGTATATGACGGCCTGTCGTCTTCGTGAACCATTGTC |
| PGI1 Rv+D | AATCACTCTCCATACAGGGTTTCATACATTTCTCCACGGGACCCACAGTCGTAGATGCGTCTGAAGAAGGCATACTACGCCAAG |
| PGI1 Fw+C | ACGTCTCACGGATCGTATATGCCGTAGCGACAATCTAAGAACTATGCGAGGACACGCTAGTTCGCGACACAATAAAGTCTTCACG |
| HXK2 Rv+C | CTAGCGTGTCCTCGCATAGTTCTTAGATTGTCGCTACGGCATATACGATCCGTGAGACGTGCAAGAGAAAAAAACGAGCAATTGTTAAAAG |
| HXK2 Fw+B | CACCTTTCGAGAGGACGATGCCCGTGTCTAAATGATTCGACCAGCCTAAGAATGTTCAACGACGGCACCGGGAAATAAACC |
| PGK1 Fw+I | GTGCCTATTGATGATCTGGCGGAATGTCTGCCGTGCCATAGCCATGCCTTCACATATAGTCCTGCATTTAAAGATGCCGATTTGG |
| PGK1 Rv+A | GTAGACGGATAGGTATAGCCAGACATCAGCAGCATACTTCGGGAACCGTAGGCATTTTAGCGTAAAGGATGGGGAAAGAG |
| For the construction of pUDC073 | |
| SCEI-Fw | GCTGCCACTAGTATAATGCATCAAAAAAACCAGGTAATG |
| SCEI-Rv | TTATCACTCGAGTTATTACTTAAGGAAAGTTTCGGAGGAGATAG |
| For fusion-PCR of the IsceI-URA3-cassette | |
| URA3-Fw | GAGCCATCCATTCGTAATTCACTACTGCCTGAGGGTTGTTCTCAGAAGCTCATCGAACTGTCATC |
| URA3-Rv | CCATTCTGTAGCCACCTTATCCATGACCGTTTTATTAATTATTTCATAGCACTTGTAATTATATTACCCTGTTATCCCTAGCGAAGTGAGTGTTGCACCGTGCCAATG |
| SCEI-Fw(2) | GCTGCATCCTTCCCATGCAAAGTGTCTTCGTATTTAGTGATGTTTTGTTAGCGACACAAAGCTAGGGATAACAGGGTAATATGCAGTGAGCGCAACGCAATTAATG |
| SCEI-Rv(2) | AACAACCCTCAGGCAGTAGTGAATTACGAATGGATGGCTCCGACTCACTATAGGGCGAATTGG |
| FUS1 | GCTGCATCCTTCCCATGCAAAGTG |
| FUS2 | CCATTCTGTAGCCACCTTATCC |
| SCEI+URA-Fw | GCAGTGAGCGCAACGCAATTAATG |
| SCEI+URA-Rv | GAAGTGAGTGTTGCACCGTGCCAATG |
| Tag F-REC-fw | CCATTCTGTAGCCACCTTATCCATGACCGTTTTATTAATTATTTCATAGCACTTGTAATTTGCCGAACTTTCCCTGTATGAAGCGATCTGACCAATCCTTTGCCG |
| Tag F-REC-rv | CCTGCATTGGCACGGTGCAACACTCACTTCGCTAGGGATAACAGGGTAATATCATACGTTGAAACTACGGCAAAGGATTGGTCAGATCGCTTCATACAGGG |
| Tag G-REC-fw | GCTGCATCCTTCCCATGCAAAGTGTCTTCGTATTTAGTGATGTTTTGTTAGCGACACAAAGCCAGAGGTATAGACATAGCCAGACCTACCTAATTGGTGCATC |
| Tag G-REC-rv | GTAACTCACATTAATTGCGTTGCGCTCACTGCATATTACCCTGTTATCCCTAGCAAGGGCCATGACCACCTGATGCACCAATTAGGTAGGTCTGGCTATGTCTATACC |
| For construction of the fragments targeting the CAN1 locus | |
| H1-Fw | TTCTAGGTTCGGGTGACGTGAAG |
| H1-Rv | AAGGGCCATGACCACCTGATGCACCAATTAGGTAGGTCTGGCTATGTCTATACCTCTGGCATTACCCTGTTATCCCTATTAATCACATTCCCACGCCATTTCG |
| Y1 | CATACGTTGAAACTACGGCAAAGGATTGGTCAGATCGCTTCATACAGGGAAAGTTCGGCATAGGGATAACAGGGTAATGCTCATTGATCCCTTAAACTTTCTTTTCGGTGTATGAC |
| Y2 | CCAGTTTTCAATCTGTCGTCAATCGAAAGTTTATTTTAATCACATTCCCACGCCATTTCGCATTCTCACCCTCATAAGTCATACACCGAAAAGAAAGTTTAAGGGATCAATGAGC |
| H2-Fw | AATAAACTTTCGATTGACGACAGATTG |
| H2-Rv | GTTTCCGGGTGAGTCATACG |
| FUS3 | CATACGTTGAAACTACGGCAAAGG |
| For analytical PCR: glycolytic genes integrated in the CAN1 locus | |
| G- fw | CCGTCATCGGAGTCGTTATCAG |
| G- rv | GCTCTTTTCTTCTGAAGGTCAATG |
| F-fw | GACGCCATTTGGAACGAAAAAAAG |
| F-rv | TAACGGCAAACAGCAAAGGC |
| H-fw | GTTACGTGCTCAGTTGTTAGATATG |
| H-rv | GCAGAAGTGTCTGAATGTATTAAGG |
| I-fw | TGAGCCACTTAAATTTCGTGAATG |
| I-rv | TTTCTCTTTCCCCATCCTTTACG |
| A-fw | AAGGATTCGCGCCCAAATCG |
| A-rv | CTTCCCAAGATTGTGGCATGTC |
| B-fw | TGGCTATCGCTGAAGAAGTTGG |
| B-rv | ACGGAATAGAACACGATATTTGC |
| C-fw | TCACGGGATTTATTCGTGACG |
| C-rv | CCCACGATGCTTCTACCAAC |
| D-fw | ACTCGCCTCTAACCCCACG |
| D-rv | AATCATGTTGATGACGACAATGG |
| J-fw | GCTTAATCTGCGTTGACAATGG |
| J-rv | CAATAAACGTCCCTCGGATCTC |
| For multiplex PCR: glycolytic genes integrated in the SPR3 locus | |
| G- fw | CTTGGCTCTGGATCCGTTATCTG |
| G- rv | GCTCTTTTCTTCTGAAGGTCAATG |
| F-fw | GACGCCATTTGGAACGAAAAAAAG |
| F-rv | TTGGGCTGGACGTTCCGACATAG |
| H-fw | GTTACGTGCTCAGTTGTTAGATATG |
| H-rv | GCAGAAGTGTCTGAATGTATTAAGG |
| I-fw | TGAGCCACTTAAATTTCGTGAATG |
| I-rv | TTTCTCTTTCCCCATCCTTTACG |
| A-fw | AAGGATTCGCGCCCAAATCG |
| A-rv | CTTCCCAAGATTGTGGCATGTC |
| B-fw | TGGCTATCGCTGAAGAAGTTGG |
| B-rv | ACGGAATAGAACACGATATTTGC |
| C-fw | TCACGGGATTTATTCGTGACG |
