

Abstract
Normal pancreatic epithelium progresses through various stages of pancreatic intraepithelial neoplasms (PanINs) in the development of pancreatic ductal adenocarcinoma (PDAC). Transcriptional regulation of this progression is poorly understood. In mouse, the Hnf6 transcription factor is expressed in ductal cells and at lower levels in acinar cells of the adult pancreas, but not in mature endocrine cells. Hnf6 is critical for terminal differentiation of the ductal epithelium during embryonic development and for pancreatic endocrine cell specification. We previously showed that, in mice, loss of Hnf6 from the pancreatic epithelium during organogenesis results in increased duct proliferation and altered duct architecture, increased periductal fibrosis and acinar-to-ductal metaplasia. Here we show that decreased expression of HNF6 is strongly correlated with increased severity of PanIN lesions in samples of human pancreata and is absent from >90% of PDAC. Mouse models in which cancer progression can be analyzed from the earliest stages that are seldom accessible in humans support a role for Hnf6 loss in progression from early to late stage PanIN and PDAC. In addition, gene expression analyses of human pancreatic cancer reveal decreased expression of HNF6 and its direct and indirect target genes compared to normal tissue and up-regulation of genes that act in opposition to HNF6 and its targets. The negative correlation between HNF6 expression and pancreatic cancer progression suggests that HNF6 maintains pancreatic epithelial homeostasis in humans, and that its loss contributes to the progression from PanIN to ductal adenocarcinoma. Insight on the role of HNF6 in pancreatic cancer development could lead to its use as a biomarker for early detection and prognosis.
Keywords: acinar-to-ductal metaplasia, exocrine HNF6, Pancreatic ductal adenocarcinoma, PanIN
Pancreatic ductal adenocarcinoma (PDAC) is the fourth leading cause of cancer mortality in the U.S. [1]. Despite improvements in survival rates of other types of cancer, the five-year survival rate for PDAC is only 6% and the vast majority of patients die within a year of diagnosis. Ninety-five percent of all pancreatic cancers are adenocarcinomas arising from the exocrine tissue. Diagnosis, analysis and treatment of human pancreatic cancer is impeded by the advanced stage at which most patients present with the disease, the inaccessibility of the organ to biopsy, and the fact that the disease is refractory to chemotherapy [2]. The progression of lesions found in PDAC have been codified into stages of pancreatic intraepithelial neoplasia (PanIN) based on architectural and cytological criteria [3].
The exocrine pancreas, which produces and transports digestive enzymes to the rostral duodenum, is composed of two cell types (acinar and ductal) and accounts for about 98% of adult pancreatic mass. During vertebrate embryogenesis, a coordinated network of transcription factors and signaling molecules tightly controls the differentiation and morphogenesis of the pancreas [4, 5]. Gene inactivation studies in mice have identified transcription factors that affect the differentiation of all or a subset of pancreatic cell types. The Hepatic nuclear factor 6 (Hnf6; also known as ONECUT-1 or OC-1) transcription factor was first identified in the liver, although it is also expressed in other endodermally-derived tissues, including the pancreas [6–8]. Hnf6 is expressed early in embryonic development in multi-potent pancreatic progenitor cells [9], but becomes down-regulated in the islet endocrine lineage once hormones are expressed [7, 9]. Its expression is maintained in mature ducts and at lower levels in acini postnatally. Global inactivation of Hnf6 (Hnf6−/−) in mice results in early postnatal lethality in 75% of animals due to liver defects [10]. Hnf6−/− animals are also severely diabetic due to a dramatic decrease in endocrine cell differentiation. In the pancreas, Hnf6 acts upstream of the critical pancreatic/βcell transcription factor, pancreatic and duodenal homeobox 1 (Pdx1) [11] and activates expression of the pro-endocrine transcription factor neurogenin 3 (Ngn3) [10].
The continued expression of Hnf6 in ductal epithelial cells suggests that it plays a role in mature exocrine tissue. Acinar tissue appears morphologically normal in Hnf6 null mutant mice, possibly due to compensation by a closely related homolog, OC-2 [10, 12]. However, embryonic duct morphology is perturbed in the absence of Hnf6, most likely due to the loss of primary cilia [9, 13]. Hnf6 tissue-specific deletion in the pancreas caused ductal cysts, acinar- to-ductal metaplasia, increased duct proliferation, and loss of duct markers such as Prox1. Molecular markers of human PDAC, including MMP7 and CTGF, were elevated in Hnf6 mutant pancreata [9].
In mouse models, transgenic over-expression of activated Kras in exocrine pancreas has produced disparate results in PanIN and PDAC development [14–17]. Cre-mediated expression of activated Kras from the endogenous Kras locus within either duct cells or acinar cells resulted in PanIN lesions but these lesions formed much more efficiently when the mutation arose in acinar cells [18, 19] Thus, acinar-to-ductal metaplasia may be a precursor to PDAC and markers for metaplasia may help predict the course of disease.
Hnf6 regulates a set of genes involved in primary cilia formation including HNF1β, polycystin, and cystin [13, 20]. Studies from the Hebrok lab showed that mutations in structural components of primary cilia in the mouse result in pancreatic ductal hyperplasia [21] and we have shown loss of primary cilia in mouse pancreas in the absence of Hnf6 [9]. Studies in human patients revealed that PanIN lesions and PDAC cells lack primary cilia, possibly due to repression of ciliogenesis by activated Kras; inhibition of Kras restored cilia formation in a mouse pancreatic cancer cell line [22]. The absence of expression of the Prox1 transcription factor in Hnf6 mutant pancreatic ducts is also of interest given the fact that loss of Prox1 is also observed in human pancreatic cancer [23, 24].
Loss of HNF6 expression and that of some of its known direct and indirect target genes has been shown to occur in human PDAC [25], however, the timing of this loss in the course of tumor progression is not known. We hypothesize that HNF6 is required to maintain exocrine homeostasis, and that it acts as a tumor suppressor. Thus, we predicted that loss of HNF6 in exocrine cells would be observed early in the course of pancreatic cancer progression and would correlate with development of PanINs and PDAC in humans. To this end, we examined expression of HNF6 mRNA and protein expression in samples of normal human pancreas and of pancreatic cancer. HNF6 protein expression was analyzed at different stages throughout human pancreatic cancer progression. In agreement with what we observe in normal mouse pancreas, HNF6 expression was consistently detected in normal human ductal epithelium and at lower levels in acinar cells. While HNF6 expression is maintained in acinar-to-ductal metaplasia (ADM), diminished HNF6 expression strongly correlated with increasing severity of PanIN lesions and HNF6 was not detected in the tumor samples. In addition, gene expression analyses of human pancreatic cancer compared to normal pancreatic tissue samples revealed decreased expression of known HNF6 target genes consistent with a loss of HNF6 transcriptional activity.
