

Abstract
BI 224436 is an HIV-1 integrase inhibitor with effective antiviral activity that acts through a mechanism that is distinct from that of integrase strand transfer inhibitors (INSTIs). This 3-quinolineacetic acid derivative series was identified using an enzymatic integrase long terminal repeat (LTR) DNA 3′-processing assay. A combination of medicinal chemistry, parallel synthesis, and structure-guided drug design led to the identification of BI 224436 as a candidate for preclinical profiling. It has antiviral 50% effective concentrations (EC50s) of <15 nM against different HIV-1 laboratory strains and cellular cytotoxicity of >90 μM. BI 224436 also has a low, ∼2.1-fold decrease in antiviral potency in the presence of 50% human serum and, by virtue of a steep dose-response curve slope, exhibits serum-shifted EC95 values ranging between 22 and 75 nM. Passage of virus in the presence of inhibitor selected for either A128T, A128N, or L102F primary resistance substitutions, all mapping to a conserved allosteric pocket on the catalytic core of integrase. BI 224436 also retains full antiviral activity against recombinant viruses encoding INSTI resistance substitutions N155S, Q148H, and E92Q. In drug combination studies performed in cellular antiviral assays, BI 224436 displays an additive effect in combination with most approved antiretrovirals, including INSTIs. BI 224436 has drug-like in vitro absorption, distribution, metabolism, and excretion (ADME) properties, including Caco-2 cell permeability, solubility, and low cytochrome P450 inhibition. It exhibited excellent pharmacokinetic profiles in rat (clearance as a percentage of hepatic flow [CL], 0.7%; bioavailability [F], 54%), monkey (CL, 23%; F, 82%), and dog (CL, 8%; F, 81%). Based on the excellent biological and pharmacokinetic profile, BI 224436 was advanced into phase 1 clinical trials.
INTRODUCTION
The primary function of HIV integrase is to integrate newly synthesized viral cDNA into the host genome, which is an obligate process for viral replication (1, 2). To fulfill this function, integrase performs two related reactions. Following the reverse transcription of viral RNA into cDNA, integrase catalyzes the hydrolysis of a dinucleotide from the 3′ end of each viral long terminal repeat (LTR) (a 3′-processing reaction). The viral preintegration complex then translocates to the nucleus, where the host cell factor lens epithelial derived growth factor (LEDGF) binds to integrase, tethering it to the host chromatin and targeting the integration of viral DNA (3–6). The two recessed 3′-LTR DNA ends are then used by integrase to mediate nucleophilic attacks on the target DNA phosphodiester backbones, resulting in the ligation of the viral DNA into the two strands of the host genomic DNA (strand transfer reaction). Each of these two integrase activities and the IN-LEDGF interaction have the potential to provide distinct targets for antiviral intervention (7).
Since the first proof of principle for the diketo acid integrase inhibitors in 2000, HIV integrase has been intensively studied as a target for drug discovery (8). The first integrase inhibitor to receive FDA approval, in October 2007, was raltegravir. Raltegravir and other members of its class bind to an integrase complex that consists of the post-3′-processed viral DNA and is proposed to bind the Mg2+ metal cofactor at the catalytic site of HIV integrase, as it competitively inhibits target DNA binding to integrase (9–12). Therefore, this class of compounds is referred to as the integrase strand transfer inhibitors (INSTIs). A second INSTI, elvitegravir, exhibits a cross-resistance profile with raltegravir, i.e., resistance mutations that emerge against one drug confer resistance to the other (13–16). Elvitegravir recently was approved as part of a single-tablet regimen of elvitegravir/cobicistat/emtricitabine/tenofovir disoproxil fumarate. A third drug from this class of integrase inhibitors, the recently approved dolutegravir, is reported to have a distinct resistance profile and possibly a higher barrier to resistance, indicating that it could be used to treat patients who have failed therapy with either raltegravir or elvitegravir (17).
With these marketed drugs and drugs in clinical development targeting integrase, it has become increasingly challenging to identify compounds with superior profiles within the existing classes of HIV-1 inhibitors due to cross-resistance and lack of differentiation in their profiles. As a result, our research efforts shifted toward the identification of compounds with new mechanisms of inhibition.
Using an integrase LTR DNA 3′-processing assay in a high-throughput format, we identified a 3-quinolineacetic acid series that specifically inhibited the 3′-processing step catalyzed by integrase (18, 19). After preliminary optimization in hit-to-lead chemistry, biochemical assays showed that in addition to inhibiting the 3′-processing step, this series also inhibited the LTR DNA interaction with integrase and the interaction between integrase and LEDGF without inhibiting the strand transfer activity of integrase (S. Mason and C. Fenwick, unpublished data). Crystallography studies with the catalytic core domain of integrase showed that the 3-quinolineacetic acid series bound into a conserved allosteric pocket on integrase. Concomitant with these studies, this pocket also was shown to serve as the binding site for LEGDF (20). Since these compounds were distinct from the integrase strand transfer inhibitors and interfered with several important functions of integrase in binding to an allosteric pocket, we refer to the 3-quinolineacetic acid compounds as non-catalytic-site integrase inhibitors (NCINIs). This series generated inhibitors with low-nanomolar potency in antiviral assays and excellent absorption, distribution, metabolism, and excretion (ADME) properties.
Given that the interaction between integrase and the host cell factor LEDGF has been proposed to be critical for HIV-1 viral replication, others have developed biochemical assays to probe for inhibitors of the integrase-LEDGF interaction (21, 22). A virtual screen of the LEDGF binding pocket on integrase with follow-up profiling using an integrase-LEDGF interaction assay identified a chemical series analogous to the NCINIs, termed LEDGINs, that were optimized to exhibit low μM antiviral potency (23).
Medicinal chemistry optimizations of the lead series led to the identification of BI 224436. This inhibitor has an effective antiviral profile against different laboratory strains of HIV-1 that was increased by only 2.1-fold in the presence of 50% human serum (HS). One of the characteristic features of the NCINI series is a steep concentration-response curve Hill slope of ∼4 in antiviral assays {% inhibition = [Imax × (inhibitor)n]/[(inhibitor)n + EC50n]}, where n is the Hill slope, Imax is the maximum inhibition, and EC50 is the 50% effective concentration. This results in antiviral EC95 values that are only about ∼2.4-fold higher than their EC50s. In vitro passage of virus in the presence of BI 224436 selected for mutations that encode either A128T, A128N, or L102F primary resistance substitutions in integrase. These substitutions confer a 2.9 (A128T)-, 64 (A128N)-, and 61 (L102F)-fold reduced susceptibility to BI 224436, and all map to a conserved allosteric pocket on the catalytic core of integrase. When tested against recombinant viruses encoding mutations that confer resistance to INSTIs, such as raltegravir and elvitegravir, BI 224436 retains antiviral activity without a loss of potency compared to wild-type virus. In two-drug combination studies, BI 224436 also exhibited at least additive in vitro antiviral activity when administered in combination with all marketed antiviral classes. Drug-like in vitro ADME properties of BI 224436 and favorable pharmacokinetic properties in preclinical species contributed to an excellent overall profile, which led to the advancement of this first-in-class novel-mechanism integrase inhibitor into phase 1 clinical trials.
MATERIALS AND METHODS
BI 224436, which has a molecular mass of 442.51 g/mol, was synthesized as reported previously (24). The structure for BI 224436, which has the chemical name (CAS) (αS,4R)-4-(2,3,-dihydropyranol [4,3,2-de]quinolon-7-yl)-α(1,1,-dimethylethoxy)-2-methyl-3-quinolineacetic acid, is shown in Table 1.
TABLE 1.
Antiviral potency of BI 224436 against HIV-1 with different integrase-containing viral strains used to infect PBMCs
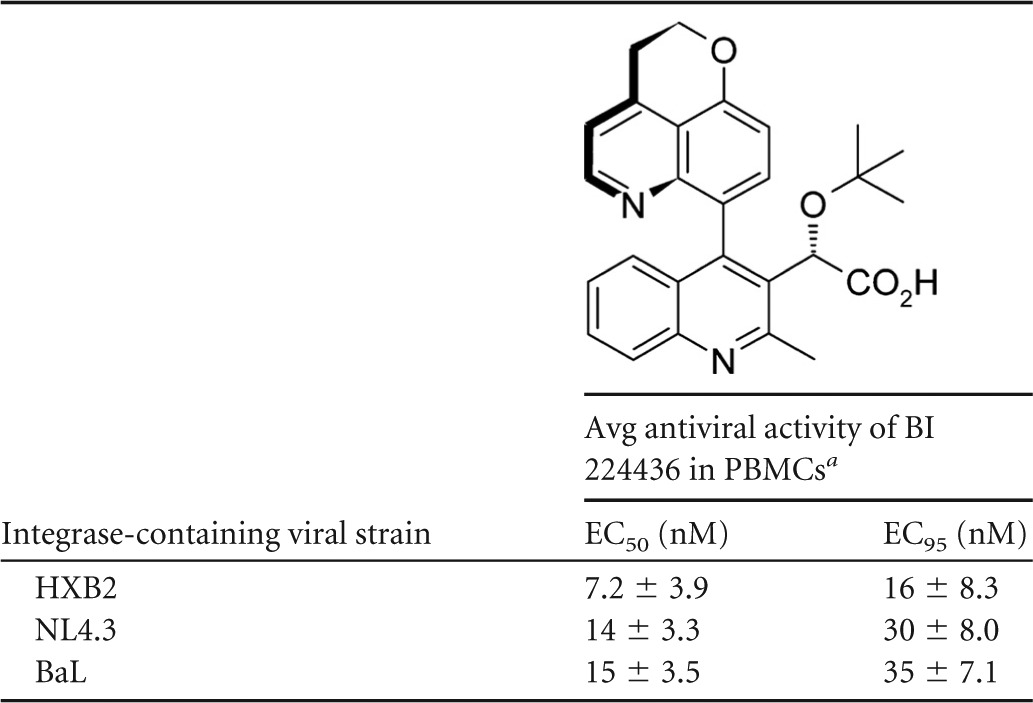
Mean EC50 and EC95 values with standard deviations represent average values from a minimum of three independent determinations.
Biochemical assays. (i) Expression and purification of recombinant proteins.
HIV-1 integrase was expressed as a hexahistidine fusion protein in BL21 bacterial cells and purified by metal-chelating chromatography on a Ni2+ column followed by SP-Sepharose ion-exchange chromatography and is based on a published purification protocol (25). The preparation was homogeneous, as revealed by SDS-PAGE and Coomassie colloidal blue protein staining. Integrase was stored at −80°C in storage buffer (25 mM HEPES, pH 7.4, 500 mM NaCl, 0.5 mM TCEP [tris(2-carboxyethyl)phosphine]).