| C-rv | CCCACGATGCTTCTACCAAC |
| D-fw | ACTCGCCTCTAACCCCACG |
| D-rv | AATCATGTTGATGACGACAATGG |
| J-fw | GCTTAATCTGCGTTGACAATGG |
| J-rv | CAATAAACGTCCCTCGGATCTC |
Construction of a platform strain for I-SceI-assisted integration
The plasmids used in this study are listed in Table 3. Plasmid pUDC073 was obtained by cloning the SCEI ORF into pAG416GAL-ccdB. The SCEI ORF was amplified from Biobrick BBa_K175041 (http://parts.igem.org/Part:BBa_K175041) with primers SCEI-Fw and SCEI-Rv. The resulting fragment was restricted by SpeI and XhoI and ligated into pAG416GAL-ccdB, yielding pUDC073.
Table 3.
Plasmids used in this study
| Plasmid | Characteristic | Source |
|---|---|---|
| pUG72 | PCR template for Kluyveromyces lactis URA3 (KlURA3) | Gueldener et al. (2002) |
| pUG73 | PCR template for Kluyvermyces lactis LEU2 (KlLEU2) | Gueldener et al. (2002) |
| pUGamdSYM | PCR template for amdSYM under the control of the AgTEF2 promoter | Solis-Escalante et al. (2013) |
| pUDE158 | PCR template for amdSYM under the control of the TDH3 promoter | Solis-Escalante et al. (2013) |
| pAG416GAL-ccdB | CEN6/ARS4 ori, URA3, PGAL1-ccdB-TCYC1 | Alberti et al. (2007) |
| pUDC073 | CEN6/ARS4 ori, URA3, PGAL1-SCEI-TCYC1 | This study |
The integration site was constructed by integration of the TagG-SCEI-URA3-TagF cassette into the yeast genome. Construction of the TagG-SCEI-URA3-TagF cassette was performed in multiple steps. First, SCEI was amplified from pUDC073 with primers SCEI-Fw(2) and SCEI-Rv(2), and KlURA3 was amplified from pUG72 with primers URA-Fw and URA-Rv. The resulting cassettes were gel-purified, and 100 ng of each cassette was used for fusion-PCR (Shevchuk et al., 2004) using primers FUS1 and FUS2. Cycling parameters were 98 °C for 1 min, then eight cycles of 98 °C for 30 s, 58 °C for 30 s, and 72 °C for 120 s, followed by 27 cycles of 98 °C for 30 s, 70 °C for 30 s, and 72 °C for 120 s, followed by a 10-min incubation at 72 °C. The intermediate strain IMX212 was constructed by integration of the resulting product at the SPR3 locus of CEN.PK102-3A, yielding strain IMX212 (Fig. 1a1). Secondly, genomic DNA of IMX212 served as a template for amplification of the SCEI/URA3 cassette with primers SCEI+URA-Fw and SCEI+URA-Rv, resulting in fragment X1 (Fig. 1a2). Thirdly, the flanking fragments X2 and X3 carrying 3 regions: (1) the regions homologous to the SPR3 locus necessary for integration of the final cassette, (2) the I-SceI recognition site, and (3) the F and G synthetic homologous recombination sequences (SHR-sequences) required for integration of cassettes during I-SceI-assisted integration were obtained by PCR. Fragment X2 (Fig. 1a2) was obtained by annealing oligonucleotides TagG-REC-Fw and TagG-REC-Rv in a 50 μL PCR mix at a concentration of 1 μM. This mix was subjected to 10 cycles in a thermocycler using the following conditions: 98 °C for 30 s, 65 °C for 30 s, and 72 °C for 15 s, followed by a 10-min incubation at 72 °C. Fragment X3 was obtained by the same procedure using oligonucleotides TagF-REC-Fw and TagF-REC-Rv (Fig. 1a2). Fourthly, the DNA fragments X1, X2, and X3 were gel-purified and fused by fusion-PCR using primers FUS1 and FUS2, using the same cycling parameters as for the previous fusion-PCR (Fig. 1a2). The resulting product was the TagG-SCEI-URA3-TagF cassette (Fig. 1a3), which was gel-purified and transformed using the LiAc/ssDNA method (Gietz & Woods, 2002) to CEN.PK113-5D, leading to IMX221, and to CEN.PK102-3A, leading to IMX222. In both strains, the regions containing the I-SceI recognition sites and the SHR-sequences F and G were PCR-amplified and sequenced using Sanger sequencing (BaseClear, Leiden, the Netherlands).