Because of its biological and clinical relevance to human cancer, we examined the timing and pattern of HNF6 loss in pancreatic cancer progression to gain crucial scientific insight on the malignant progression of the disease. We also show here that two mouse models of PDAC mirror the progressive loss of Hnf6 expression in developing lesions, supporting these models as reflective of what goes on in the in vivo human situation. Thus, use of these models in correlation with human data could potentially aid in the development of clinical diagnostic tests for early detection and improved survival outcomes in patients with pancreatic cancer.
Materials and Methods
Meta-analyses of gene expression in human pancreatic cancer
Using T-test followed by Step-up False Discovery Rate procedure [26], differential expression of ten genes known to be co-regulated with HNF6 was examined in published DNA microarray datasets. Pei, et al. performed gene expression analyses of 36 pancreatic tumors and 16 normal tissue samples (GSE 16515) [27]. Badea, et al. performed gene expression analyses of 36 pancreatic tumors and matching 36 normal tissue samples (GSE 15471) [28]. Data from Arumugam, et al. includes expression of HNF6 and its associated genes in nine human pancreatic cancer cell lines with known sensitivity to the chemotherapeutic agent gemcitabine compared to an immortalized “normal” human pancreatic epithelial line (HPDE; GSE 15550) [29, 30].
Gene expression analysis of normal and diseased human tissue
Five pancreatic ductal adenocarcinomas were selected from the Vanderbilt University Pathology Archives. Five 5 µm unstained slides from each sample were prepared from formalin-fixed paraffin embedded tissue. Microdissection was performed to achieve a neoplastic cellularity of at least 50% with minimal residual benign pancreatic tissue. Twenty five-µM corresponding normal pancreatic tissue was also prepared from each case. Total RNA was extracted from these samples. cDNA was made using the Invitrogen SuperScript III kit and quantitative reverse-transcriptase qRT-PCR was performed using the Bio-Rad IQ SYBERGreen Supermix on a Bio-Rad CFX Real time PCR instrument for HNF6, direct and indirect targets of HNF6, and genes known to be inversely correlated with HNF6 expression (see Table 1 for PCR primer sequences). All reactions were performed in triplicate and the mean was compared to GapDH expression (see Table 2 for PCR protocol). Values for each individual were then plotted for each gene where normal pancreas could be compared with PDAC tumor. Significance was determined by a T-test with Welch’s correction.
Table 1.
Primers used for quantitative reverse-transcriptase PCR analysis
| Gene | Direction | Sequence |
|---|---|---|
| GapDH | Forward | TCAACGACCACTTTGTCAAGCT |
| Reverse | AGCCAAATTGGTTGTGTCATACCA | |
| Hnf6 | Forward | GAGTTCCAGCGCATGTCC |
| Reverse | TGTTGCCTCTATCCTTCCCA | |
| Pdx1 | Forward | GTCCAGCTGCCTTTCCCAT |
| Reverse | TCCGCTTGTTCTCCTCCG | |
| FoxA2 | Forward | CTACTATGCAGAGCCCGAGG |
| Reverse | CGGCGTTCATGTTGCTCAC | |
| PKHD1 | Forward | TGATGGTTTGGAGTTGGGTG |
| Reverse | CACCACCATGTTCACGTTCA | |
| Tgfβ | Forward | TCAAGTTAAAAGTGGAGCAGCA |
| Reverse | CGGTTGCTGAGGTATCGC | |
| Ctgf | Forward | GACGAGCCCAAGGACCAAA |
| Reverse | CAAACGTGTCTTCCAGTCGG | |
| MMP7 | Forward | ACAGGCTCAGGACTATCTCA |
| Reverse | TGGCTTCTAAACTGTTGGCA |
Table 2.
Reaction specifications for quantitative reverse-transcriptase PCR specifications.
| Step | Temperature (°C) | Time | Cycles |
|---|---|---|---|
| 1 | 95 | 3 min | 1 |
| 2 | 95 | 10 sec | 39 |
| 3 | 58 | 10 sec | |
| 4 | 72 | 30 sec | |
| 5 | 95 | 10 sec | 1 |
| 6 | 65 | 5 sec | 1 |
| 1 | 1 | 1 | 1 |
Tissue microarrays
Samples from surgically resected pancreatic adenocarcinomas from human patients were used to construct a tissue microarray containing lesions of all histological grades of the pancreatic adenocarcinoma progression model [31–33]. These samples have all been de- identified and include normal ductal epithelium, PanINs, true adenocarcinomas, and metastatic tumors. The individual determining whether HNF6 protein expression was positive or negative was unaware of the disease status of the samples. Once HNF6 immunolabeling was completed, we determined the correlation between HNF6 expression and stage of the disease. A pathologist (C.S.) specializing in human pancreatic cancer determined stage of disease.
Mice
All experiments were done with the approval of the Vanderbilt Institutional Care and Use Committee. Pdx1-sHBEGF mice [34], Ptf1aCre mice [35] and LSL-KrasG12D (Krastm4Tmj) mice [36] were described previously. All alleles were bred for at least 10 generations onto the C57BL6/J background.
Tissue processing and immunolabeling
Tissue sections were deparaffinized in xylene and rehydrated to deionized water. Sections were exposed to antigen retrieval using 10 mM sodium citrate buffer in a pressure cooker for 15 minutes and then left to cool to room temperature for 2 hours, followed by washing in PBS. Endogenous peroxidase activity was quenched by exposing slides to 3% hydrogen peroxide for 20 minutes. Slides were blocked for 1 hour at room temperature in 5% normal donkey serum (NDS) and 1% BSA in PBS. Rabbit anti-HNF6 primary antibody (Santa Cruz Biotechnology, Inc.) was added at 1:300 (human tissue) or 1:500 (mouse tissue) in 1% BSA in PBS overnight at 4°C. Sections were washed in PBS and incubated with secondary antibody (biotinylated goat anti-rabbit; Abcam Inc. or Vector Labs, Burlingame, CA) at 1:500 in 1% BSA in PBS for 1 hour at room temperature. Protein localization was visualized using the Vectastain ABC and DAB Peroxidase Substrate kits (Vector Laboratories) according to the manufacturer’s instructions. Tissues were counterstained with eosin or hematoxylin for contrast and mounted with Permount (Fisher). Double immunofluorescence was performed using TSA-Plus kits (Perkin Elmer LAS, Boston, MA) with 1:500 rabbit anti-Mist1 (a kind gift of Dr. Steven Konieczny, Pudue University) and rabbit anti-HNF6, and counterstained with Toto3 (Molecular Probes, Eugene, OR). Because primary antibodies were the same species, slides were boiled after the first antibody’s TSA reaction, then labeling was repeated with the second primary antibody. Controls always included slides that lacked either the first or second primary antibody to validate that boiling removed all of the first antibody.
Microscope imaging
Human tissue samples were viewed under bright-field illumination at both 10× and 20× magnifications using an Olympus BX41 microscope and digital camera with the Magnafire program (Optronics, Inc.). Mouse tissues were visualized with an Axioskop 40 microscope and Axio Vision camera and software (Carl Zeiss Microimaging, Thornwood, NY).