The LEDGF construct used in this assay was cloned using basic molecular biology techniques. Briefly, the integrase binding domain of LEDGF (IBD; consisting of the nucleotides coding for residues 319 to 430 of LEDGF/p75) was amplified by PCR from Jurkat T-cell cDNA. The 3′ primer for this LEDGF gene amplification also introduced a Flag tag at the amino terminus of the IBD LEDGF construct. The DNA product was then cut with BamHI and XhoI restriction enzymes (New England BioLabs) and ligated into a pGEX4T vector precut with the same enzymes. The sequence of the resulting construct was verified by DNA sequencing. The glutathione S-transferase (GST)-Flag-LEGDF IBD construct was expressed in BL21 bacterial cells, with the cells lysed by sonication. After centrifugation at 20,000 × g for 30 min, GST-Flag-LEDGF IBD in the supernatant was bound to a 5-ml GSTrap HP column (from GE), washed extensively with phosphate-buffered saline (PBS), and eluted with 50 mM reduced glutathione. The purity of the GST-Flag-tagged LEDGF IBD protein was determined to be >85% by SDS-PAGE analysis. The protein was then dialyzed against storage buffer (25 mM HEPES, pH 7.4, 500 mM NaCl, 0.5 mM TCEP) and stored at −80°C.
(ii) Integrase LTR DNA 3′-processing assay.
The integrase LTR DNA 3′-processing assay measures the enzymatic activity of HIV-1 integrase to perform the essential 3′-processing reaction. Integrase binds to the viral DNA LTR ends at the CAGT-3′ sequence and catalyzes the removal of the two terminal nucleotides. In this homogeneous assay, the HIV-1 LTR DNA substrate consists of two annealed oligonucleotides, a 31-mer modified at the 3′ end with a black hole quencher (BHQ) and a 31-mer modified at the 5′ end with rhodamine red-X N-hydroxysuccinimide (NHS) ester (5RhoR-XN). Enzymatic cleavage by integrase releases the terminal dinucleotides and black hole quencher, which allows the rhodamine fluorescence to be detected (26). Briefly, 200 nM integrase was incubated with various concentrations of BI 224436, followed by the addition of 12.5 nM LTR substrate for a final volume of 10 μl and buffer composition of 25 mM morpholinepropanesulfonic acid (MOPS), pH 7.3, 25 mM NaCl, 2.5% polyethylene glycol (PEG), 4.2 mM dithiothreitol (DTT), 2% dimethylsulfoxide (DMSO), and 8 mM MgCl2. Samples were incubation for 2 h at 37°C, followed by the addition of 5 μl EDTA (0.5 M, pH 8) to stop the reaction, with fluorescence detection on a Victor2 V plate reader. Sequences used were the following: top strand, 5′-CTTTTAGTCAGTGTGGAAAATCTCTAGCAGT-3BHQ-2/3′; bottom strand, 5′-/5RhoR-XN/ACTGCTAGAGATTTTCCACACTGACTAAAAG-3′.
(iii) HIV-1 integrase strand transfer assay.
The integrase strand transfer assay was based on a previously published assay (8, 10). HIV integrase catalyzes a strand transfer reaction, resulting in the covalent attachment of a biotinylated target DNA molecule to a previously immobilized LTR-like oligonucleotide. The strand transfer product is detected by time-resolved fluorescence (TRF) using europium cryptate-labeled streptavidin, which binds to the biotinylated target DNA. In order to evaluate only the strand transfer enzymatic activity, integrase is first incubated with the immobilized LTR oligonucleotide to cleave off a dinucleotide at the LTR end, generating a 3′-recessed hydroxyl group. In the strand transfer reaction, integrase then uses that hydroxyl group as a nucleophile to attack the target DNA and covalently attach the LTR to target DNA. Compounds are added during this second reaction to test for inhibitors of strand transfer activity in this assay. An alternative version of the assay uses an immobilized LTR oligonucleotide that already has a 3′-recessed hydroxyl. The HIV U5 LTR-like DNA is prepared by annealing two complementary oligonucleotides, one of which has a primary amine group attached to the 5′ end via a 12-carbon linker. This primary amine permits covalent immobilization of the LTR to the DNA to bind to the amide-reactive N-oxysuccinimide plate (Corning) used for the assay. The sequences of the LTR oligonucleotides were the following: top strand (34-mer), 5′-amino-C12-ACC CTT TTA GTC AGT GTG GAA AAT CTC TAG CAG T-3′; bottom strand (31-mer), 5′-ACT GCT AGA GAT TTT CCA CAC TGA CTA AAA G-3′.
(iv) Integrase-LEDGF interaction assay.
The integrase-LEDGF interaction assay measures the protein-protein interaction between HIV-1 integrase and the LEDGF IBD using a homogeneous time-resolved fluorescence detection method. The integrase-LEDGF complex can be formed in the presence or absence of inhibitors. Integrase has a hexahistidine fusion tag which binds to an anti-His-tagged, XL665-labeled antibody, and the GST-Flag-tagged LEDGF IBD binds to the anti-GST, europium cryptate-labeled antibody.
Upon excitation at 340 nm, the europium cryptate will be excited and emit at 615 nm. If the XL665-labeled anti-His tag antibody is in close proximity to the europium cryptate-labeled anti-GST antibody, then there will be efficient energy transfer and XL665 will emit at 665 nm. Efficient energy transfer between the europium cryptate and XL665 will take place only if integrase and LEDGF form a bound complex (27).
This assay was performed in 384-well black flat-bottom, low-volume NBS plates (3676; Corning) and was divided into three steps. For step 1, compound dilutions were added into the reaction plate with 2 μl of each dilution added to the wells and included blank wells without compound. For step 2, 8 μl of premixed His-integrase (45 nM) and GST-LEDGF (15 nM) were added to the assay plate. Plates were incubated at 22°C for 30 min, followed by the addition of 10 μl of premixed antibodies (anti-His XL665 [50 nM] and anti-GST europium cryptate [1 nM]) so that the final concentration of reagents is 18 nM His-integrase, 6 nM GST-LEDGF, 2.5% DMSO, 25 nM XL665, and 0.5 nM europium cryptate. The plate containing the assay components was incubated for 1 h at 22°C. For step 3, the plate was read on a Victor2 V reader.
Antiviral assays. (i) Viral stock production.
Cells and reagents obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH, included λHXB2 (28), pNL4.3 (29), and BaL (catalog no. 510; NIH AIDS Reagent Program). The C8166 cell line was obtained from J. L. Sullivan, University of Massachusetts Medical Centre.
Human PBMCs were obtained from the laboratory of Rafick Sékaly and were isolated from donor 434. Cells were grown in RPMI 1640 medium plus 10% fetal bovine serum (FBS), containing 10 µg/ml gentamicin and 10 µM β-mercaptoethanol. The PBMC medium also contained 3 μg PHA/ml, 50 U recombinant human interleukin-2 (rhIL-2)/ml, 100 U of penicillin, and 100 μg streptomycin.
Viral stocks were produced by transfecting the molecular clones into 293 T cells with Fugene 6. After 3 days of incubation, the supernatant was collected and centrifuged for 10 min at 1,000 × g at 4°C to remove cells. Aliquots of 1 ml were kept at −80°C.
Titers of viruses were determined as previously described without any modifications (30). The stock solution of virus was titrated by log dilution, and the 50% tissue culture infectious dose (TCID50) of each viral stock was determined by monitoring the macroscopic formation of syncytia in the C8166 cell line at day 3. For PBMCs, the TCID50 value was determined at day 7 by p24 analysis. Calculations of TCID50 values were made using the Spearman-Karber method (31).
(ii) HIV-1 inhibitory assay.
PBMCs were stimulated with 3 μg phytohemagglutinin (PHA)/ml and 50 U rhIL-2/ml for 3 days prior to infection. PBMCs were infected at a multiplicity of infection (MOI) of 0.001 with viruses in complete RPMI 1640 for 1.5 h on a rotating rack in a 5% CO2 incubator at 37°C. The cells were then centrifuged for 5 min at 400 × g and washed with complete RPMI 1640 medium. Cells were resuspended at 1 × 105 PBMCs/175 μl complete RPMI 1640 and were transferred to 96-well cell microtiter plates (175 μl/well) already containing 25 μl of 8× inhibitor. Eight replicates were done for each dilution (i.e., one plate/compound). After the 7-day incubation, 100-μl aliquots of supernatant from each replicate were pooled for each inhibitor dilution for a total of 800 μl in a 1-ml 96 DeepWell block. The blocks were centrifuged for 10 min at 400 × g, and the top 400 μl was transferred to a new block. The PerkinElmer HIV-1 p24 antigen assay kit was used according to the manufacturer's instructions, with no modifications, to determine the extracellular p24 protein concentration. Cytotoxicity assays were performed in parallel using the tetrazolium salt 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) metabolic assay (32) to determine the concentration of inhibitor that results in 50% cell death (CC50) of lymphocytes.
Selection of HIV-1 variants resistant to BI 224436.
In vitro passage experiments to select for viral variants that conferred resistance to BI 224436 were performed as previously described (33). Briefly, C8166 cells were infected with the NL4.3 virus harboring HXB2 integrase at an MOI of 0.1 with inhibitor concentrations of 2- or 5-fold the EC50. At each passage, a visual evaluation of the viral cytopathic effect (CPE) was performed, and the cell culture supernatants were used to infect fresh C8166 cells. Inhibitor concentrations were either maintained or increased with the passage of virus, depending upon the evidence of viral replication as observed through the CPE. At passages 4 and 14, the genomic DNA was isolated from the C8166 cells using the DNeasy blood and tissue kit (Qiagen). The integrase coding region was amplified by PCR with the fragment cloned into the Zero blunt TOPO cloning vector (Invitrogen) and sequenced on an ABI Prism 377 DNA sequencer.
Replication capacity of recombinant viruses.
The replication capacity of recombinant viruses encoding amino acid substitutions in HIV integrase was evaluated using a Jurkat LTR-Luciferase cell line as previously described (33). Briefly, cells were infected with recombinant virus at an MOI of 0.02 for 2 h at 37°C, washed, seeded in 96-well microtiter plates at 1 × 105 cells/well in a volume of 200 μl RPMI 1640 medium supplemented with 10% fetal bovine serum and 10 μg/ml gentamicin, and incubated in cell culture for up to 14 days. Luciferase levels were determined by adding 50 μl/well of BrightGlo (Promega) at days 7, 10, 12, and 14 postinfection, and chemiluminescent readings were performed on the LUMIstar galaxy plate reader (BMG).
Cloning of the recombinant viruses encoding amino acid substitutions that confer resistance to INSTIs, NCINIs, and non-nucleoside reverse transcriptase inhibitors (NNRTIs).