Fig. 1.

Construction of the TagG-SCEI-KlURA3-TagF and the H2 cassette. (a) First, the SCEI/URA3 cassette was obtained by PCR on genomic DNA of IMX212 with primers SCEI+URA-Fw and SCEI+URA-Rv, resulting in fragment X1 (a1). Fragment X2 was obtained by fusing oligos TagG-REC-Fw and TagG-REC-Rv in an independent PCR. Fragment X3 was obtained in the same way, using oligos TagF-REC-fw and TagF-REC-Rv (a2). Fragments X1, X2, and X3 were fused in a fusion-PCR with primers FUS1 and FUS2, resulting in the TagG-SCEI-KlURA3-TagF cassette (a3). (b) Fragment Z2 was obtained by PCR on genomic DNA of CEN.PK113-7D with primers H2-fw and H2-rv (b1). Fragment Z1 was obtained by fusing oligos Y1 and Y2 in a PCR (b2). Fragments Z1 and Z2 were fused in a fusion-PCR with oligos FUS3 and H2-rv, resulting in fragment H2, which contains SHR-sequence F and 300 bp homology to the CAN1 locus (b3).
Preparation of fragments for in vivo assembly
Fragments for in vivo assembly were obtained by PCR from either genomic or plasmid template DNA. The amplified fragments were stocked in TE buffer (10 mM Tris, pH8, 1 mM EDTA). Fragments amplified from plasmid templates were subjected to gel extraction to prevent false-positive transformants that might arise from contamination with linearized template plasmid. Fragment amdSYMAB carrying the counter selectable amdS marker behind the TDH3 promoter was amplified from pUDE158 (Table 3) in the experiment targeting the CAN1 locus using primers Amds-GPD-Fw+A and Amds-GPD-Rv+B. In the other experiments, an amdSYMAB cassette carrying the amdS marker behind the AgTEF2 promoter was used, to eliminate sequence homology between the marker cassette and the yeast genome. This cassette was amplified from pUGamdSYM with primers Amds-TEF-Fw+A and Amds-TEF-Rv+B. The marker fragment KlLEU2AB was obtained from pUG73 with primers KlLEU2-Fw+A and KlLEU2-Rv+B. The fragments containing the glycolytic genes were all amplified from CEN.PK113-7D genomic DNA (Nijkamp et al., 2012) using the primers with the corresponding glycolytic gene names listed in Table 2. Fragments H1 and H2 homologous to the CAN1 locus and used for targeted integration of in vivo-assembled constructs at that locus were obtained by PCR amplification from CEN.PK113-7D genomic DNA. H1 was amplified with primers H1-Fw and H1-Rv. Fragment H2 was obtained in two steps (Fig. 1b). First fragment Z1 was obtained by fusion of oligos Y1 and Y2 in the same way as described for fragment X2 (Fig. 1b2). Fragment Z2 was obtained by PCR on genomic DNA using primers H2-Fw and H2-Rv (Fig. 1b1). Fragment Z1 and Z2 were gel-purified and fused by fusion-PCR using the same method as described before using primers H2-rv and FUS3, resulting in fragment H2 (Fig. 1b3). Fragments H1 and H2 were gel-purified before addition to the transformation mix.
Results
Poor efficiency of simultaneous assembly and targeted integration of seven glycolytic genes into the CAN1 locus
We demonstrated previously that S. cerevisiae can assemble nine fragments very efficiently and with high fidelity into a 21-kb plasmid carrying six glycolytic genes (Kuijpers et al., 2013). To test whether combined assembly and targeted integration at a specific locus of a multiple gene construct could be achieved, the six previously designed glycolytic gene fragments (Kuijpers et al., 2013) and one additional glycolytic gene fragment were used to assemble and integrate a total of seven glycolytic genes in a single step (Fig. 2). A set of ten fragments was obtained by adding two flanking fragments designed to target the CAN1 locus (Whelan et al., 1979) and one marker fragment carrying the amdS dominant marker [Fig. 2, (Solis-Escalante et al., 2013)] to the seven glycolytic gene cassettes. All fragments were designed to overlap by 60-bp synthetic homologous recombination sequences (from now on referred to as SHR-sequences) that do not share homology with the yeast genome (Kuijpers et al., 2013). After transformation with these ten fragments, yeast cells were grown on glucose synthetic medium. To identify transformants in which genomic integration of amdS had occurred, acetamide was used as the sole nitrogen source. Thirty-five transformants were obtained, of which 20 were picked and plated on medium containing L-canavanine, to select for integration of the assembled construct in the CAN1 locus. Only two of the 20 transformants were able to grow in the presence of L-canavanine, indicating that CAN1 was disrupted in only 10% of the tested transformants. Of these two L-canavanine-resistant transformants, only one was correctly assembled and carried all transformed fragments (Fig. 3). Although simultaneous assembly and targeted integration of a multigene construct in a single chromosomal locus was achieved, it was extremely inefficient (one of 20 of the tested transformants). As we previously established efficiencies of plasmid assembly of 95% with the same overlapping sequences (Kuijpers et al., 2013), these results pointed at the integration step as the main bottleneck for the CATI approach.