Results
Examination of HNF6 gene expression in human pancreatic tumors and immortalized cell lines
We previously showed that loss of Hnf6 in the embryonic pancreatic epithelium resulted in acinar-to-ductal metaplasia and fibrosis in mice [9]. To determine whether loss of HNF6 gene expression also correlates with pancreatic neoplasia in humans, we first performed a meta-analysis of two published DNA microarray datasets for differential expression of ten selected genes, the expression of which are known to positively or negatively correlate with HNF6 in liver and/or pancreas. Some of these genes, including PDX1, forkhead box (Fox) A2, and HNF4α, have been identified as direct transcriptional targets of HNF6 in pancreas [11, 37, 38], while others, are positively (PKHD1) or negatively (TGF-β, CTGF, MMP7) indirectly regulated by HNF6 [9, 39–41]. Similar results were found in both published datasets. Pei, et al. performed gene expression analyses of 36 pancreatic tumors and 16 normal tissue samples [27]. HNF6 expression was lower in tumors compared to normal pancreatic tissues with statistical significance (p=0.007, Table 3). Badea, et al. performed gene expression analyses of 36 pancreatic tumors and 36 normal tissue samples [28]. Again, HNF6 expression was significantly lower in tumors compared to normal pancreatic tissues (p=0.004, Table 4). In accordance with our mouse model of Hnf6 gene inactivation, expression of TGF-β, CTGF and MMP7 was up-regulated in the human tumor samples. In addition, several known direct and indirect HNF6 target genes were down-regulated in the tumor samples [PDX1, FoxA2, hepatic nuclear factor (HNF) 4α, and polycystic kidney and hepatic disease (PKHD) 1] [20] [40].
Table 3.
Differential expression of HNF6 and related genes (Pei, et al. Cancer Cell, 2009, GSE 16515). Green box: False Discovery Rate <0.01
| Probeset ID | Gene Title | Gene Symbol | p-value (T vs. N) |
Fold-Change Description |
|---|---|---|---|---|
| 208559_at | pancreatic and duodenal homeobox 1 | PDX1 | 0.00145187 | T down vs N |
| 214312_at | forkhead box A2 | FOXA2 | 0.00193461 | T down vs N |
| 210745_at | one cut homeobox 1 | ONECUT1 | 0.00686838 | T down vs N |
| 241694_at | polycystic kidney and hepatic disease 1 | PKHD1 | 0.0138692 | T down vs N |
| 228739_at | cystin 1 | CYS1 | 0.0143826 | T down vs N |
| 214851_at | hepatocyte nuclear factor 4, alpha | HNF4A | 0.0513435 | T down vs N |
| 205313_at | transcription factor 2, hepatic; LF-B3 | TCF2 (HNF1B) | 0.388387 | T down vs N |
| 204259_at | matrix metallopeptidase 7 | MMP7 | 0.000251983 | T up vs N |
| 203085_s_at | transforming growth factor, beta 1 | TGFB1 | 0.0098017 | T up vs N |
| 209101_at | connective tissue growth factor | CTGF | 0.465259 | T up vs N |
Table 4.
Differential expression of HNF6 and related genes (Badea, et al. Hepato-gastroenterology, 2008, GSE 15471) Green box: False Discovery Rate <0.01
| Probeset ID | Gene Title | Gene Symbol | p-value (T vs. N) |
Fold-Change Description |
|---|---|---|---|---|
| 208559_at | pancreatic and duodenal homeobox 1 | PDX1 | 2.96E-07 | T down vs N |
| 216889_s_at | hepatocyte nuclear factor 4, alpha | HNF4A | 1.07E-06 | T down vs N |
| 214312_at | forkhead box A2 | FOXA2 | 2.24E-05 | T down vs N |
| 210745_at | one cut homeobox 1 | ONECUT1 | 0.00421182 | T down vs N |
| 1553003_at | polycystic kidney and hepatic disease 1 | PKHD1 | 0.00619324 | T down vs N |
| 208135_at | transcription factor 2, hepatic; LF-B3 | TCF2 (HNF1B) | 0.0465427 | T down vs N |
| 228739_at | cystin 1 | CYS1 | 0.906878 | T down vs N |
| 204259_at | matrix metallopeptidase 7 | MMP7 | 4.58E-11 | T up vs N |
| 203085_s_at | transforming growth factor, beta 1 | TGFB1 | 1.21E-07 | T up vs N |
| 209101_at | connective tissue growth factor | CTGF | 4.11E-06 | T up vs N |
We next examined whether expression of HNF6 in human pancreatic cancer correlated with sensitivity to a commonly used chemotherapy in pancreatic cancer, gemcitabine. In pancreatic cancer patients, loss of sensitivity to gemcitabine strongly correlates with poor prognosis [42]. Using a published dataset [29] in which nine human pancreatic cancer cell lines with known sensitivity to gemcitabine were compared to an immortalized “normal” human pancreatic epithelial line (HPDE; [30]) we examined expression of HNF6 and its associated genes. Gemcitabine-sensitive pancreatic cancer cell lines included SU.86.86, CFPAC-1, BxPC3, and L3.6pl. Resistant cell lines included Panc-1, Hs766t, MPanc-96, AsPC1, and MIA-PaCa. HNF6 expression did not correlate with gemcitabine sensitivity in these cell lines. However, HNF6 showed a reciprocal relationship with CTGF expression in the majority of these cell lines (6/9 of cell lines examined), similar to what we observe in vivo [9, 41]. In five out of nine of the cell ines examined, expression of the direct HNF6 target gene, HNF4α,and the PKHD1 gene (indirectly regulated by HNF6 through its activation of the direct target, HNF1α) followed the expression pattern of HNF6 (Figure 1).
Figure 1. Examination of HNF6 expression and associated genes in human pancreatic cell lines.

Hierarchical clustering of HNF6 (ONECUT1; red text) and associated genes expression ratio of nine pancreatic cancer cell lines with known gemcitabine sensitivity and one immortalized “normal” human pancreatic epithelial line (HPDE). Cell lines in blue are gemcitabine-sensitive; cell lines in black are gemcitabine-resistant. Shades of red indicate increased expression compared with HPDE cells and shades of green indicate decreased expression compared with HPDE cells. (dataset from [29]).