Amino acid substitutions that have been reported to lead to reduced susceptibility to integrase strand transfer inhibitors (34) (T66I/S153Y, E92Q, G140S/Q148H, and N155S amino acid substitutions in the integrase coding region) and noncatalytic integrase inhibitors (A128T, A128N, L102F, and N222K) were introduced into an HXB2 integrase-containing vector using the QuikChange site-directed mutagenesis kit from Stratagene according to the manufacturer's protocol. DNA sequencing was used to confirm the appropriate sequence for each clone. HXB2 integrase encoding each of the single- or multiple-amino-acid substitutions was cloned into pNL4.3 using the AgeI-SalI restriction sites.
HIV-1 pNL4.3 BH10 RT, encoding the K103N/Y181C double substitution in the reverse transcriptase (RT) gene, was generated as previously described (35, 36). HIV-1 inhibitory assays with wild-type and recombinant viruses resistant to INSTIs, NCINIs, or NNRTIs were performed as previously described using C8166 LTR luciferase reporter cells (33, 37).
Determination of antiviral EC50 shift in the presence of human serum.
C8166 LTR luciferase reporter cells were infected with wild-type HIV-1 NL4.3 virus as previously described (33, 36). Briefly, cells were infected at an MOI of 0.015 in complete RPMI 1640 for 1.5 h on a rotating rack in a 5% CO2 incubator at 37°C. The cells were centrifuged for 5 min at 400 × g, the supernatant removed, and the cells resuspended at 2.5 × 104 cells/150 μl complete RPMI 1640 or one of the five human serum-supplemented culture medium stock solutions. The cells were transferred to a 96-well cell microtiter plate (150 μl/well) containing 50 μl of 4× inhibitor. With this dilution, the final concentration of human serum in the wells is 10%, 20%, 30%, 40%, or 50% for the EC50 determinations of BI 224436. Triplicates were done for each dilution (i.e., two plates/compound). A total of 25,000 uninfected C8166-LTR luciferase cells/well in 200 μl complete RPMI (with 0% to 50% human serum) was added to the last row for background control. Cells were incubated at 37°C in a 5% CO2 incubator for 3 days. After the 3-day incubation, 50 μl Steady Glo (luciferase substrate; half-life [t1/2], 5 h) was added to each well of the 96-well plate with luminescence monitored on a PerkinElmer EnVision plate reader. The luminescence, determined in counts per second (cps) in each well of the culture plate, was a measure of the amount of HIV replication in the presence of the various concentrations of inhibitor. The level of inhibition (% inhibition) of each well containing inhibitor was calculated with the following equation: % inhibition = 100 − [100 × cps (inhibitor)/cps (control without inhibitor)].
In vitro combination studies of BI 224436 with a broad panel of antiviral agents (nucleoside reverse transcriptase inhibitors [NRTIs], NNRTIs, protease inhibitors [PIs], INSTIs, and entry inhibitors) against wild-type HIV-1 virus.
C8166 LTR luciferase reporter cells were infected with wild-type HIV-1 NL4.3 virus as described above. All test compounds were prepared from a powdered drug substance, which was dissolved in 100% DMSO to a final concentration of 5, 10, or 20 mM. The DMSO stock solution of inhibitor was then diluted in complete RPMI 1640 to an 8× concentration of the final test solution (0.1% DMSO final concentration). Each combination experiment included 6 doses of antiviral drug alone, 8 doses of BI 224436, and 48 drug combinations. BI 224436 and the combination partner tested were serially diluted 2-fold in cell culture medium, starting at approximately 6-fold the EC50 of the individual inhibitors. Drug combinations were prepared in a matrix format using the same range of concentrations as that used for the drugs alone. In each experiment, combinations were tested in triplicate for activity in the HIV replication assay.
The degree of HIV replication determined by the reporter luciferase signal was used to calculate the percentage of inhibition for each condition tested. The results obtained for each drug combination were subsequently transformed with Prichard and Shipmann's MacSynergy II software using the Bliss independence model (38). This method uses a nonparametric three-dimensional approach to quantify areas where observed effects are significantly greater (synergy) or less (antagonism) than those predicted from the data obtained from the individual dose-response curves. All data points generated from the matrix of drug concentrations were used in the analysis. Triplicate data sets were used to perform statistical analysis at the 95% confidence level and can be interpreted as previously described (38, 39). Mean synergy volumes at 95% confidence (in nM2%) are described at the following levels: >100, strong synergy; 50 to 100, moderate synergy; 25 to 50, minor amount of synergy; 25 to −25, additive; −25 to −50, minor amount of antagonism; −50 to −100, moderate antagonism; and <−100, strong antagonism.
ADME profiling. (i) Caco-2 cell permeability assay.
Caco-2 cells were seeded on Costar Transwell inserts (12-well plate) and allowed to grow and differentiate for 23 to 25 days. Culture medium (DMEM supplemented with 10% FBS, 1% nonessential amino acids, and penicillin-streptomycin) was removed from both sides of the transwell insert, and cells were rinsed twice with warm Hanks' balanced salt solution (HBSS). At the last rinse step, the chambers were filled with warm transport buffer (the apical-side buffer was HBSS, 25 mM morpholineethanesulfonic acid [MES], 0.25% bovine serum albumin [BSA], pH 6.0; the basolateral-side buffer was HBSS, 25 mM HEPES, 0.25% BSA, pH 7.4). The plates were incubated at 37°C for 30 min prior to assay. The donor fluid (apical side for A-to-B assay, basolateral side for B-to-A assay) was removed and replaced with compound at a final solution concentration of 10 μM. At designated time points (0, 1, 2, and 3 h), fluid from the receiver chamber was removed and replaced with the appropriate fresh transport buffer. Samples were quenched with cold acetonitrile-methanol (50:50, vol/vol), centrifuged to pellet protein, and then submitted for liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis. Reported permeability represents averages from 2 wells.
(ii) Cytochrome P450 inhibition assay.
cDNA-expressed human cytochrome P450 (Supersome) isozymes were purchased from Gentest Corporation. All CYP450 isozymes were received frozen and stored at −80°C. Isozymes tested were thawed immediately prior to the initiation of the experiment and kept at 4°C until use.
BI 224436 was dissolved in acetonitrile-methanol (50:50, vol/vol) to achieve a concentration of 1.5 mM. Standard positive controls, namely, furafylline (CYP1A2), sulfaphenazole (CYP2C9), tranylcypromine (CYP2C19), quinidine (CYP2D6), and ketoconazole (CYP3A4), at the required concentration ranges were also included in every plate for each isozyme.
Phosphate buffer (pH 7.4), cofactor, and test substance or isoform-selective inhibitors were added to 96-well plates and were prewarmed to 37°C for 10 min. Cofactor concentrations were 1.3 mM NADP, 3.3 mM glucose-6-phosphate, and 0.4 U/ml glucose-6-phosphate dehydrogenase. Reactions were initiated by the addition of prewarmed (37°C) enzyme and substrate. Reaction mixtures were incubated at 37°C and terminated by the addition of 0.038 ml of 40:40:20 (vol/vol) methanol–acetonitrile–0.5 M Tris buffer. Formation of the fluorescent metabolites {7-ethoxy-3-cyanocoumarin for CYP1A2 and CYP2C19, 7-methoxy-4-trifluoromethylcoumarin for CYP2C9, 3-[2-(N,N-diethyl-N-methylamino)ethyl]-7-methoxy-4-methylcoumarin for CYP2D6, and 7-hydroxy-4-trifluoromethylcoumarin for CYP3A4} was measured using a SpectraMax Gemini XS microplate spectrofluorometer at specific excitation and emission wavelengths. The 50% inhibitory concentration (IC50) was determined using the 96-well 32 procedure supplied with the SAS software.
(iii) In vitro metabolism in the presence of LM and hepatocytes.
A liver microsome (LM) stability assay was performed with four time points of incubation at 0, 10, 20, and 30 min with a 2 μM BI 224436 initial concentration. Male human liver microsomes were obtained from Gentest. Metabolic studies in cryopreserved male human hepatocytes were performed in 96-well plates with samples collected at five time points (0, 15, 30, 60, and 90 min). Hepatocytes (purchased from In Vitro Technologies) at a density of 1 × 106 cells/ml were incubated with 2 μM BI compound at 37°C under a O2 and CO2 (95 and 5%, respectively) atmosphere. Half-lives were determined by the ratio of ln2 over the first-order rate of disappearance of the parent compound (0.693/−k). Intrinsic clearance (CLint) was calculated as CLint = 0.693/t1/2 × volume of incubation (ml)/protein concentration in the assay (mg) × LM yield (mg of microsomes/g of liver) × liver weight (g/kg). CLLM or hepatic = QH × CLint/(QH+ CLint), where QH is liver blood flow (%QH = CLhepatic/liver blood flow rate × 100).
(iv) Plasma protein binding.
Plasma protein binding was determined across species by equilibrium dialysis (40). The dialysis apparatus consists of a two-compartment chamber separated by a cellulose membrane. One side of the membrane is filled with plasma spiked with the compound of interest, while the other side is filled with buffer. The amount of compound collected on the buffer side after the 6-h incubation indicates how much of the compound is unbound, from which a value for the percent plasma protein binding is determined according to the following equation: % protein binding = [(plasma side concentration − buffer side concentration)/(plasma side concentration)] × 100%.
(v) Solubility determination.
A 0.5-ml aliquot of the selected aqueous medium was added into a vial which contained a minimum of 0.5 mg solid drug substance. The vial was shaken in an orbital shaker at 250 rpm and 25°C for predefined time periods (typically 24 h). After shaking, the solution or suspension was filtered using an appropriate filter membrane. The amount of dissolved drug substance was determined by LC-MS. Buffers included 50 mM phosphate buffer, pH 2.0, and 50 mM phosphate buffer, pH 6.8.
PK experiments.
All protocols involving animal experimentation were reviewed and approved by the respective Animal Care and Use Committee of each test facility. In-life procedures were in compliance with the Guide for the Care and Use of Laboratory Animals from the Canadian Council of Animal Care. All rat and mouse pharmacokinetics (PK) studies were performed at Boehringer Ingelheim (Canada) Ltd., R&D. PK studies in dogs and monkeys were performed at ITR Laboratories Canada, Inc., Montreal, Québec, Canada. All chemicals used were reagent grade or better.
(i) Formulation for oral and intravenous (i.v.) dosing.
For oral PK studies, BI 224436 was administered in a suspension of 0.5% (wt/vol) methyl cellulose (MC), 0.3% (vol/vol) Tween 80, and 1% (vol/vol) N-methyl-2-pyrrolidine (MP) in water. The appropriate amount of BI 224436 was placed in a mortar and ground with MP and then with Tween 80 until a smooth paste was obtained. The 0.5% methyl cellulose solution in water was added while stirring. The concentration in the dosing formulation was 0.04 mg/ml (90.4 μM) for rat and mouse studies and 0.2 mg/ml (45.2 μM) for dog and monkey studies.