Fig. 2.
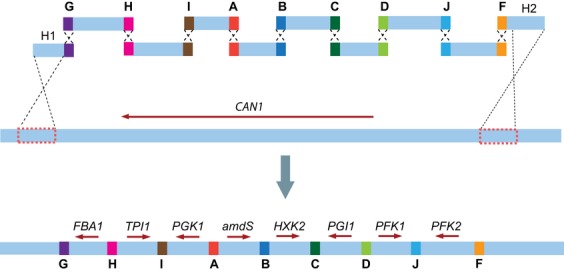
Combined assembly and integration of seven glycolytic genes in the CAN1 locus of Saccharomyces cerevisiae. Ten overlapping DNA fragments, containing seven glycolytic genes, the amdS selection marker, and the two flanking fragments H1 and H2, carrying 300-bp sequences homologous to the CAN1 integration locus, were cotransformed to S. cerevisiae and assembled in yeast via homologous recombination into a single large integration cassette. 60-bp SHR-sequences were used to promote in vivo assembly of the fragments.
Fig. 3.
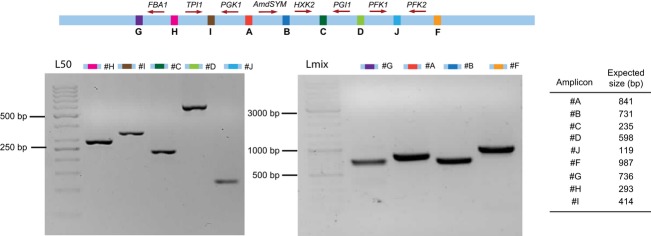
PCR analysis of a positive transformant after cotransformation of ten overlapping fragments to Saccharomyces cerevisiae. The PCRs were designed to produce amplicons covering the indicated junctions. PCR products covering junctions H, C, D, J, and I were separated on a 2% agarose gel, and PCR products covering junctions A,F,G, and B were separated on a 1% agarose gel by electrophoresis. In the lane labeled ‘L50,’ a 50-bp Gene Ruler ladder was loaded; in the lane labeled ‘Lmix,’ a Gene Ruler Mix ladder was loaded; sizes are indicated. All amplicons matched the expected size, thereby indicating correct assembly and integration of seven glycolytic genes in the CAN1 locus.
Substantial improvement of integration efficiency of an amdS marker cassette into the SPR3 locus using I-SceI-induced double-strand DNA breaks
The cellular function of homologous recombination is to repair double-strand DNA breaks (DSBs). While in vivo assembly supplies DNA fragments with open ends, readily accessible for the homologous recombination machinery, chromosomal integration of these fragments requires recombination of DNA fragments with intact genomic DNA and is therefore far less likely to occur (Orr-Weaver et al., 1981; Leem et al., 2003). A way to enhance the efficiency of integration would therefore be to artificially introduce a DSB at the integration site. Rare-cutting endonucleases can be used to introduce DSBs, thereby drastically increasing the efficiency of integration by homologous repair (Storici et al., 2003; Wingler & Cornish, 2011). The well-studied I-SceI meganuclease, originally encoded by the S. cerevisiae mitochondrial gene SCEI, has a 18-bp unique recognition sequence and has been previously functionally expressed in the nucleus of S. cerevisiae (Plessis et al., 1992). To investigate whether introduction of DSBs might eliminate or alleviate the bottleneck in chromosomal integration, a platform was engineered to combine in vivo assembly with I-SceI-facilitated integration. IMX221 was constructed by integration of a cassette containing SCEI under the control of the inducible GAL1 promoter and a uracil marker in the SPR3 locus of the uracil auxotroph S. cerevisiae CEN.PK113-5D (Fig. 4a). The resulting locus carried two 22-bp I-SceI recognition sequences flanked by 60-bp SHR-sequences G and F (Fig. 4a). A single cassette, carrying the amdS marker and SHR-sequences G and F at its 5′ and 3′ ends, respectively, was constructed to integrate at this synthetic locus (Fig. 4b). This cassette was transformed to IMX221 preincubated in galactose medium to induce expression of SCEI. To quantify the effect of the I-SceI-induced DSB, IMX221 cells not induced on galactose were transformed with the same amdSYM cassette and used as a negative control. Previous reports indicated that incubation of strains containing SCEI under control of the GAL1 promoter in the presence of galactose resulted in induction of DSBs within several hours (Storici et al., 2003). In the present study, SCEI was induced by growing the yeast cells for four hours in galactose medium prior to transformation. While transformation of I-SceI-expressing cells resulted in c. 104 transformants, the negative control yielded only 15 transformants. PCR analysis of three colonies of each of the I-SceI-expressing and nonexpressing transformants revealed that they all contained the amdSYM cassette, correctly integrated at the SPR3 locus. One correct clone resulting from the transformation of induced cells was named IMX224. Sequencing of the SPR3 locus of IMX224 showed that the region between the SHR-sequences G and F was successfully replaced by the amdSYM cassette without leaving any scar of the I-SceI recognition sequences. These results demonstrate that induction of a DSB is a critical step for integration of DNA fragments in yeast chromosomes and suggested that I-SceI-assisted integration should improve the efficiency of CATI.