These meta-analyses of the published literature show that loss of HNF6 is detected in several human pancreatic cancer cell lines. To more directly determine whether loss of HNF6 gene expression is associated with pancreatic cancer in humans, we extracted RNA from normal human pancreas samples and pancreatic tumor samples and quantified HNF6 expression and expression of known direct (FOXA2, PDX1) and indirect (PKHD1) target genes, as well as genes whose expression inversely correlates with HNF6 in other situations (TGF-β, CTGF, MMP7). As shown in Figure 2, expression of HNF6 and its positively associated genes was decreased in all of the human pancreatic cancer samples compared with the controls. In one tumor sample, HNF6 expression was completely undetectable. Genes reported to be inversely correlated with HNF6 in other tissues, and in our meta-analysis, were either unchanged in these human tumor samples (CTGF, MMP7) or were slightly decreased (TGF-β). Thus, genes that rely on HNF6 for activation seem to be more consistently affected by HNF6 loss than those that maybe negatively regulated by HNF6 indirectly. In contrast with the primary human tumor samples analyzed here and in the meta-analysis, expression of FoxA2 in pancreatic cancer cell lines tended to be inversely correlated with HNF6 expression (7/9 cell lines examined; Figure 1).
Figure 2. Expression of HNF6 and co-regulated genes in normal pancreas and PDAC tumors.
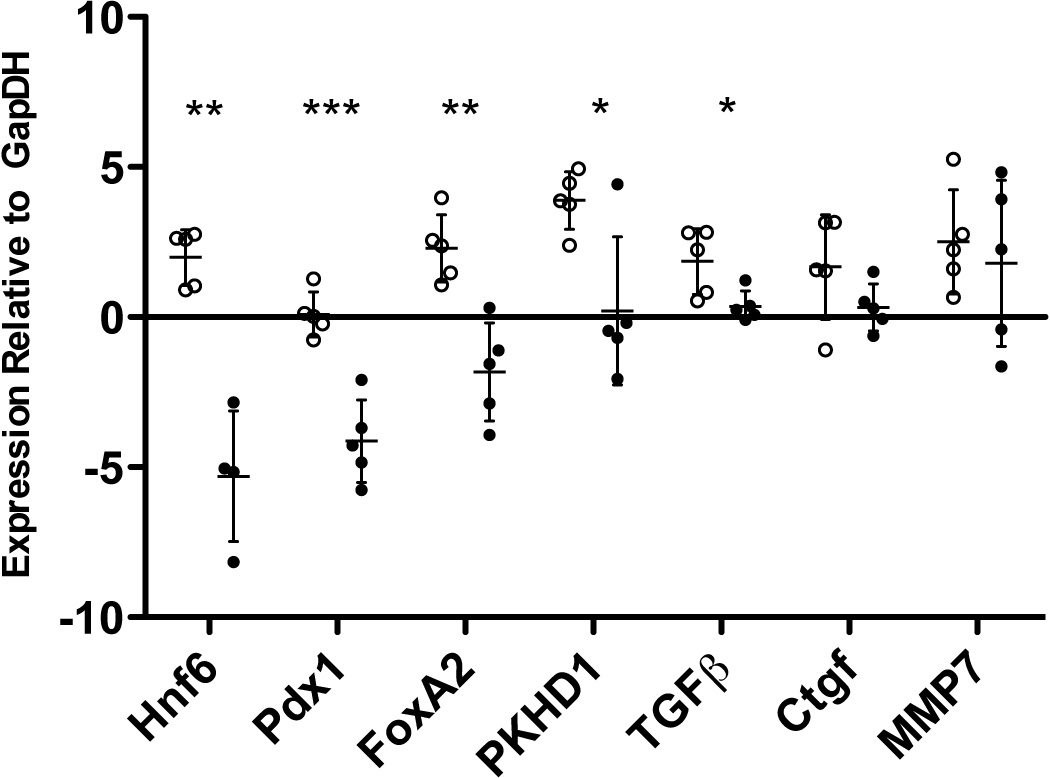
Gene expression analysis of normal (open circles) and PDAC tumor (closed circles) tissue samples (n = 5 for each). qRT-PCR was performed on RNA extracted from samples and expression was normalized to GapDH. HNF6 expression was significantly decreased in all of the PDAC tumors, and was undetectable in one sample. Expression of direct (FOXA2, PDX1) and indirect (PKHD1, TGF-β) HNF6 target genes, were also significantly decreased in PDAC tumor samples compared with normal pancreas. (*, p<0.05; **, p<0.005; ***, p<0.0005)
Loss of HNF6 protein is correlated with PanIN and PDAC in human tissues
To examine the correlation between HNF6 protein expression and the progression to pancreatic cancer in humans, immunohistochemistry labeling of HNF6 was performed on tissue samples taken from patients with pancreatic cancer to assay for the presence of the HNF6 protein (Figures 3–5). The microscopic images were examined to determine a correlation of HNF6 protein expression with pancreatic morphology. Analyses of images suggested that there was a general trend for decreasing HNF6 expression with increasing severity of disease. HNF6 protein was clearly observed in normal human pancreatic ducts and at lower levels in acinar cells (Figure 3A, C and Figure 4A; n = 11). The lower level of HNF6 in acinar cells can be better appreciated when compared with expression of the acinar-specific transcription factor, MIST1 (Figure 4B, C). Compared with normal exocrine tissue, late stage PanINs and pancreatic adenocarcinoma samples showed a dramatic loss of HNF6 protein (Figure 3B, D; n = 13).
Figure 3. HNF6 Expression in normal and diseased human pancreas tissue.

Representative examples of human pancreatic tissue immunolabeled for HNF6 protein expression. HNF6 (brown) is present in normal human ducts and acini (A,C), but absent from high grade human PanINs (B,D). Sections were counterstained with hematoxylin. Bars, 50 µm.
Figure 5. Survey of HNF6 expression in normal and diseased human pancreas tissue using tissue microarrays.

Representative examples from human tissue microarrays immunolabeled for HNF6 protein expression (brown) against eosin contrast. HNF6 expression is observed in normal pancreas (A), acinar-to-ductal metaplasia (B), and PanIN1 (C). PanIN2 (D) and PanIN3 (E) showed reduced HNF6 expression, while PDAC (F) lacked HNF6 protein expression. Magnification: 200×.
Figure 4. HNF6 is detected at consistently high levels in human pancreatic ducts but at low and variable levels in acinar cells.