For i.v. dosing, BI 224436 was dissolved in 70% PEG 400–30% water (vol/vol). The appropriate amount of BI 224436 was dissolved in PEG 400 with sonication. Water was slowly added, and the preparation was sonicated to obtain a homogeneous solution. The concentration of the intravenous dosing solution was 0.2 mg/ml (452 μM) for rat and mouse studies and 2 mg/ml (4.52 mM) for dog and monkey studies.
(ii) Animals used for PK studies.
Six male Sprague-Dawley rats (250 to 300 g; Charles River, St-Constant, Québec, Canada) were used for the study. Animals were fasted overnight with access to 10% dextrose in water. Groups of three animals were used per dosing regimen. Thirty-eight female CD-1 mice (21 to 30 g; Charles River, St-Constant, Québec, Canada) were used for the study. Animals were allowed food and water ad libitum. Groups of three to six animals were used at each time point per dosing regimen.
Three male beagle dogs (8.4 to 10.1 kg) or three male cynomolgus monkeys (3.2 to 3.7 kg), all housed at ITR Laboratories Canada, Inc., Montreal, Québec, Canada, were used for the study. Animals were fasted overnight prior to dosing and fed 4 h after dosing. Water was available at all times.
(iii) Dosing and sampling of animals.
The rats received a single i.v. dose of 0.2 mg/kg of body weight (1 ml/kg) via the jugular vein as a bolus or received a single oral dose of 0.4 mg/kg (10 ml/kg) administered by gavage. Blood samples were obtained at 0, 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, and 32 h after dosing (additional samples were obtained at 0.033 and 0.083 h for the i.v. studies) from the carotid cannula and were collected into tubes containing K2EDTA. At each time point, the blood from three rats was pooled and immediately placed on ice and then centrifuged (4,000 × g, 10 min) at 4°C. The plasma was separated and stored at −20°C until extraction and LC-MS/MS analysis. The mice received either a single intravenous dose of 0.2 mg/kg (4 ml/kg) via the tail vein as a bolus or a single oral dose of 0.4 mg/kg (10 ml/kg) administered by gavage. Blood samples were obtained at the same time points as those described for rats. Animals were bled at three different time points, twice by retro-orbital sampling (150 μl each in heparinized capillary tubes), and the terminal bleeding was performed by cardiac puncture using a heparinized syringe. At each time point, the blood was immediately placed on ice and then centrifuged (4,000 × g, 10 min) at 4°C. The plasma was separated and stored at −20°C until extraction and LC-MS/MS analysis.
Three male beagle dogs or three male cynomolgus monkeys were dosed by i.v. bolus injection at a dose of 1 mg/kg. Blood samples were obtained at 0, 5, 15, 30, and 60 min and 2, 4, 6, 8, 12, 24, and 48 h after dosing from the left or right jugular vein and collected into tubes containing K2EDTA. After a washout period of 1 week, the same animals were dosed orally at 2 mg/kg. Blood samples were collected at similar time points, except that the samples at 5 min were omitted. All plasma samples were collected after centrifugation (1,500 × g, 10 min) at 4°C and were stored at −20°C until extraction and LC-MS/MS analysis. A similar protocol is described in Beaulieu et al. (41).
(iv) Bioanalysis of plasma samples.
Plasma sample extraction was performed on the Hamilton SPE workstation using a Waters Oasis HLB extraction plate. Fifty μl of plasma was mixed with 0.15 ml Milli-Q water, 50 μl of acetonitrile, and 5 μl 20% phosphoric acid (vol/vol) in water. Samples were loaded on an Oasis HLB 30-mg/1-ml 96-well extraction plate that had previously been conditioned with 1 ml of 5% (vol/vol) ammonium hydroxide in acetonitrile, followed by 1 ml of acetonitrile and then 1 ml of Milli-Q water. Cartridges were washed with 1 ml of Milli-Q water followed by 1 ml of 5% (vol/vol) methanol in water and finally by 1 ml of 2% (vol/vol) acetic acid in methanol-acetonitrile-water (1:1:8) prior to elution. The compound was twice eluted with 500 μl of 5% (vol/vol) ammonium hydroxide in acetonitrile. The organic phase was evaporated under a nitrogen stream at 60°C using a Zymark TurboVap 96. The residue was reconstituted in 500 μl of 0.2% (vol/vol) ammonium hydroxide in acetonitrile-water (1:1). Two μl of sample was injected for LC-MS/MS analysis using an Atlantis dC18 2.1- by 30-mm, 5-μm-volume column at 40°C with a flow rate of 0.4 ml/min. A high-performance liquid chromatography (HPLC) gradient of 20 to 98% B from 0.2 to 1.4 min and 98% B from 1.4 to 2.3 min was used for compound separation (for the mobile phase, A contained water and 0.1% formic acid; B contained acetonitrile and 0.1% formic acid) with MS/MS analysis in positive electrospray ionization mode (spray voltage, 3,700 V; capillary temperature, 350°C; sheath gas pressure, 40 lb/in2; capillary offset, 35 V; auxiliary gas pressure, 20 lb/in2; scan width, 0.01 s; scan time, 0.10 s; q2 collision pressure, 1.5 mTorr; collision energy, 28 V; BI 224436 parent m/z 443.1; product m/z 172.0). The limit of quantification was determined to be 9 ng/ml (20 nM) by injecting standard solutions of BI 224436. Pharmacokinetic calculations were performed using WinNonlin 5.0 (Pharsight, CA).
RESULTS
Activity in biochemical assays.
The initial hit from the NCINI series was identified using a fluorescence-based assay with an LTR DNA probe to measure the 3′-processing enzymatic activity of HIV-1 integrase. Medicinal chemistry optimization combined with rational design based on our understanding of the binding mode of our inhibitors led to the identification of our first development candidate, BI 224436 (18). A mean IC50 of 15 ± 4 nM was obtained for BI 224436 with the LTR 3′-processing assay (see Fig. S1a in the supplemental material). In contrast, BI 224436 displayed minimal inhibition of the integrase strand transfer enzymatic activity with a mean IC50 of >50 μM. Raltegravir, tested in parallel with BI 224436, has a mean IC50 of 19 ± 4 nM, which is consistent with the IC50 of 15 nM reported previously (42).
X-ray crystallography studies showed that BI 224436 and the NCINI series binds a conserved allosteric pocket on HIV-1 integrase that serves as the interaction site for LEDGF (18 and data not shown), a host cell cofactor that is important for viral replication. The integrase-LEDGF interaction was evaluated using a homogeneous time-resolved fluorescence assay, which measures the extent of association between the two proteins. This interaction was inhibited in the presence of BI 224436 with a mean IC50 of 11 ± 1 nM (see Fig. S1b in the supplemental material). As such, in binding to the allosteric pocket at the integrase catalytic core dimer interface, BI 224436 inhibits both the LTR 3′-processing enzymatic activity and the integrase-LEDGF interaction with equivalent potencies.
Activity in cellular assays.
Activity in cellular antiviral assays is an important indicator of a potential in vivo effect of our inhibitor series in HIV-1-infected patients. The in vitro antiviral activity of BI 224436 was investigated in peripheral blood mononuclear cells (PBMCs) infected with HIV-1 expressing integrase from different laboratory strains. The antiviral activity was evaluated by monitoring for extracellular HIV-1 capsid core protein (p24) in the presence of different inhibitor concentrations. In the PBMCs, BI 224436 had EC50s of 7.2 nM, 14 nM, and 15 nM observed for HXB2, NL4.3, and BaL integrase-containing viruses, respectively (Table 1). These antiviral activities observed in the cell-based assays are consistent with the IC50s obtained for BI 224436 in both the 3′-processing and integrase-LEDGF interaction biochemical assays.
In determining the antiviral activity of early compounds from the NCINI class (18, 19), we noted that the Hill coefficient was consistently about 4. As shown in Fig. 1, BI 224436 displays a steep dose-response curve compared to raltegravir, with Hill coefficients of 4 and 1, respectively. This results in an EC95-to-EC50 ratio of <2.5-fold for BI 224436 in all in vitro antiviral cell-based assays that we tested. This has also been reported by others for structural analogues of the NCINI class of compounds (43–47).
FIG 1.
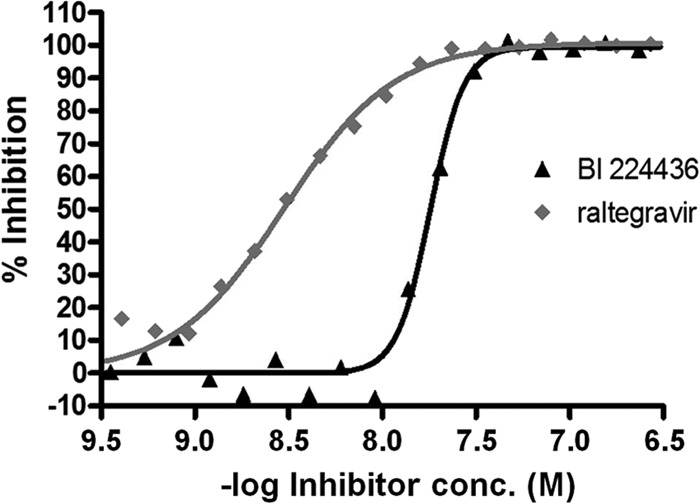
Comparison of the antiviral inhibition curves of integrase strand transfer and non-catalytic-site integrase inhibitors. The HIV-1 NL4.3 virus was used to infect PBMCs in the presence of increasing concentrations (conc.) of either BI 224436 or raltegravir, representing the NCINI and INSTI classes of integrase inhibitors, respectively. The antiviral concentration-response curves were generated with 1.5-fold compound dilutions in order to more precisely evaluate the Hill slope values of approximately 4 for BI 224436 and 1 for raltegravir.
Cytotoxicity resulting in 50% death of cells (CC50) was determined using the tetrazolium salt MTT metabolic assay (32). The mean CC50 value for BI 224436 was 97 μM after 3 days of incubation for C8166 cells (see Fig. S2 in the supplemental material) and >120 μM for PBMCs after 7 days. Given the antiviral potency of 15 nM for BI 224436 in the least-susceptible HIV-1 laboratory strain tested, the in vitro selectivity index of BI 224436 is 6,500-fold in C8166 cells and >8,000-fold in PBMCs.
Antiviral potency in the presence of HS.
The antiviral potency reported in Table 1 was assessed in medium containing 10% FBS but in the absence any human serum proteins. In order to assess the compound's potency in human blood (48, 49), the antiviral activity of BI 224436 was determined in experiments in which the cell culture medium was supplemented with 10%, 20%, 30%, 40%, or 50% human serum (HS). Inhibition curves were performed with 1.5-fold compound dilutions around the EC50 for BI 224435 in order to increase the precision of both the EC50 determinations and the calculated fold shift in view of the high Hill slope that had been determined with BI 224436 (Fig. 2). As HS was increased from 0% to 40% in the in vitro HIV-1 viral replication assay, the fold change in EC50s for BI 224436 increased until it approached a maximum of approximately 2.1-fold in the presence of 40% and 50% HS. This maximal fold change in EC50s suggests that binding of BI 224436 to the proteins present in HS has reached saturation. Extrapolating to 100% HS, the inhibition of HIV-1 viral replication by BI 224436 increased by 2.14- ± 0.20-fold with a 95% confidence window relative to EC50s obtained in the absence of HS. The Hill slope of 4 was also observed in antiviral inhibition curves for BI 224436 in the presence of 10% to 50% human serum. Using the serum shift correction, BI 224436 is predicted to have an EC50 of 32 nM and an EC95 of 75 nM in 100% human serum against the least-susceptible laboratory strain of HIV-1 tested based on the results shown in Table 1.