Fig. 4.
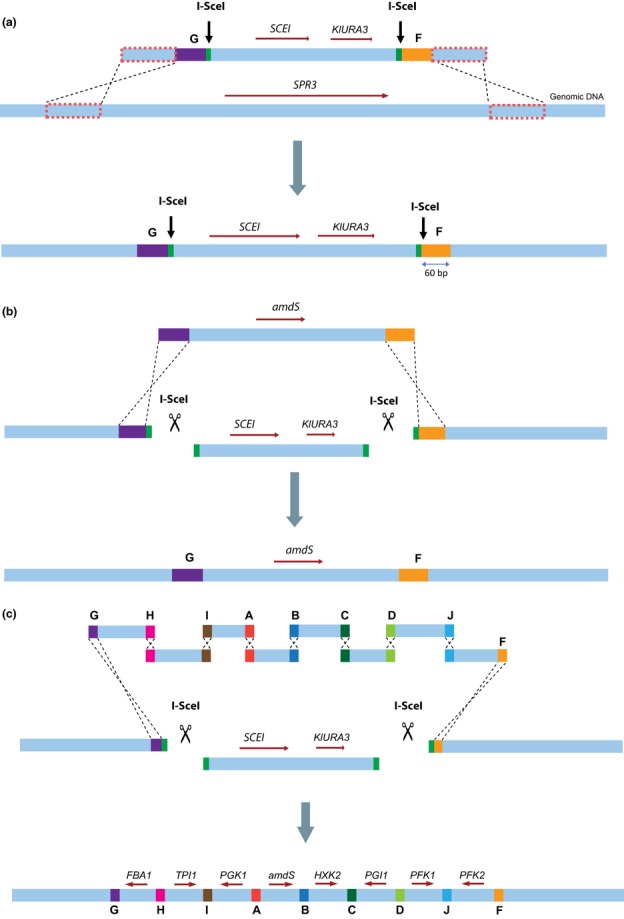
Design of the I-SceI-facilitated CATI method. (a) First, the platform strain was obtained by introducing a cassette containing SCEI and KlURA3 flanked by three regions, the I-SceI recognition site, synthetic recombination sequences G and F, and flanking regions homologous to the targeted locus SPR3. (b) Induction of plasmid-borne SCEI in the platform strain prior to transformation, causing excision of the SCEI/URA3 fragment and leaving the 60-bp SHR-sequences F and G exposed for recombination. Transformation of the induced yeast cells with the amdSYM cassette flanked by SHR-sequences G and F led to integration of the cassette at the I-SceI-restricted locus. (c) Integration of multiple overlapping fragments, using the same integration approach described in (b), leading to I-SceI-assisted integration of seven glycolytic genes and a KlLEU2 marker cassette into the SPR3 locus.
I-SceI-assisted integration of seven glycolytic genes at a synthetic chromosomal locus
To test whether I-SceI-assisted integration could be combined with in vivo assembly of multiple genes, the same seven overlapping glycolytic gene cassettes used in the first experiment and the amdSYM marker cassette were cotransformed to I-SceI-expressing cells of the platform strain IMX221 (Fig. 4c). Two control sets of fragments were also tested. An incomplete set of cassettes (lacking HXK2BC) was used to estimate the occurrence of nonhomologous recombination events within the construct. Secondly, a single cassette carrying the selection marker but without homology to the integration site was used to evaluate the possible integration of nonhomologous fragments. Transformation of I-SceI-expressing cells with these two control sets of fragments did not yield transformants. Conversely, transformation of I-SceI-expressing cells with the complete set of fragments resulted in 336 transformants capable of using acetamide as sole nitrogen source. Analysis by multiplex PCR of ten randomly picked clones demonstrated the integration of a full set of fragments in the SPR3 locus of S. cerevisiae for nine clones (Fig. 5a). To evaluate the robustness of the CATI approach, the same experiment was repeated by replacing the dominant amds marker by the widely used auxotrophic selection marker LEU2. A new platform strain was constructed by introducing the same synthetic locus used in IMX221 in the leucine auxotrophic strain S. cerevisiae CEN.PK102-3A, resulting in strain IMX222. Subsequently, the above-described cassettes carrying the seven glycolytic genes were cotransformed to IMX222 together with the Kluyveromyces lactis LEU2 orthologous marker cassette. The KlLEU2-based CATI resulted in 470 clones, of which ten clones were analyzed by multiplex PCR. These ten clones harbored all expected amplicons, indicating the correct integration of all eight fragments at the targeted locus (Fig 5b). These results demonstrate the high efficiency (c. 95%) and robustness of I-SceI-assisted simultaneous assembly and chromosomal integration of eight overlapping DNA cassettes, comprising a 22-kb construct, in S. cerevisiae.