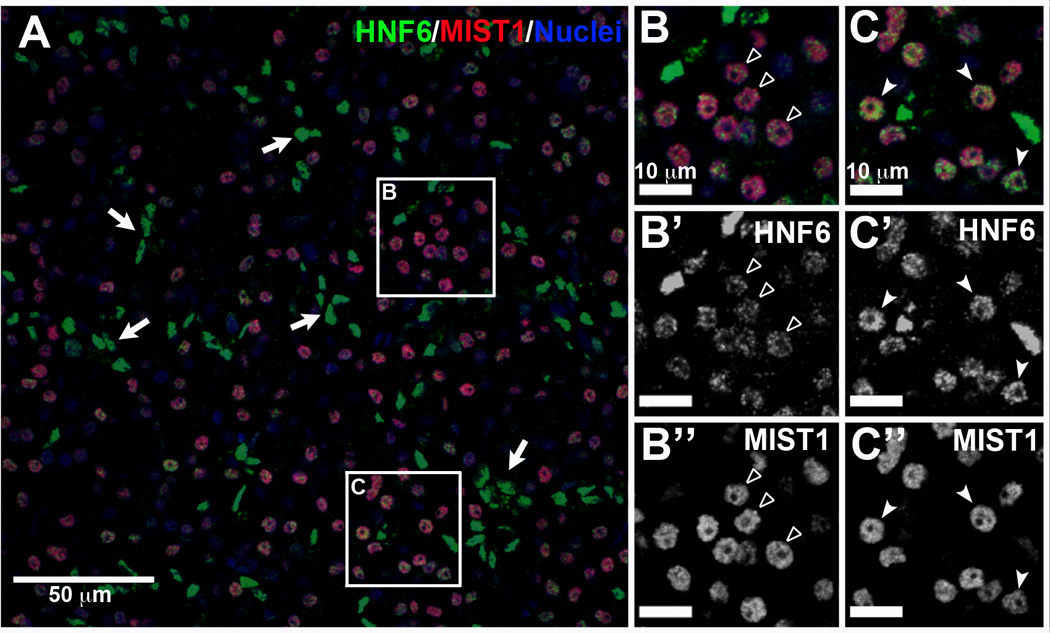
Normal human pancreas was immunolabeled for HNF6 (green) and MIST1 (red); nuclei were counterstained in blue. (A) Duct cells (MIST1 negative) have high levels of HNF6 labeling (arrows). (B, C) MIST1-positive acinar cells have HNF6 levels ranging from barely detectable (open arrowheads) to intermediate (closed arrowheads) but consistently lower than duct levels. B and C are enlargements of areas indicated in panel A. B’ and C’, HNF6 labeling alone for B and C, respectively. B” and C”, MIST1 labeling alone for B and C.
To analyze expression of HNF6 during pancreatic disease progression in more detail, we examined HNF6 protein by immunolabeling of human tissue microarrays containing ADM, non-invasive precursor (PanIN) lesions, and invasive adenocarcinomas (Figure 5). Acinar expression of HNF6 was clearly detected in only 21% of the normal tissue microarray resection samples in this dataset, although ductal expression was evident in all samples (Figure 5A). HNF6 expression was more easily detected in acinar cells in the setting of ADM (50%; Figure 5B; n = 14), in agreement with recently published findings from Jacquemin and colleagues showing increased HNF6 expression in ADM [43]. However, 70% of PanIN1 (Figure 5C; n = 10), 50% of PanIN2 (Figure 5D; n = 6), 78% of PanIN3 (Figure 5E; n = 9), and 91% of cancer samples (Figure 5F; n = 34) were negative for HNF6 protein expression. In the few tumor samples in which HNF6 protein was detected (3/34), this was only in a subset of cells within the tumor (Figure 6). Thus, loss of HNF6 protein strongly correlates with more severe PanIN lesions and PDAC.
Figure 6. Human pancreatic cancer showing some HNF6 protein expression.

Three of thirty-four human PDAC samples contained variable numbers of HNF6-positive cells. Two examples are shown here (HNF6 in brown). Arrows, HNF6+ PDAC cells; arrowhead, HNF6+ normal duct cells.
HNF6 expression is inversely correlated with severity of lesions in mouse models of PanIN and PDAC
Because early stages of pancreatic cancer are seldom observed in humans due to the inaccessibility of the organ and the diffuse nature of symptoms, we examined mouse models to determine when Hnf6 expression was altered in the course of disease progression. Ptf1aCre; LSL-KrasG12D (henceforth KrasG12D) mice are a well-established model of pancreatic cancer that closely mimics that seen in humans [36]. While these mice have a normal pancreas at birth, as they age they accumulate PanIN lesions and progress to pancreatic ductal adenocarcinoma with low frequency and long latency. We examined young KrasG12D mice at 4–6 weeks of age, when PanINs are first beginning to develop. At this stage, Hnf6 was abundant in the cytoplasm and nuclei of acinar cells that had characteristics of early acinar-to-ductal metaplasia (reduced apical cytoplasm and lumen formation) (Figure 7A). In regions of established acinar-to-ductal metaplasia, Hnf6 was still abundant but primarily localized to nuclei (Figure 7B). To determine if Hnf6 expression in acinar-to-ductal metaplasia was specific to cells expressing the activated KrasG12D allele, we examined pancreata from another mouse model of acinar-to-ductal metaplasia: transgenic over-expression of the growth factor HB-EGF [34]. These mice also showed high levels of Hnf6 in areas of acinar-to-ductal metaplasia (Figure 7C).
Figure 7. Tumorigenesis in a mouse model reflects altered expression of Hnf6 as was seen in human disease.

A. Five week old Ptf1aCre;LSL-KrasG12D (KrasG12D) mouse pancreas labeled with Hnf6 antibody (brown). At this age, most of the pancreas is normal and labeling is observed at a high level in ducts and a lower and variable level in normal acini. In acini undergoing early changes associated with acinar-to-ductal metaplasia, a high level of Hnf6 is observed in both cytoplasm and nucleus. B. As acinar-to-ductal metaplasia develops, labeling becomes localized to nuclei. C. Hnf6 is also upregulated in acinar-ductal metaplasia initiated by transgenic overexpression of the active form of the growth factor HB-EGF. D, E. In 12 month old KrasG12D pancreas, ducts with normal, low cuboidal architecture maintain a high level of Hnf6, but PanIN lesions exhibit lower levels, particularly in more advanced lesions that have begun to undergo cellular atypia. F. In KrasG12D mice that also over-express HB-EGF, advanced PanINs are common and exhibit little to no Hnf6 labeling. Closed arrowheads, normal ductal epithelium; open arrowheads, acinar-to-ductal metaplasia; arrows, PanIN epithelium. Nuclei are counterstained with hematoxylin (blue). Scale bar, 50 µm; all images at the same magnification.
In one year old KrasG12D mice with established PanINs of different stages, we found that Hnf6 expression inversely correlated with the degree of severity of the lesion (Figure 7D, E). Duct cells with normal, cuboidal architecture maintained a high level of Hnf6 while PanIN1 lesions had a slightly lower but variable level. PanIN2 lesions, characterized by loss of polarity and other cellular atypia, had little or no detectable Hnf6. Combination of HB-EGF overexpression with KrasG12D expression leads to rapid development of advanced PanINs and locally invasive lesions [44]. In these mice, PanIN3 lesions, characterized by cribriform architecture, luminal budding and cellular atypia, as well as PanIN2 lesions similarly had no detectable Hnf6 protein (Figure 7F and data not shown) mirroring the results from human patient samples.