FIG 2.
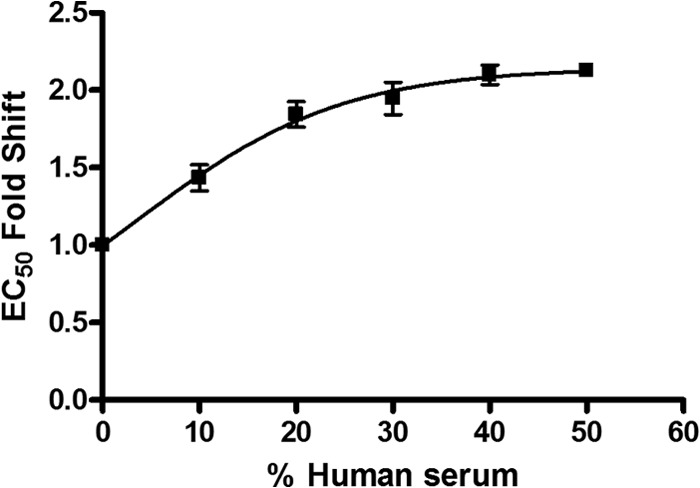
Fold decrease in mean EC50s in the presence of different percentages of human serum with the in vitro antiviral activity determined using the C8166 LTR luciferase reporter cell line. The HIV-1 NL4.3 virus was used to infect the reporter cell line in RPMI medium containing 10% FBS that was supplemented with 0, 10, 20, 30, 40, or 50% human serum. Data represent the averages from three separate experiments with the standard deviations shown. Extrapolation to 100% human serum gives a 2.14-fold increase in the EC50 for BI 224436.
Selection of HIV-1 variants resistant to BI 224436.
To help establish that BI 224436 inhibits integrase through a mechanism distinct from that of INSTIs, HIV-1 virus was serially passaged in the presence of increasing concentrations of BI 224436 in cell culture. Viral infections were initiated at inhibitor concentrations of approximately 2- or 5-fold the EC50 of BI 224436. Upon each passage, a macroscopic evaluation of the cells was performed to determine if drug concentration would be maintained or increased if syncytium formation was observed as evidence of viral replication. At passage 4, the A128T amino acid substitution was selected in HIV integrase (see Table S1 in the supplemental material). After 14 passages, viral variants encoding either the A128N or L102F substitution were sequenced and were found in combination with the N222K variant. Recombinant viruses encoding the single-amino-acid substitutions observed in the in vitro passage experiments were generated by site-directed mutagenesis and used in the cell culture antiviral assay with BI 224436. In these studies, the substitutions resulted in a 2.9-fold (A128T), 64-fold (A128N), 61-fold (L102F), and 2.8-fold (N222K) decrease in antiviral potency of BI 224436 relative to the wild-type virus (Table 2). In contrast, recombinant viruses that conferred resistance to BI 224436 were susceptible to the INSTIs raltegravir and elvitegravir with no loss in antiviral potency (Table 2). The replication capacity of these recombinant viruses was 30% (A128T), 6% (A128N), <1% (L102F), and 90% (N222K) compared to wild-type virus when propagated in Jurkat T cells.
TABLE 2.
EC50 fold changes for inhibition of recombinant HIV-1 virus replication by BI 224436a
| Test compound | EC50 fold change in inhibition of: |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Active-site integrase inhibitor-resistant mutant |
NCINI-resistant mutant |
NNRTI-resistant mutant | |||||||
| N155S | G140S/Q148H | T66I/S153Y | E92Q | A128T | A128N | L102F | N222K | K103N/Y181C | |
| BI 224436 | 1.0 | 1.2 | 0.5 | 1.0 | 2.9 | 64 | 61 | 2.8 | 0.9 |
| Elvitegravir | 77 | NA | 81 | 16 | 1.1 | 0.9 | 1.1 | 1.6 | 0.5 |
| Raltegravir | 8.5 | 290 | 0.4 | 3.1 | 0.9 | 1.0 | 1.0 | 1.0 | 0.7 |
Shown are EC50 fold changes for inhibition by BI 224436 of replication of recombinant HIV-1 viruses containing mutations that confer resistance to either NNRTIs or the active-site inhibitors raltegravir and elvitegravir. Single or double point mutations were introduced into the HXB2 integrase-containing virus, which was used to infect the C8166 LTR luciferase reporter cell line. The EC50s were calculated in a minimum of three separate experiments with the mean fold shift in EC50 calculated relative to the EC50 of the wild-type virus.
To correlate the reduced susceptibility of BI 224436 with virus encoding the mutations in integrase selected during in vitro passage experiments, recombinant integrase proteins with A128T and L102F substitutions were expressed and purified. Integrase proteins with these substitutions were found to be catalytically active in the LTR DNA 3′-processing assay, and the inhibitor potential of BI 224436 was evaluated (see Fig. S3 in the supplemental material). Compared to the IC50 for BI 224436 (15 nM), the A128T integrase had a decreased susceptibility of 2.9-fold and the L102F integrase had a >500-fold decreased susceptibility to BI 224436. These reduced susceptibilities in the integrase biochemical assay are consistent with the shift in antiviral potency observed in the cell culture assay.
Antiviral profile of BI 224436 against recombinant viruses with resistance to INSTIs and NNRTIs.
In order to establish that BI 224436 will maintain its antiviral activity against viruses that emerge during treatment failure with other inhibitor classes and to confirm the specificity of its mechanism of action, BI 224436 was tested against recombinant viruses encoding amino acid substitutions that have been shown to confer resistance to either NNRTIs (K103N/Y181C) or active-site integrase inhibitors (T66I/S153Y, E92Q, G140S/Q148H, and N155S). The same recombinant HIV-1 viruses were also tested against the active-site integrase inhibitors raltegravir and elvitegravir. As shown in Table 2, the NNRTI-resistant viruses remained susceptible to BI 224436, raltegravir, and elvitegravir, with no change in the EC50s for any of these inhibitors relative to the wild-type control virus. As expected, raltegravir and elvitegravir lost activity against INSTI-resistant viruses; however, these viruses remained fully susceptible to BI 224436.
In vitro combination studies of BI 224436 with a broad panel of antiviral agents (NRTIs, NNRTIs, PIs, INSTIs, and entry inhibitors) against wild-type HIV-1 virus.
Current therapy for HIV infections involves the administration of a combination of several antiretroviral drugs in order to effectively suppress viral replication. Therefore, it is essential to determine whether any given combination of drugs is antagonistic and whether the drugs interfere with each other's antiviral activity as opposed to inhibiting viral replication in an additive or, preferably, synergistic manner. Two-drug combination studies were performed on wild-type HIV-1 testing with BI 224436 in combination with several other antiretroviral agents, including NRTIs, NNRTIs, PIs, INSTIs, and entry inhibitors. The degree of interaction was determined according to the Bliss independence model using MacSynergy II software to analyze the results. Volumes of statistically significant synergy and antagonism at 95% confidence were determined with combination volumes (CV) of >25 nM2% interpreted as evidence for synergy, CV between 25 and −25 nM2% as evidence for additive effect, and CV of <−25 nM2% as evidence for antagonism. Overall results of this study suggest that only slight to moderate interactions occur between BI 224436 and most of the antiviral agents tested, as shown in Table 3. Combination studies between BI 224436 and the NRTI tenofovir showed strong synergy (CVsynergy, 124 ± 18 nM2%; CVantagonism, −5.1 ± 4.1 nM2%) in all experiments. Moderate synergy was observed in combination studies between BI 224436 and the protease inhibitors amprenavir, atazanavir, and nelfinavir. Studies performed with combinations of BI 224436 and the NRTI abacavir show a tendency toward minor antagonism; however, additivity is within the range of the standard deviations (CVsynergy, 12 ± 5.6 nM2%; CVantagonism, −44 ± 28 nM2%). Indications of strong or medium antagonism were not detected with any of the combinations involving BI 224436. In targeting the same viral enzyme, combination studies between BI 224436 and the INSTIs raltegravir and dolutegravir were evaluated for signs of synergy or antagonism. As shown in Fig. 3, two-drug combinations of BI 224436 and raltegravir had a clear additive antiviral inhibition profile.
TABLE 3.
Mean synergy and antagonism volumes for pairwise combination studies performed between BI 224436 and HIV antiviral inhibitorsa
| Inhibitor class and drug | Volumeb (nM2%; ±SD) |
Combination effect | |
|---|---|---|---|
| Synergy | Antagonism | ||
| NRTI | |||
| Lamivudine | 39 ± 7.4 | −5.8 ± 3.2 | Additive |
| Abacavir | 12 ± 5.6 | −44 ± 28 | Minor antagonism |
| Zidovudine | 26 ± 26 | −14 ± 15 | Additive |
| Tenofovir | 124 ± 18 | −5.1 ± 4.1 | Synergy |
| Emtricitabine | 19 ± 5.0 | −0.36 ± 0.63 | Additive |
| Didanosine | 15 ± 19 | −4.3 ± 6.4 | Additive |
| Stavudine | 17 ± 15 | −9.0 ± 6.6 | Additive |
| NNRTI | |||
| Nevirapine | 0.2 ± 0.3 | −12 ± 13 | Additive |
| Efavirenz | 20 ± 16 | −4.7 ± 4.1 | Additive |
| Etravirine | 43 ± 70 | −12 ± 13 | Additive |
| Protease inhibitor | |||
| Amprenavir | 72 ± 51 | −12 ± 12 | Moderate synergy |
| Atazanavir | 72 ± 33 | −26 ± 23 | Moderate synergy |
| Darunavir | 25 ± 18 | −24 ± 10 | Additive |
| Indinavir | 24 ± 24 | −19 ± 17 | Additive |
| Lopinavir | 4.6 ± 8.0 | −12 ± 20 | Additive |
| Nelfinavir | 67 ± 33 | −30 ± 10 | Moderate synergy |
| Ritonavir | 42 ± 48 | −28 ± 12 | Additive |
| Saquinavir | 3.4 ± 3.0 | −16 ± 22 | Additive |
| Tipranavir | 37 ± 34 | −17 ± 3.9 | Additive |
| Integrase inhibitor | |||
| Raltegravir | 11 ± 12 | −5.9 ± 7.9 | Additive |
| Dolutegravir | 23 ± 44 | −12 ± 14 | Additive |
| Entry inhibitors | |||
| Maraviroc | 19 ± 11 | −3.2 ± 4.9 | Additive |
| Enfuvirtide | 4.5 ± 1.4 | −5.9 ± 6.3 | Additive |
Results are mean synergy and antagonism volumes at 95% confidence intervals, as calculated by MacSynergy II software, for pairwise combination studies performed between BI 224436 and HIV antiviral inhibitors. Mean volumes were calculated based on a minimum of three separate experiments. Cytotoxicity assays were performed in parallel at the highest drug concentrations tested, and no increase in cytotoxicity was observed in drug combinations with BI 224436.