Fig. 5.

Characterization of positive clones isolated after I-SceI-assisted CATI of ten fragments by multiplex PCR. (a) PCR patterns of ten clones resulting from cotransformation of the glycolytic genes with the amdS selection marker, (b) PCR patterns of ten clones obtained by replacing amdS by the KlLEU2 selection marker in the cotransformation with the glycolytic genes. Transformants were randomly picked and analyzed by multiplex PCR producing amplicons covering the indicated junctions. PCR products were separated on a 2% agarose gel by electrophoresis. In lanes labeled ‘L’, a 50-bp Gene Ruler ladder was loaded; sizes are indicated. From these 20 tested clones, a single one [(a), transformant number 10, amplicon C] did not display the expected pattern.
Discussion
The current demand from the biotechnology industry for strains able to produce complex synthetic pathways with ever increasing productivity and robustness requires the construction of strains simultaneously expressing dozens of homologous and heterologous genes (Ro et al., 2006; Koopman et al., 2012). Genetic tools enabling the fast and efficient construction of chromosome-borne large synthetic constructs are therefore urgently needed. Comparatively little attention has been given to the development of such methods to date. One study explored the possibilities of combining recombination-based cloning with chromosomal integration in S. cerevisiae (Shao et al., 2009) and provided a clear proof of principle. Eight genes were assembled in vivo into a 23.7-Kb construct and successfully integrated into a δ-site. Using this approach, variable number of clones (30–50 clones) and efficiencies (10% to 70%) were obtained depending on the length of the overlapping sequences used, the lowest efficiency being obtained using short overlaps of 50 bp. In the present study, implementation of I-SceI-assisted combined in vivo assembly and targeted chromosomal integration in S. cerevisiae led to consistently high efficiencies of c. 95% for a construct of a similar size using 60-bp overlapping sequences and a similar number of fragments. Furthermore, while transformation to δ-sites inherently randomizes the location and number of integration sites, the present work is, to the best of our knowledge, the first published report of targeted integration of an in vivo-assembled multigene construct in S. cerevisiae. The SHR-sequences used in the presented platform for the assembly of the fragments add versatility to the system, which allows for parallel construction of replicative and integrative constructs. These achievements present a new step toward reliable and robust high-throughput strain construction. We are currently using the I-SceI-assisted CATI approach to assemble and integrate complete pathways up to 35 kb from 15 fragments, and the efficiency of correct assembly is similar to the efficiencies in this study. While the number of transformants seems to decrease with the number of assembled fragments (c. 400 clones with nine fragments and c. 200 clones with 15 fragments), the number of transformants obtained is sufficiently high to be compatible with high-throughput strain construction programs.
The present study indicates that the critical step in chromosomal integration and the key to the high efficiencies obtained for combined assembly and integration of a multifragment pathway is the introduction of a double-strand DNA break at the integration locus targeted. Because of its high specificity and resulting lack of toxicity in most tested organisms, I-SceI has been employed in many systems to induce site-specific double-strand breaks, such as induction of homologous recombination in higher eukaryotes (Rouet et al., 1994; Choulika et al., 1995), seamless gene modifications in yeast (Noskov et al., 2010; Khmelinskii et al., 2011), and sequential pathway engineering in yeast (Wingler & Cornish, 2011). In recent years, several approaches have been explored to engineer synthetic endonucleases for any recognition sequence of interest: the zinc-finger nucleases (ZFNs; Kandavelou et al., 2005), the TAL effector nucleases (TALENs; Christian et al., 2010), and the recent RNA-guided CRISPR/Cas nucleases (Cong et al., 2013). Those synthetic ‘genomic scissors’ could greatly contribute to further development of the CATI method for chromosomal modifications. Screening of those synthetic endonucleases for site-specific nuclease activity in S. cerevisiae might reveal even more efficient DSB inducers, thereby further improving the targeting efficiency for integration. Furthermore, design of synthetic meganucleases for specific recognition sequences already present in the yeast genome could abolish the need for a defined synthetic locus to target for integration. While in the present approach the meganuclease was expressed by the host organism, it has been previously shown that endonucleases can enter yeast cells and reach the nucleus during transformation (Schiestl & Petes, 1991). Cotransformation of cells with the desired fragments and custom-made meganucleases would make the presented method applicable to any strain without any prior modification of the genome. We therefore anticipate that the coming years will see further increases in the flexibility and ease of endonuclease-assisted CATI for strain engineering.
While S. cerevisiae is known for its high efficiency of homologous recombination (Schiestl & Petes, 1991; Gibson et al., 2008), the present work demonstrates that integration of DNA fragments in its genome can be substantially increased by introduction of a DSB. Microorganisms in which it is notoriously difficult to achieve highly efficient targeted integration of DNA fragments, such as the yeast Kluyveromyces lactis and filamentous fungi (Kooistra et al., 2004; Ninomiya et al., 2004; Krappmann et al., 2006; Snoek et al., 2009), may similarly benefit from endonuclease-assisted integration. Although the presented method has been engineered for S. cerevisiae, the principle could be applied to any organism with an efficient homologous repair mechanism. Alternatively, S. cerevisiae and its outstanding recombination efficiency could be exploited to assemble the in vivo DNA constructs prior to transformation to the final host.