Discussion
These studies examine the link between HNF6, a critical regulator of embryonic pancreas development, and pancreatic cancer progression. The original hypothesis in this study was that inactivation of HNF6 would occur in a progression from acinar-to-ductal metaplasia, to PanIN stages, and ultimately invasive carcinoma. Our observation that the percentage of HNF6-negative tissue samples increases with increasing disease stage supports this hypothesis. However, our results also indicated that loss of HNF6 in pancreatic cancer progression does not begin during acinar-to-ductal metaplasia but occurs in later stages, beginning with PanIN lesion formation, suggesting that HNF6 expression is inconsistent with more transformed phenotypes such as cellular atypia and expression of genes such as CTGF [9]. The lack of HNF6 protein immunolabeling in the majority of late stage PanIN lesions and nearly all of the pancreatic cancer tissue samples, strongly reiterates the negative correlation between HNF6 expression and cancer. The progressive loss of HNF6 expression in human pancreatic disease development outlined here may potentially indicate the course of the development of PDAC, which is a subject that currently remains unclear.
While HNF6 protein was lost from late stage PanINs and from PDAC, HNF6 was elevated in acinar cells beginning to undergo acinar-to-ductal metaplasia. It is now well-established that acinar cells can alter their cellular identity through transdifferentiation to become duct cells [45–47] and this may explain the increase in HNF6 to levels seen in ducts. We found that elevation of HNF6 occurs early in this process while cells still maintain some acinar properties. While the cell of origin for pancreatic cancer remains a highly debated topic, recent work in a mouse model demonstrated that acinar cells can give rise to PanIN lesions presumably through acinar-to-ductal metaplasia [19]. Because HNF6 is elevated as cells go from an acinar to ductal phenotype in both premalignant and benign disease, but is lost as cells undergo atypia during cancer progression, we speculate that HNF6 may function in establishment and/or maintenance of normal ductal epithelium. Loss of HNF6 disrupts this process, such that acinar-to-ductal metaplasia is altered, leading to abnormal ducts and chronic disease, such as PanIN/ductal adenocarcinoma when cells contain a Kras mutation, or benign lesions when they do not. Some of this dependence on HNF6 may be through its repression of CTGF expression [9, 41]. When HNF6 is lost, acinar-to-ductal metaplasia may be more likely to lead to fibrosis via CTGF production. This fibrotic microenvironment may then promote cancer development in some cases or chronic pancreatitis in others.
Since HNF6 is expressed only in duct (high) and acinar cells (low) in healthy adult pancreata [8], a future goal is to determine the role of HNF6 in maintenance of the differentiated state specifically in each of these cell types using conditional gene inactivation. Results from these studies would be compared with those from the pancreatic epithelial-specific Hnf6 mutant [9], to help determine the potential cell of origin for the cysts and metaplasia observed in this line. These studies may, in turn, help to confirm the cell-type of origin of PDAC. We propose that loss of HNF6 expression could be used as a biomarker for staging pancreatic disease in the progression toward pancreatic cancer. Interestingly, HNF6 expression appeared to increase in acinar clusters undergoing acinar-to-ductal metaplasia, similar to observations recently made by another group [43].
Experimental animal models that mimic human pancreatic disease are essential to better understand the pathophysiology of these diseases and to evaluate potential therapeutic agents. The ability to specifically alter the expression of a gene(s) of interest in a particular cell type makes the mouse the best available model system to dissect the molecular regulation of pancreas organogenesis and mature organ function. To date, there has been no examination of HNF6 function in promoting pancreatic neoplasia. Our data indicate that decreased HNF6 expression correlates strongly with severity of pancreatic disease in humans. Our findings are supported by a previous study showing decreased expression of HNF6 and some of its known target genes in human pancreatic cancer compared with normal ductal tissue [25], although an analysis of the timing of HNF6 loss in human pancreatic cancer progression was not reported. In that study, reintroduction of HNF6 into a human pancreatic cancer cell line reduced invasiveness, but not tumor growth, in an in vitro assay, suggesting that HNF6 can reestablish some aspects of normal cell behavior even in a long-standing immortalized cell line.
In the liver, HNF6 inhibits TGF-β signaling [48]. We found that pancreata from HNF6⊗panc animals show characteristics of TGF-β pathway up-regulation including up-regulation of CTGF and MMP7 in and around pancreatic ducts [9]. Thus, HNF6 may inhibit the TGF-β pathway in the pancreas as it does in the liver. Both CTGF and the TGF-β type II receptor are over- expressed in human pancreatic cancer and increased TGF-β signaling is found in pancreatic cancer in humans and is associated with decreased survival [49]. Therefore, loss of HNF6 may promote pancreatic ductal hyperplasia/metaplasia via TGF-β activation. CTGF expression is induced in response to TGF-β in several cell types [50–53] and it interacts with TGF-β extracellularly to enhance signaling [54]. Increased CTGF expression in pancreatic tumor cells is correlated with enhanced growth of these cells, while siRNA inhibition of CTGF results in decreased tumor growth and increased apoptosis [55]. In addition, increased TGF-β and CTGF production is important for the desmoplastic stroma (proliferating fibroblasts, inflammatory cells, and excess collagen) present in human pancreatic cancer [56]. Because many pancreatic adenocarcinomas have loss of Smad4, an obligate component of TGF-β signaling, the effects of increased TGF-β production by the tumor may have its most profound effects on the tumor microenvironment. Understanding the mechanisms underlying pancreatic cancer progression, including increased production of TGF-β and CTGF, is crucial in developing new therapeutic targets.
Acknowledgements
We thank members of the Gannon laboratory for critical reading of the manuscript and Dr. Nagaraj Nagathihalli for helpful discussions. We also thank Dr. Steve Konieczny for the Mist1 antibody and Ms. Keeli Lewis for RNA isolation from human tissue samples. X.M. would like to thank Dr. Angela M. Eeds for her mentorship and expertise and the Vanderbilt University School for Science and Math support. This work was conducted in part using the Tissue Procurement Shared Resource and Tissue Morphology cores at Vanderbilt University. Mr. Peter Kropp is a recipient of a pre-doctoral T32 award from the Vanderbilt University Training Program in Stem Cell and Regenerative Developmental Biology (NIH/NICHD #). This study was supported by a Pilot and Feasibility award from the Vanderbilt DDRC to M.G. (P30 DK058404); and by NIH grants CA123061 and CA98322 to ALM.
Abbreviations
- CTGF
connective tissue growth factor
- Hnf6
Hepatic nuclear factor 6
- MMP7
matrix metalloproteinase 7
- Ngn3
neurogenin 3
- OC-1
onecut 1
- PanIN
pancreatic intraepithelial neoplasia
- PDAC
Pancreatic ductal adenocarcinoma
- Pdx1
pancreatic and duodenal homeobox 1
- Ptf1a
pancreas transcription factor 1a
Footnotes
The authors declare no conflict of interest.