Mean synergy volumes at 95% confidence (in nM2%) are described at the following levels: >100, strong synergy; 50 to 100, moderate synergy; 25 to 50, minor amount of synergy; 25 to −25, additive; −25 to −50, minor amount of antagonism; −50 to −100, moderate antagonism; and <−100, strong antagonism.
FIG 3.
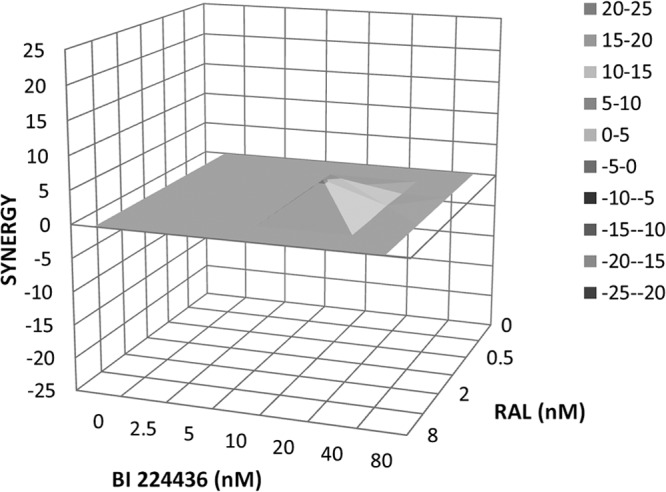
Three-dimensional plot of synergy and antagonism at 95% confidence for the pairwise combination of BI 224436 with raltegravir (RAL). Analysis was performed with MacSynergy II software and is shown as a representative plot of the data shown in Table 3. The synergy volume is 10 nM2%, and the antagonism volume is 0 nM2%.
The cellular cytotoxicity levels evaluated for all pairwise combinations of BI 2244336 and test compounds are similar to those observed with the inhibitors tested alone at the concentrations used in these experiments. Any differences in cytotoxicity observed were less than <5% (data not shown) and reflect variations in the cytotoxicity assay. Therefore, there is no evidence of synergistic cytotoxicity of agents when used in combination with BI 224436.
In vitro ADME profile of BI 224436.
BI 224436 demonstrated good solubility of the hydrochloride crystalline form (>780 μg/ml or >1.76 mM in aqueous buffers at pH 2.0, 4.5, and 6.8), as shown in Table 4. The Caco-2 A-to-B permeability was 14 × 10−6 cm/s with a B-to-A rate of 3.9 × 10−6 cm/s, consistent with a high fraction of compound absorbed, as demonstrated in preclinical species. BI 224436 demonstrated little or no activity when tested as an inhibitor of the 5 major CYP isozymes tested. The IC50s against the most important isozymes, 3A4, the most abundant isozyme, and 2D6, a polymorphic isozyme responsible for a large individual variation in the metabolism of several marketed drugs, were 23 and >30 μM, respectively. The lowest IC50, against the 2C9 isozyme, was 20 μM. In vitro liver microsome and hepatocyte stability studies showed low metabolic clearance of <14% of hepatic blood flow (QH) in most species tested, with the exception of the dog, which had a hepatic clearance of 32% QH. Clearance by phase II metabolism in liver microsomes is slower than phase I metabolism in all species tested. BI 224436 was highly protein bound in mouse and rat plasma (>97%), and between 75.5 to 84.3% protein was bound in human, dog, and monkey plasma samples, as determined by equilibrium dialysis.
TABLE 4.
In vitro ADME profile of BI 224436
| Assay or parameter | Value(s) for BI 224436 |
|---|---|
| Solubility (μg/ml) at pH 2, 4.5, 6.8 (crystalline) | 840, >780, >1,000 |
| Caco-2 cell permeability, A to B and B to A (10−6 cm/s) | 13 and 3.9 |
| P450 IC50 (μM) for 2C9, 3A4BFC, 1A2, 2C19, 2D6 | 20, 23, >30, >30, >30 |
| PK parameter (H, M, R, Mnk, D)a | |
| Phase I LM CL (%QH) | 14, 6.4, 7, 11, 16 |
| Hepatic CL (%QH) | 13, 12, 9, 13, 32 |
| Phase II LM CL (%QH) | <11, <6, <6, <7, 20 |
| Plasma protein binding (%) | 84.3, 97.3, 98.2, 78.0, 75.5 |
H, M, R, Mnk, and D correspond to human, mouse, rat, monkey, and dog, respectively.
Pharmacokinetics in animals.
The clearance of BI 224436 was very low in the mouse (0.7 ml/min/kg) and rat (0.5 ml/min/kg), low in the dog (2.5 ml/min/kg), and appreciably higher in the monkey (11 ml/min/kg) (see Fig. 5). The volume of distribution was low and somewhat variable across the species tested (0.2 to 0.88 liters/kg). The terminal half-life was moderate in the mouse (2.6 h), rat (8.9 h), and dog (6.0 h) but short in the monkey (1.4 h). Following the oral dose of 2 mg/kg, the maximum concentration of drug in the serum (Cmax) was 4.8 to 15 μM in the four species, and area-under-the-curve (AUC) values were highest in mouse and rat (99 and 75 μM · h, respectively), intermediate in dog (24 μM · h), and lowest of the four species in monkey (4.8 μM · h). Oral bioavailability (F) of BI 224436 ranged from 54 to 100% in the animal species tested.
DISCUSSION
BI 224436 is an effective inhibitor of HIV-1 viral replication through the inhibition of integrase function. A number of biochemical and cellular studies were carried out to verify that the observed cell-based activity of BI 224436 was due to its action on integrase. In contrast to active-site integrase inhibitors, such as raltegravir and elvitegravir, BI 224436 does not inhibit the strand transfer activity of integrase. However, BI 224436 inhibits both the LTR DNA 3′-processing reaction and integrase-LEDGF interaction at levels that are equivalent to the activity observed in the cell-based antiviral assay. BI 224436 exerts this inhibition by binding to a highly conserved allosteric pocket on the catalytic core of integrase, which also serves as a binding pocket for LEDGF. The binding of BI 224436 to the integrase LEDGF binding pocket has been confirmed by crystallography studies with the integrase core domain (data not shown). Similar findings have been reported for this chemical series by our group (18), researchers at Gilead Sciences (45, 50), and for an analogous chemical series referred to as LEDGINs (23).
In HIV-1 viral replication assays using human peripheral blood mononuclear cells (PBMCs), BI 224436 has antiviral EC50s ranging from 7.2 to 15 nM when tested against several laboratory strains of HIV-1, including CXCR4 (HXB2 and NL4.3)- and CCR5-tropic (BaL) viruses (51). BI 224436 also exhibited low cellular cytotoxicity, leading to an in vitro selectivity index of >8,000-fold in PBMCs. The assessment of antiviral potency was determined in medium containing 10% FBS in the absence of any human serum proteins. Since compound binding to plasma proteins in human serum can have a pronounced impact on the free drug available to inhibit viral replication, the antiviral activity of BI 224436 was determined in experiments in which the cell culture medium was supplemented with up to 50% human serum. BI 224436 exhibits a low fold decrease in antiviral potency in the presence of human serum. The antiviral potency of BI 224436 in human blood was estimated at an EC50 of 32 nM and an EC95 of 75 nM versus the least-susceptible laboratory strain of HIV-1 tested.
The steep dose-response curve slope observed for BI 224436 and other NCINIs (not shown) results in EC95 values being only ∼2.4-fold higher than the EC50s. A Hill slope of ∼4 was observed in all antiviral assays tested, including a single-cycle infection assay (data not shown). The importance of a steep inhibitor Hill slope is that at clinically relevant compound concentrations, these inhibitors can have a more pronounced effect on viral suppression. This has been evaluated in terms of the instantaneous inhibition potential (IIP) for HIV-1 antiviral agents. Analysis of randomized, controlled trials suggests a more favorable clinical outcome with a treatment regimen which included antiviral agents with sustained high IIPs (52). Based on IPP potential calculations {IIPCmin = log [1 + (Cmin/ssEC50)]slope} (where Cmin is the minimum concentration of drug during the dosing regimen and ssEC50 is the serum-shifted EC50 of the drug in the presence of human serum) at an inhibitory quotient (IQ) of 15 to 30 (where IQ = Cmin/ssEC50), BI 224436 would have an IIP of ∼5 to 6, comparable to that of HIV protease inhibitors that provide the highest IIP values of all HIV antiviral classes (52). Although IIP is not the sole parameter to predict the clinical efficacy of an HIV-1 antiviral, a Hill slope value of 4 can be viewed as a positive feature for BI 224436 and the NCINI series of inhibitors.
The primary viral variants that were selected by in vitro passage experiments with BI 224436 all map to the vicinity of an allosteric pocket on the catalytic core of integrase. Residue L102 is situated at the bottom of this pocket, while A128 is on the edge of the LEDGF binding site. The integrase A128T substitution emerged first at early viral passages as a result of a single-nucleotide change. This substitution conferred a modest 2.9-fold reduced susceptibility to BI 224436 and has a 30% replication capacity relative to that of the wild-type virus. The integrase L102F substitution was identified at later passages and is also the result of a single-nucleotide substitution. Although this substitution confers a significant 61-fold reduced susceptibility, a reduced replication capacity of <1% relative to the wild-type virus may explain the emergence of this variant at later passages. The A128N substitution requires two nucleotide mutations from the wild-type virus, confers a significant 64-fold reduced susceptibility to BI 224436, and has a moderately low replication capacity of 6% relative to the wild-type virus. The N222K substitution was identified only at later passages in combination with A128N and L102F substitutions. Since this substitution is located outside the allosteric pocket targeted by BI 224436 and confers a low 2.8-fold decrease in susceptibility, it is possible that N222K represents a compensatory mutation that confers an improved replication capacity on viruses encoding the A128N or L102F substitution in integrase. In keeping with these results, other groups have reported similar mutations that map to the same pocket with structural analogues of the NCINI series (44–46).