The presented I-SceI-assisted CATI approach has drastically changed the strain engineering procedures in our laboratory and opened new possibilities for large-scale metabolic engineering of S. cerevisiae. It is our hope that this work will further contribute to the development of S. cerevisiae as a valuable platform for the production of many industrially relevant compounds.
Acknowledgments
We thank the TU Delft iGEM team 2009 for supplying the I-SceI BioBrick. We thank Dr. Kirsten Benjamin (Amyris, Emeryville CA) for helpful discussions. We thank the Netherlands Organisation for Scientific Research (NWO) for financial support (VIDI grant).
References
- Alberti S, Gitler AD, Lindquist S. A suite of Gateway cloning vectors for high-throughput genetic analysis in Saccharomyces cerevisiae. Yeast. 2007;24:913–919. doi: 10.1002/yea.1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit RM, Wilhelm RN, Scherer-Becker D, Ostermeier C. An improved method for fast, robust, and seamless integration of DNA fragments into multiple plasmids. Protein Expr Purif. 2006;45:66–71. doi: 10.1016/j.pep.2005.09.022. [DOI] [PubMed] [Google Scholar]
- Choulika A, Perrin A, Dujon B, Nicolas JF. Induction of homologous recombination in mammalian chromosomes by using the I-SceI system of Saccharomyces cerevisiae. Mol Cell Biol. 1995;15:1968–1973. doi: 10.1128/mcb.15.4.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian M, Cermak T, Doyle EL, Schmidt C, Zhang F, Hummel A, Bogdanove AJ, Voytas DF. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics. 2010;186:757–761. doi: 10.1534/genetics.110.120717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis T, Adie T, Baldwin GS. DNA assembly for synthetic biology: from parts to pathways and beyond. Integr Biol (Camb) 2011;3:109–118. doi: 10.1039/c0ib00070a. [DOI] [PubMed] [Google Scholar]
- Entian KD, Kotter P. Yeast genetic strain and plasmid collections. In: Stansfield I, Stark MJR, editors. Method Microbiol. Vol. 36. Amsterdam, The Netherlands: Academic Press; 2007. pp. 629–666. [Google Scholar]
- Gibson DG, Benders G, Axelrod KC, Zaveri J, Algire M, Moodie M, Montague MG, Venter JC, Smith HO, Hutchison C. One-step assembly in yeast of 25 overlapping DNA fragments to form a complete synthetic Mycoplasma genitalium genome. P Natl Acad Sci USA. 2008;105:20404–20409. doi: 10.1073/pnas.0811011106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang RY, Venter JC, Iii CAH, Smith HO, America N. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6:12–16. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- Gietz RD, Woods R. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 2002;350:87–96. doi: 10.1016/s0076-6879(02)50957-5. [DOI] [PubMed] [Google Scholar]
- Gueldener U, Heinisch J, Koehler GJ, Voss D, Hegemann JH. A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res. 2002;30:e23. doi: 10.1093/nar/30.6.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandavelou K, Mani M, Durai S, Chandrasegaran S. “Magic” scissors for genome surgery. Nat Biotechnol. 2005;23:686–687. doi: 10.1038/nbt0605-686. [DOI] [PubMed] [Google Scholar]
- Khmelinskii A, Meurer M, Duishoev N, Delhomme N, Knop M. Seamless gene tagging by endonuclease-driven homologous recombination. PLoS ONE. 2011;6:e23794. doi: 10.1371/journal.pone.0023794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooistra R, Hooykaas PJ, Steensma HY. Efficient gene targeting in Kluyveromyces lactis. Yeast. 2004;21:781–792. doi: 10.1002/yea.1131. [DOI] [PubMed] [Google Scholar]
- Koopman F, Beekwilder J, Crimi B, van Houwelingen A, Hall RD, Bosch D, van Maris AJ, Pronk JT, Daran JM. De novo production of the flavonoid naringenin in engineered Saccharomyces cerevisiae. Microb Cell Fact. 2012;11:155. doi: 10.1186/1475-2859-11-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krappmann S, Sasse C, Braus GH. Gene targeting in Aspergillus fumigatus by homologous recombination is facilitated in a nonhomologous end-joining-deficient genetic background. Eukaryot Cell. 2006;5:212–215. doi: 10.1128/EC.5.1.212-215.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuijpers NGA, Solis-Escalante D, Bosman L, van den Broek M, Pronk JT, Daran JM, Daran-Lapujade P. A versatile, efficient strategy for assembly of multi-fragment expression vectors in Saccharomyces cerevisiae using 60 bp synthetic recombination sequences. Microb Cell Fact. 2013;12:47. doi: 10.1186/1475-2859-12-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leem SH, Noskov VN, Park JE, Kim SI, Larionov V, Kouprina N. Optimum conditions for selective isolation of genes from complex genomes by transformation-associated recombination cloning. Nucleic Acids Res. 2003;31:e29. doi: 10.1093/nar/gng029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MZ, Elledge SJ. Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nat Methods. 2007;4:251–256. doi: 10.1038/nmeth1010. [DOI] [PubMed] [Google Scholar]