References
- 1.Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362:1605–1617. doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- 2.Furukawa T, Sunamura M, Horii A. Molecular mechanisms of pancreatic carcinogenesis. Cancer Sci. 2006;97:1–7. doi: 10.1111/j.1349-7006.2005.00134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hruban RH, Adsay NV, Albores-Saavedra J, et al. Pathology of genetically engineered mouse models of pancreatic exocrine cancer: consensus report and recommendations. Cancer Res. 2006;66:95–106. doi: 10.1158/0008-5472.CAN-05-2168. [DOI] [PubMed] [Google Scholar]
- 4.Jensen J. Gene regulatory factors in pancreatic development. Dev Dyn. 2004;229:176–200. doi: 10.1002/dvdy.10460. [DOI] [PubMed] [Google Scholar]
- 5.Pan FC, Wright C. Pancreas organogenesis: from bud to plexus to gland. Dev Dyn. 2011;240:530–565. doi: 10.1002/dvdy.22584. [DOI] [PubMed] [Google Scholar]
- 6.Samadani U, Costa RH. The transcriptional activator hepatocyte nuclear factor 6 regulates liver gene expression. Mol Cell Biol. 1996;16:6273–6284. doi: 10.1128/mcb.16.11.6273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rausa F, Samadani U, Ye H, et al. The cut-homeodomain transcriptional activator HNF-6 is coexpressed with its target gene HNF-3 beta in the developing murine liver and pancreas. Dev Biol. 1997;192:228–246. doi: 10.1006/dbio.1997.8744. [DOI] [PubMed] [Google Scholar]
- 8.Landry C, Clotman F, Hioki T, et al. HNF-6 is expressed in endoderm derivatives and nervous system of the mouse embryo and participates to the cross-regulatory network of liver- enriched transcription factors. Dev Biol. 1997;192:247–257. doi: 10.1006/dbio.1997.8757. [DOI] [PubMed] [Google Scholar]
- 9.Zhang H, Ables ET, Pope CF, et al. Multiple temporal-specific roles for HNF6 in pancreatic endocrine and ductal differentiation. Mech Dev. 2009;126:958–973. doi: 10.1016/j.mod.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jacquemin P, Durviaux SM, Jensen J. Transcription factor hepatocyte nuclear factor 6 regulates pancreatic endocrine cell differentiation and controls expression of the proendocrine gene ngn3. Mol Cell Biol. 2000;20:4445–4454. doi: 10.1128/mcb.20.12.4445-4454.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]; 1) Jacquemin P, Lemaigre FP, Rousseau GG. The Onecut transcription factor HNF-6 (OC-is required for timely specification of the pancreas and acts upstream of Pdx-1 in the specification cascade. Dev Biol. 2003;258:105–116. doi: 10.1016/s0012-1606(03)00115-5. [DOI] [PubMed] [Google Scholar]
- 11.Vanhorenbeeck V, Jenny M, Cornut JF, et al. Role of the Onecut transcription factors in pancreas morphogenesis and in pancreatic and enteric endocrine differentiation. Dev Biol. 2007;305:685–694. doi: 10.1016/j.ydbio.2007.02.027. [DOI] [PubMed] [Google Scholar]
- 12.Pierreux CE, Vanhorenbeeck V, Jacquemin P, et al. The transcription factor hepatocyte nuclear factor-6/Onecut-1 controls the expression of its paralog Onecut-3 in developing mouse endoderm. J Biol Chem. 2004;279:51298–51304. doi: 10.1074/jbc.M409038200. [DOI] [PubMed] [Google Scholar]
- 13.Grippo P, Nowlin PS, Demeure MJ, et al. Preinvasive pancreatic neoplasia of ductal phenotype induced by acinar cell targeting of mutant Kras in transgenic mice. Cancer Res. 2003;63:2016–2019. [PubMed] [Google Scholar]
- 14.Brembeck FH, Schreiber FS, Deramaudt TB, et al. The mutant K-ras oncogene causes pancreatic periductal lymphocytic infiltration and gastric mucous neck cell hyperplasia in transgenic mice. Cancer Res. 2003;63:2005–2009. [PubMed] [Google Scholar]
- 15.Tuveson D, Zhu L, Gopinathan A, et al. Mist1-KrasG12D knock-in mice develop mixed differentiation metastatic exocrine pancreatic carcinoma and hepatocellular carcinoma. Cancer Res. 2006;66(1):242–247. doi: 10.1158/0008-5472.CAN-05-2305. [DOI] [PubMed] [Google Scholar]
- 16.Ji B, Tsou L, Wang H, et al. Ras activity levels control the development of pancreatic diseases. Gastroenterology. 2009;137(3):1072–1082. 1082 e1–1082 e 6. doi: 10.1053/j.gastro.2009.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ray KC, Bell KM, Yan J, et al. Epithelial tissues have varying degrees of susceptibility to Kras(G12D)-initiated tumorigenesis in a mouse model. PLoS One. 2011;6:e16786. doi: 10.1371/journal.pone.0016786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kopp JL, von Figura G, Mayes E, et al. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;22:737–750. doi: 10.1016/j.ccr.2012.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gresh L, Fischer E, Reimann A, et al. A transcriptional network in polycystic kidney disease. Embo J. 2004;23:1657–1668. doi: 10.1038/sj.emboj.7600160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cano DA, Sekine S, Hebrok M. Primary cilia deletion in pancreatic epithelial cells results in cyst formation and pancreatitis. Gastroenterology. 2006;131:1856–1869. doi: 10.1053/j.gastro.2006.10.050. [DOI] [PubMed] [Google Scholar]
- 21.Seeley ES, Carrière C, Goetze T, et al. Pancreatic cancer and precursor pancreatic intraepithelial neoplasia lesions are devoid of primary cilia. Cancer Res. 2009;69:422–430. doi: 10.1158/0008-5472.CAN-08-1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang J, Kilic G, Aydin M, et al. Prox1 activity controls pancreas morphogenesis and participates in the production of "secondary transition” pancreatic endocrine cells. Dev Biol. 2005;286:182–194. doi: 10.1016/j.ydbio.2005.07.021. [DOI] [PubMed] [Google Scholar]
- 23.Schneider M, Büchler P, Giese N, et al. Role of lymphangiogenesis and lymphangiogenic factors during pancreatic cancer progression and lymphatic spread. Int J Oncol. 2006;28:883–890. [PubMed] [Google Scholar]
- 24.Jiang X, Zhang W, Kayed H, et al. Loss of ONECUT1 expression in human pancreatic cancer cells. Oncol Rep. 2008;19:157–163. [PubMed] [Google Scholar]
- 25.Somerville PN. Calculation of critical values for Somerville's FDR procedure. J Stat Software. 2007;21:6. [Google Scholar]
- 26.Pei H, Li L, Fridley BL, et al. FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell. 2009;16:259–266. doi: 10.1016/j.ccr.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Badea L, Herlea V, Dima SO, et al. Combined gene expression analysis of whole-tissue and microdissected pancreatic ductal adenocarcinoma identifies genes specifically overexpressed in tumor epithelia. Hepatogastroenterology. 2008;55:2016–2027. [PubMed] [Google Scholar]