Biochemical LTR DNA 3′-processing assays performed with recombinant integrase encoding the A128T or L102F amino acid substitutions provides additional evidence that BI 224436 exerts its antiviral activity through targeting the allosteric pocket on the integrase catalytic core. The fold decrease in IC50 for the A128T substitution integrase is similar in both the LTR DNA 3′-processing and antiviral assays (2.9-fold in each). However, the L102F substitution in integrase confers a >500-fold reduced susceptibility to BI 224436 in the LTR DNA 3′-processing assay compared to a 61-fold shift in the viral replication assay. This discrepancy may be explained by the significantly reduced replication capacity of the virus encoding the L102F integrase substitution (<1% relative to wild-type virus), which would diminish the benefit associated with the reduced susceptibility to BI 224436.
Our studies show that BI 224436 remains potent against recombinant viruses that encode substitutions conferring resistance to either NNRTIs (K103N/Y181C) or integrase strand transfer inhibitors (T66I/S153Y, E92Q, G140S/Q148H, and N155S), with no shift in EC50 compared to the wild-type virus. In contrast, recombinant viruses with amino acid substitutions conferring resistance to active-site integrase inhibitors resulted in considerable cross-resistance against both raltegravir and elvitegravir. Raltegravir and elvitegravir were also found to maintain antiviral activity against recombinant viruses with reduced susceptibility to BI 224436. In combination with activity in the IN-LEDGF biochemical assay and crystallography data (18), these results are consistent with BI 224436 targeting HIV-1 integrase by a distinct mechanism of action in binding to an allosteric pocket on integrase that is distant from the active site where INSTIs bind. Taken together, these studies demonstrate that BI 224436 has a resistance profile that is distinct from and nonoverlapping with the integrase strand transfer inhibitors.
In two-drug combination studies performed using wild-type HIV-1 virus, most combinations of BI 224436 with other antiviral agents produced additive to synergistic effects. The combination of BI 224436 with integrase strand transfer inhibitors, nonnucleoside reverse transcriptase inhibitors, and entry inhibitors all exhibited additive antiviral effects in the HIV-1 viral replication assay. Combination studies performed between BI 224436 and either nucleoside reverse transcriptase inhibitors or protease inhibitors were predominantly additive, but moderate to strong synergy was observed for specific drug-drug combinations. Combination studies performed between BI 224436 and the NRTI tenofovir showed strong synergy (mean CVsynergy of 124 nM2%), while BI 224436 with the protease inhibitors amprenavir, atazanavir, and nelfinavir exhibited signs of moderate synergy (mean CVsynergy ranging from 67 to 72 nM2%). In each of these combinations, residual plots illustrating the statistically significant volumes of inhibition of HIV-1 replication show strong synergy near the EC50s of the drugs tested. Two-drug combination synergies observed between BI 224436 and either tenofovir or several of the HIV protease inhibitors may be rationalized by the observation that NCINIs exert antiviral activity at both the late and early stages of the viral replication cycle (NCINI structural analogue data are from references 42 and 43; data for BI 224436 are unpublished). NCINIs promote integrase multimerization at the stage of viral production, which results in a block in the formation of mature, infectious viral particles. This was shown by evaluating the formation of electron-dense HIV-1 cores by electron microscopy (43, 44, 53). As such, NCINIs may influence the activity of HIV protease, resulting in the more than additive antiviral inhibition observed with some protease inhibitors. Additional studies showed that viruses produced by cells in the presence of NCINIs are inhibited in their reverse transcription activity after infecting target cells. Although BI 224436 exerts antiviral activity at the same stage as the NRTI tenofovir, it is difficult to rationalize mechanistically why combination studies would be strongly synergistic with one NRTI and additive or potentially slight antagonistic with others. Further mechanistic studies will be required to investigate these differences observed in the combination of BI 224436 with NRTIs.
Two-drug combination studies showed that there were no signs of synergistic cytotoxicities up to the highest compound concentrations of antiviral agents tested when combined with BI 224436. Most importantly, moderate or strong antagonism was not detected with any combination studies performed with BI 224436, providing supporting evidence that BI 224436 could be coadministered in the clinic with all antiviral agents tested in this study.
BI 224436 has excellent solubility at the pH values from 2.0 to 6.8, covering the pH environments of both the stomach and small intestine. Experiments performed in Caco-2 cells indicate a good permeability in the A-to-B direction. Consistent with these in vitro studies, BI 224436 is well absorbed in pharmacokinetic experiments in all animal species tested, with Cmax values of >4.8 μM at the low oral dose of 2 mg/kg and oral bioavailability values ranging from 54 to 100%. In cytochrome P450 inhibition assays, BI 224436 demonstrated little activity, with IC50s of ≥20 μM with the 5 major CYP isozymes. As such, these in vitro studies indicated that BI 224436 has a low potential for major CYP-mediated drug-drug interactions.
The pharmacokinetic parameters of BI 224436 were an important part of the criteria leading to its selection for preclinical profiling. Although the PK profile of most species tested was excellent, there were some difficulties in correlating the in vitro clearance with the in vivo PK values observed in the different species (Tables 4 and 5). This discrepancy may be attributed in part to different levels of enterohepatic recirculation between species. The time-concentration profiles of BI 224436 in the rodents and dog showed a large secondary peak suggestive of enterohepatic recirculation. This takes place through acyl glucuronide modification of the carboxylic acid moiety on the inhibitor and then subsequent excretion into the bile, followed by hydrolysis and reabsorption of the parent drug. Such secondary peaks were not observed in the monkey (data not shown), and it is likely that enterohepatic recirculation contributes to the apparently low clearance in the mouse and rat and, to a lesser extent, the dog. In addition, the cross-species plasma protein binding showed a trend inversely related to in vivo clearance and volume of distribution at steady state (Vss). These cross-species differences in enterohepatic recirculation and plasma protein binding result in a degree of uncertainty with respect to the human PK and dose predictions (54). However, based on the overall favorable preclinical antiviral, in vitro ADME, and pharmacokinetic properties and animal toxicology studies (data not shown), BI 224436 was advanced into phase 1 clinical development.
TABLE 5.
PK profile of BI 224436 after 1-mg/kg i.v. and 2-mg/kg oral doses
| Test species | Protein binding (%) | CL (ml/min/kg) (% QH) | Vss (liter/kg) | t1/2 (h) | Cmax (μM) | AUC (μM · h) | F (%) |
|---|---|---|---|---|---|---|---|
| Mouse | 97.3 | 0.7 (0.8) | 0.20 | 2.6 | 15a | 99a | 100 |
| Rat | 98.2 | 0.5 (0.7) | 0.45 | 8.8 | 13a | 75a | 54 |
| Monkey | 78.0 | 11 (23) | 0.54 | 1.4 | 4.8 | 6.3 | 82 |
| Dog | 75.5 | 2.5 (8) | 0.88 | 5.9 | 12 | 24 | 81 |
A mouse and rat oral dose of 0.4 mg/kg was dose normalized to 2 mg/kg to allow for an appropriate comparison to monkey and dog PK studies. The oral formulation contained 1% MP, 0.3% Tween 80, 0.5% MC; the i.v. formulation contained 70% PEG, 30% water.
Although there are multiple drugs that have been developed for the treatment of HIV-1-infected patients, a major obstacle to long-term control of viral replication is the ability of the virus to develop resistance to current antiviral treatments. Over the years, antiretroviral therapy has improved dramatically as new antiretrovirals have come to market. However, with the extended life span of infected patients comes the increased likelihood of a need to change medications due to the development of viral resistance, adverse events of antiviral drugs in some patients, drug-specific toxicities, and potential drug-drug interactions with the additional medications taken by an aging population (55). Development of inhibitors that operate by a novel mechanism of action could expand the options for clinicians to address these unmet medical needs for the HIV-1-infected patient population. BI 224436 is an effective antiviral agent that belongs to the non-catalytic-site integrase inhibitor class. Our preclinical studies provide evidence that BI 224436 retains antiviral activity against viral isolates with resistance to integrase strand transfer inhibitors and has an additive antiviral profile in in vitro two-drug combination studies. As such, it is expected that BI 224436 could be administered before, after, or in combination with INSTIs such as raltegravir or elvitegravir in antiviral therapy. In addition, with the excellent solubility and absorption properties of BI 224436, this compound is a good candidate for further development as a component of a combination antiviral regimen for use in first-line and subsequent lines of antiviral therapy.
Supplementary Material
ACKNOWLEDGMENTS
We thank Christine Martens and Patrick Salois for their work with the integrase 3′-processing screening assay, Paul Whitehead for protein purification, Kevork Mekhssian and Céline Plouffe for integrase profiling assays, and Mary Fyfe, Sophie Desmeules, and Elizabeth Wardrop for performing supportive antiviral studies for this project.
Footnotes
Published ahead of print 24 March 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02719-13.