- Monteilhet C, Perrin A, Thierry A, Colleaux L, Dujon B. Purification and characterization of the in vitro activity of I-Sce I, a novel and highly specific endonuclease encoded by a group I intron. Nucleic Acids Res. 1990;18:1407–1413. doi: 10.1093/nar/18.6.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijkamp JF, van den Broek M, Datema E, et al. De novo sequencing, assembly and analysis of the genome of the laboratory strain Saccharomyces cerevisiae CEN.PK113-7D, a model for modern industrial biotechnology. Microb Cell Fact. 2012;11:36. doi: 10.1186/1475-2859-11-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninomiya Y, Suzuki K, Ishii C, Inoue H. Highly efficient gene replacements in Neurospora strains deficient for nonhomologous end-joining. P Natl Acad Sci USA. 2004;101:12248–12253. doi: 10.1073/pnas.0402780101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noskov VN, Segall-Shapiro TH, Chuang RY. Tandem repeat coupled with endonuclease cleavage (TREC): a seamless modification tool for genome engineering in yeast. Nucleic Acids Res. 2010;38:2570–2576. doi: 10.1093/nar/gkq099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr-Weaver TL, Szostak JW, Rothstein RJ. Yeast transformation: a model system for the study of recombination. P Natl Acad Sci USA. 1981;78:6354–6358. doi: 10.1073/pnas.78.10.6354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plessis A, Perrin A, Haber JE, Dujon B. Site-specific recombination determined by I-SceI, a mitochondrial group I intron-encoded endonuclease expressed in the yeast nucleus. Genetics. 1992;130:451–460. doi: 10.1093/genetics/130.3.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronk JT. Auxotrophic yeast strains in fundamental and applied research. Appl Environ Microbiol. 2002;68:2095–2100. doi: 10.1128/AEM.68.5.2095-2100.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ro DK, Paradise EM, Ouellet M, et al. Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature. 2006;440:940–943. doi: 10.1038/nature04640. [DOI] [PubMed] [Google Scholar]
- Rouet P, Smih F, Jasin M. Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol Cell Biol. 1994;14:8096–8106. doi: 10.1128/mcb.14.12.8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiestl RH, Petes TD. Integration of DNA fragments by illegitimate recombination in Saccharomyces cerevisiae. P Natl Acad Sci USA. 1991;88:7585–7589. doi: 10.1073/pnas.88.17.7585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Z, Zhao H, Zhao H. DNA assembler, an in vivo genetic method for rapid construction of biochemical pathways. Nucleic Acids Res. 2009;37:e16. doi: 10.1093/nar/gkn991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchuk NA, Bryksin AV, Nusinovich YA, Cabello FC, Sutherland M, Ladisch S. Construction of long DNA molecules using long PCR-based fusion of several fragments simultaneously. Nucleic Acids Res. 2004;32:e19. doi: 10.1093/nar/gnh014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata T, Watabe H, Kaneko T, Iino T, Ando T. On the nucleotide sequence recognized by a eukaryotic site-specific endonuclease, Endo.SceI from yeast. J Biol Chem. 1984;259:10499–10506. [PubMed] [Google Scholar]
- Snoek IS, van der Krogt ZA, Touw H, Kerkman R, Pronk JT, Bovenberg RA, van den Berg MA, Daran JM. Construction of an hdfA Penicillium chrysogenum strain impaired in non-homologous end-joining and analysis of its potential for functional analysis studies. Fungal Genet Biol. 2009;46:418–426. doi: 10.1016/j.fgb.2009.02.008. [DOI] [PubMed] [Google Scholar]
- Solis-Escalante D, Kuijpers NGA, Bongaerts N, Bolat I, Bosman L, Pronk JT, Daran JM, Daran-Lapujade P. amdSYM, a new dominant recyclable marker cassette for Saccharomyces cerevisiae. FEMS Yeast Res. 2013;13:126–139. doi: 10.1111/1567-1364.12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storici F, Durham CL, Gordenin DA, Resnick MA. Chromosomal site-specific double-strand breaks are efficiently targeted for repair by oligonucleotides in yeast. P Natl Acad Sci USA. 2003;100:14994–14999. doi: 10.1073/pnas.2036296100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijken JP, Bauer J, Brambilla L, et al. An interlaboratory comparison of physiological and genetic properties of four Saccharomyces cerevisiae strains. Enzyme Microb Technol. 2000;26:706–714. doi: 10.1016/s0141-0229(00)00162-9. [DOI] [PubMed] [Google Scholar]
- Verduyn C, Postma E, Scheffers W, van Dijken JP. Effect of benzoic acid on metabolic fluxes in yeasts: a continuous-culture study on the regulation of respiration and alcoholic fermentation. Yeast. 1992;8:501–517. doi: 10.1002/yea.320080703. [DOI] [PubMed] [Google Scholar]
- Watabe H, Iino T, Kaneko T, Shibata T, Ando T. A new class of site-specific endodeoxyribonucleases. Endo. Sce I isolated from a eukaryote, Saccharomyces cerevisiae. J Biol Chem. 1983;258:4663–4665. [PubMed] [Google Scholar]
- Whelan WL, Gocke E, Manney TR. The CAN1 Locus of Saccharomyces cerevisiae: fine-structure analysis and forward mutation rates. Genetics. 1979;91:35–51. doi: 10.1093/genetics/91.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingler LM, Cornish VW. Reiterative recombination for the in vivo assembly of libraries of multigene pathways. P Natl Acad Sci USA. 2011;108:15135–15140. doi: 10.1073/pnas.1100507108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Moo-Young M, Chisti Y. Plasmid stability in recombinant Saccharomyces cerevisiae. Biotechnol Adv. 1996;14:401–435. doi: 10.1016/s0734-9750(96)00033-x. [DOI] [PubMed] [Google Scholar]