- 28.Arumugam T, Ramachandran V, Fournier KF, et al. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009;69:5820–5828. doi: 10.1158/0008-5472.CAN-08-2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Furukawa T, Duguid WP, Rosenberg L, et al. Long-term culture and immortalization of epithelial cells from normal adult human pancreatic ducts transfected by the E6E7 gene of human papilloma virus 16. Am J Pathol. 1996;148:1763–1770. [PMC free article] [PubMed] [Google Scholar]
- 30.Maitra A, Adsay NV, Argani P, et al. Multicomponent analysis of the pancreatic adenocarcinoma progression model using a pancreatic intraepithelial neoplasia tissue microarray. Mod Pathol. 2003;16:902–912. doi: 10.1097/01.MP.0000086072.56290.FB. [DOI] [PubMed] [Google Scholar]
- 31.Cao D, Zhang Q, Wu LS, et al. Prognostic significance of maspin in pancreatic ductal adenocarcinoma: tissue microarray analysis of 223 surgically resected cases. Mod Pathol. 2007;20:570–578. doi: 10.1038/modpathol.3800772. [DOI] [PubMed] [Google Scholar]
- 32.Rasheed ZA, Yang J, Wang Q, et al. Prognostic Significance of tumorigenic cells with mesenchymal features in pancreatic adenocarcinoma. J Natl Cancer Inst. 2010;102:340–351. doi: 10.1093/jnci/djp535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ray KC, Blaine SA, Washington MK, et al. Transmembrane and soluble isoforms of heparin-binding epidermal growth factor-like growth factor regulate distinct processes in the pancreas. Gastroenterology. 2009;137:1785–1794. doi: 10.1053/j.gastro.2009.07.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kawaguchi Y, Cooper B, Gannon M, et al. The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat Genet. 2002;32:128–134. doi: 10.1038/ng959. [DOI] [PubMed] [Google Scholar]
- 35.Hingorani SR, Petricoin EF, Maitra A, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 36.Odom DT, Petricoin EF, Maitra A. Control of pancreas and liver gene expression by HNF transcription factors. Science. 2004;303:1378–1381. doi: 10.1126/science.1089769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stanescu DE, Raum JC, Stoffers DA, et al. Hnf6 and Pdx1 occupy common targets during pancreas development. Diabetes. 2013;(Suppl):A11. 43-OR. [Google Scholar]
- 38.Thomas J, Morlé L, Soulavie F, et al. Transcriptional control of genes involved in ciliogenesis: a first step in making cilia. Biol Cell. 2010;102:499–513. doi: 10.1042/BC20100035. [DOI] [PubMed] [Google Scholar]
- 39.Pierreux CE, Poll AV, Kemp CR, et al. The transcription factor hepatocyte nuclear factor-6 controls the development of pancreatic ducts in the mouse. Gastroenterology. 2006;130:532–541. doi: 10.1053/j.gastro.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 40.Wilding Crawford L, Tweedie Ables E, Oh YA, et al. Gene expression profiling of a mouse model of pancreatic islet dysmorphogenesis. PLoS ONE. 2008;3:e1611. doi: 10.1371/journal.pone.0001611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim MP, Gallick GE. Gemcitabine resistance in pancreatic cancer: picking the key players. Clin Cancer Res. 2008;14:1284–1285. doi: 10.1158/1078-0432.CCR-07-2247. [DOI] [PubMed] [Google Scholar]
- 42.Prevot PP, Simion A, Grimont A, et al. Role of the ductal transcription factors HNF6 and Sox9 in pancreatic acinar- to-ductal metaplasia. Gut. 2012;61:1723–1732. doi: 10.1136/gutjnl-2011-300266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ray KC, Moss ME, Franklin JL, et al. Heparin-binding epidermal growth factor-like growth factor eliminates constraints on activated Kras to promote rapid onset of pancreatic neoplasia. Oncogene. 2013 doi: 10.1038/onc.2013.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Means AL, Meszoely IM, Suzuki K, et al. Pancreatic epithelial plasticity mediated by acinar cell transdifferentiation and generation of nestin-positive intermediates. Development. 2005;132:3767–3776. doi: 10.1242/dev.01925. [DOI] [PubMed] [Google Scholar]
- 45.Strobel O, Rosow DE, Rakhlin EY, et al. Pancreatic duct glands are distinct ductal compartments that react to chronic injury and mediate Shh-induced metaplasia. Gastroenterology. 2010;138:1166–1177. doi: 10.1053/j.gastro.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Blaine SA, Ray KC, Anunobi R, et al. Adult pancreatic acinar cells give rise to ducts but not endocrine cells in response to growth factor signaling. Development. 2010;137:2289–2296. doi: 10.1242/dev.048421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Plumb-Rudewiez N, Clotman F, Strick-Marchand H, et al. Transcription factor HNF-6/OC-1 inhibits the stimulation of the HNF-3alpha/Foxa1 gene by TGF-beta in mouse liver. Hepatology. 2004;40:1266–1274. doi: 10.1002/hep.20459. [DOI] [PubMed] [Google Scholar]
- 48.di Mola FF, Friess H, Martignoni ME, et al. Connective tissue growth factor is a regulator for fibrosis in human chronic pancreatitis. Ann Surg. 1999;230:63–71. doi: 10.1097/00000658-199907000-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grotendorst GR, Okochi H, Hayashi N. A novel transforming growth factor beta response element controls the expression of the connective tissue growth factor gene. Cell Growth and Differentiation. 1996;7:469–480. [PubMed] [Google Scholar]
- 50.Eguchi T, Kubota S, Kondo S, et al. A novel cis-element that enhances connective tissue growth factor gene expression in chondrocytic cells. Biochem Biophys Res Commun. 2002;295:445–451. doi: 10.1016/s0006-291x(02)00700-3. [DOI] [PubMed] [Google Scholar]
- 51.Leask A, Holmes A, Black CM, et al. Connective tissue growth factor gene regulation Requirements for its induction by transforming growth factor-beta 2 in fibroblasts. J Biol Chem. 2003;278:13008–13015. doi: 10.1074/jbc.M210366200. [DOI] [PubMed] [Google Scholar]
- 52.Zhao Q, Chen N, Wang WM, et al. Effect of transforming growth factor-beta on activity of connective tissue growth factor gene promoter in mouse NIH/3T3 fibroblasts. Acta Pharmacol Sin. 2004;25:485–489. [PubMed] [Google Scholar]
- 53.Abreu JG, Ketpura NI, Reversade B, et al. Connective-tissue growth factor (CTGF) modulates cell signalling by BMP and TGF-beta. Nat Cell Biol. 2002;4:599–604. doi: 10.1038/ncb826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bennewith KL, Huang X, Ham CM, et al. The role of tumor cell-derived connective tissue growth factor (CTGF/CCN2) in pancreatic tumor growth. Cancer Res. 2009;69(3):775–784. doi: 10.1158/0008-5472.CAN-08-0987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Korc M. Pancreatic cancer-associated stroma production. Am J Surg. 2007;194(4 Suppl):S84–S86. doi: 10.1016/j.amjsurg.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]