REFERENCES
- 1.Asante-Appiah E, Skalka AM. 1999. HIV-1 integrase: structural organization, conformational changes, and catalysis. Adv. Virus Res. 52:351–369. 10.1016/S0065-3527(08)60306-1 [DOI] [PubMed] [Google Scholar]
- 2.Bushman FD, Craigie R. 1991. Activities of human immunodeficiency virus (HIV) integration protein in vitro: specific cleavage and integration of HIV DNA. Proc. Natl. Acad. Sci. U. S. A. 88:1339–1343. 10.1073/pnas.88.4.1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maertens G, Cherepanov P, Pluymers W, Busschots K, De Clercq E, Debyser Z, Engelborghs Y. 2003. LEDGF/p75 is essential for nuclear and chromosomal targeting of HIV-1 integrase in human cells. J. Biol. Chem. 278:33528–33539. 10.1074/jbc.M303594200 [DOI] [PubMed] [Google Scholar]
- 4.Llano M, Vanegas M, Fregoso O, Saenz D, Chung S, Peretz M, Poeschla EM. 2004. LEDGF/p75 determines cellular trafficking of diverse lentiviral but not murine oncoretroviral integrase proteins and is a component of functional lentiviral preintegration complexes. J. Virol. 78:9524–9537. 10.1128/JVI.78.17.9524-9537.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Llano M, Saenz DT, Meehan A, Wongthida P, Peretz M, Walker WH, Teo W, Poeschla EM. 2006. An essential role for LEDGF/p75 in HIV integration. Science 314:461–464. 10.1126/science.1132319 [DOI] [PubMed] [Google Scholar]
- 6.Hombrouck A, De Rijck J, Hendrix J, Vandekerckhove L, Voet A, De Maeyer M, Witvrouw M, Engelborghs Y, Christ F, Gijsbers R, Debyser Z. 2007. Virus evolution reveals an exclusive role for LEDGF/p75 in chromosomal tethering of HIV. PLoS Pathog. 3:e47. 10.1371/journal.ppat.0030047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Poeschle E. 2008. Integrase, LEDGF/p75 and HIV replication. Cell. Mol. Life Sci. 65:1403–1424. 10.1007/s00018-008-7540-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hazuda DJ, Felock P, Witmer M, Wolfe A, Stillmock K, Grobler JA, Espeseth A, Gabryelski L, Schleif W, Blau C, Miller MD. 2000. Inhibitors of strand transfer that prevent integration and inhibit HIV-1 replication in cells. Science 287:646–650. 10.1126/science.287.5453.646 [DOI] [PubMed] [Google Scholar]
- 9.Métifiot M, Maddali K, Naumova A, Zhang X, Marchand C, Pommier Y. 2010. Biochemical and pharmacological analyses of HIV-1 integrase flexible loop mutants resistant to raltegravir. Biochemistry 49:3715–3722. 10.1021/bi100130f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Espeseth AS, Felock P, Wolfe A, Witmer M, Grobler J, Anthony N, Egbertson M, Melamed JY, Young S, Hamill T, Cole JL, Hazuda DJ. 2000. HIV-1 integrase inhibitors that compete with the target DNA substrate define a unique strand transfer conformation for integrase. Proc. Natl. Acad. Sci. U. S. A. 97:11244–11249. 10.1073/pnas.200139397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grobler JA, Stillmock K, Hu B, Witmer M, Felock P, Espeseth AS, Wolfe A, Egbertson M, Bourgeois M, Melamed J, Wai JS, Young S, Vacca J, Hazuda DJ. 2002. Diketo acid inhibitor mechanism and HIV-1 integrase: implications for metal binding in the active site of phosphotransferase enzymes. Proc. Natl. Acad. Sci. U. S. A. 99:6661–6666. 10.1073/pnas.092056199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hare S, Gupta SS, Valkov E, Engelman A, Cherepanov P. 2010. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature 464:232–236. 10.1038/nature08784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goethals O, Clayton R, Van Ginderen M, Vereycken I, Wagemans E, Geluykens P, Dockx K, Strijbos R, Smits V, Vos A, Meersseman G, Jochmans D, Vermeire K, Schols D, Hallenberger S, Hertogs K. 2008. Resistance mutations in human immunodeficiency virus type 1 integrase selected with elvitegravir confer reduced susceptibility to a wide range of integrase inhibitors. J. Virol. 82:10366–10374. 10.1128/JVI.00470-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marinello J, Marchand C, Mott BT, Bain A, Thomas CJ, Pommier Y. 2008. Comparison of raltegravir and elvitegravir on HIV-1 integrase catalytic reactions and on a series of drug-resistant integrase mutants. Biochemistry 47:9345–9354. 10.1021/bi800791q [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shimura K, Kodama EN. 2009. Elvitegravir: a new HIV integrase inhibitor. Antivir. Chem. Chemother. 20:79–85. 10.3851/IMP1397 [DOI] [PubMed] [Google Scholar]
- 16.Zolopa AR, Berger DS, Lampiris H, Zhong L, Chuck SL, Enejosa JV, Kearney BP, Cheng AK. 2010. Activity of elvitegravir, a once-daily integrase inhibitor, against resistant HIV type 1: results of a phase 2, randomized, controlled, dose-ranging clinical trial. J. Infect. Dis. 201:814–822. 10.1086/650698 [DOI] [PubMed] [Google Scholar]
- 17.Mascolinli M, Kort R. 2010. Fifth International AIDS Society Conference on HIV Pathogenesis, Treatment and Prevention: summary of key research and implications for policy and practice–clinical sciences. J. Int. AIDS Soc. 13(Suppl 2):S3. 10.1186/1758-2652-13-S2-S3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fader LD, Malenfant E, Parisien M, Carson R, Bilodeau F, Landry S, Pesant M, Brochu C, Morin S, Chabot C, Halmos T, Bousquet Y, Bailey MD, Kawai SH, Coulombe R, LaPlante S, Jakalian A, Bhardwaj PK, Wernic D, Schroeder P, Amad M, Edwards P, Garneau M, Duan J, Cordingley M, Bethell R, Mason SW, Bös M, Bonneau P, Poupart M-A, Faucher A-M, Simoneau B, Fenwick C, Yoakim C, Tsantrizos Y. 2014. Discovery of BI 224436, a noncatalytic site integrase inhibitor (NCINI) of HIV-1 ACS Med. Chem. Lett. 5:422–427. 10.1021/ml500002n [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsantrizos YS, Boes M, Brochu C, Fenwick C, Malenfant E, Mason S, Pesant M, Boehringer Ingelheim Pharma November 2007. Inhibitors of human immunodeficiency virus replication. Patent WO/2007/131350
- 20.Cherepanov P, Sun ZY, Rahman S, Maertens G, Wagner G, Engelman A. 2005. Solution structure of the HIV-1 integrase-binding domain in LEDGF/p75. Nat. Struct. Mol. Biol. 12:526–532. 10.1038/nsmb937 [DOI] [PubMed] [Google Scholar]
- 21.Hou Y, McGuinness DE, Prongay AJ, Feld B, Ingravallo P, Ogert RA, Lunn CA, Howe JA. 2008. Screening for antiviral inhibitors of the HIV integrase-LEDGF/p75 interaction using the AlphaScreen luminescent proximity assay. J. Biomol. Screen. 13:406–414. 10.1177/1087057108317060 [DOI] [PubMed] [Google Scholar]
- 22.Christ F, Voet A, Marchand A, Nicolet S, Desimmie BA, Marchand D, Bardiot D, Van der Veken NJ, Van Remoortel B, Strelkov SV, De Maeyer M, Chaltin P, Debyser Z. 2010. Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nat. Chem. Biol. 6:442–448. 10.1038/nchembio.370 [DOI] [PubMed] [Google Scholar]
- 23.De Luca L, Barreca ML, Ferro S, Christ F, Iraci N, Gitto R, Monforte AM, Debyser Z, Chimirri A. 2009. Pharmacophore-based discovery of small-molecule inhibitors of protein-protein interactions between HIV-1 integrase and cellular cofactor LEDGF/p75. Chem. Med. Chem. 4:1311–1316. 10.1002/cmdc.200900070 [DOI] [PubMed] [Google Scholar]
- 24.Tsantrizos YS, Bailey MD, Bilodeau F, Carson RJ, Coulombe R, Fader L, Halmos T, Kawai S, Landry S, Laplante S, Morin S, Parisien M, Poupart M-A, Simoneau B. 2009. Inhibitors of human immunodeficiency virus replication. Patent application WO 2009/062285
- 25.Taganov KD, Cuesta I, Daniel R, Cirillo LA, Katz RA, Zaret KS, Skalka AM. 2004. Integrase-specific enhancement and suppression of retroviral DNA integration by compacted chromatin structure in vitro. J. Virol. 78:5848–5855. 10.1128/JVI.78.11.5848-5855.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hawkins ME, Pfleiderer W, Mazumder A, Pommier YG, Balis FM. 1995. Incorporation of a fluorescent guanosine analog into oligonucleotides and its application to a real time assay for the HIV-1 integrase 3′-processing reaction. Nucleic Acids Res. 23:2872–2880. 10.1093/nar/23.15.2872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou G, Cummings R, Hermes J, Moller DE. 2001. Use of homogeneous time-resolved fluorescence energy transfer in the measurement of nuclear receptor activation. Methods 25:54–61. 10.1006/meth.2001.1215 [DOI] [PubMed] [Google Scholar]
- 28.Shaw GM, Hahn BH, Arya SK, Groopman JE, Gallo RC, Wong-Staal F. 1984. Molecular characterization of human T-cell leukemia (lymphotrophic) virus type III in the acquired immune deficiency syndrome. Science 226:1165–1170. 10.1126/science.6095449 [DOI] [PubMed] [Google Scholar]
- 29.Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 59:284–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson VA, Byington RE. 1990. Quantitative assays for virus infectivity: infectivity assay (virus yield assay), p 71–76 In Aldovini A, Walker BD. (ed), Techniques in HIV research. Stockton Press, New York, NY [Google Scholar]
- 31.Schmidt NJ, Emmons RW. 1989. General principles of laboratory diagnostic methods for viral, rickettsial and chlamydial infections, p 1–35 In Schmidt NJ, Emmons RW. (ed), Diagnostic procedures for rickettsial and chlamydial infections, 6th ed. American Public Health Association, Washington, DC [Google Scholar]
- 32.Mosmann T. 1983. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 65:55–63. 10.1016/0022-1759(83)90303-4 [DOI] [PubMed] [Google Scholar]
- 33.Lemke CT, Titolo S, von Schwedler U, Goudreau N, Mercier JF, Wardrop E, Faucher AM, Coulombe R, Banik SS, Fader L, Gagnon A, Kawai SH, Rancourt J, Tremblay M, Yoakim C, Simoneau B, Archambault J, Sundquist WI, Mason SW. 2012. Distinct effects of two HIV-1 capsid assembly inhibitor families that bind the same site within the N-terminal domain of the viral CA protein. J. Virol. 86:6643–6655. 10.1128/JVI.00493-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jegede O, Babu J, di Santo R, McColl DJ, Weber J, Quiñones-Mateu ME. 2008. HIV type 1 integrase inhibitors: from basic research to clinical implications. AIDS Rev. 10:172–189 [PubMed] [Google Scholar]
- 35.O'Meara JA, Yoakim C, Bonneau PR, Bös M, Cordingley MG, Déziel R, Doyon L, Duan J, Garneau M, Guse I, Landry S, Malenfant E, Naud J, Ogilvie WW, Thavonekham B, Simoneau B. 2005. Novel 8-substituted dipyridodiazepinone inhibitors with a broad spectrum of activity against HIV-1 strains resistant to non-nucleoside reverse transcriptase inhibitors. J. Med. Chem. 48:5580–5588. 10.1021/jm050255t [DOI] [PubMed] [Google Scholar]
- 36.Doyon LD, Croteau G, Thibeault D, Poulin F, Pilote L, Lamarre D. 1996. Second locus involved in human immunodeficiency virus type 1 resistance to protease inhibitors. J. Virol. 70:3763–3769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rajotte D, Tremblay S, Pelletier A, Salois P, Bourgon L, Coulombe R, Mason S, Lamorte L, Sturino CF, Bethell R. 2013. Identification and characterization of a novel HIV-1 nucleotide-competing reverse transcriptase inhibitor series. Antimicrob. Agents Chemother. 57:2712–2718. 10.1128/AAC.00113-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prichard MN, Shipman C. 1990. A three-dimensional model to analyze drug-drug interactions. Antiviral Res. 14:181–206. 10.1016/0166-3542(90)90001-N [DOI] [PubMed] [Google Scholar]
- 39.Prichard MN, Prichard LE, Baguley WA, Nassiri MR, Shipman C. 1991. Three-dimensional analysis of the synergistic cytotoxicity of ganciclovir and zidovudine. Antimicrob. Agents Chemother. 35:1060–1065. 10.1128/AAC.35.6.1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.White PW, Llinàs-Brunet M, Amad M, Bethell RC, Bolger G, Cordingley MG, Duan J, Garneau M, Lagacé L, Thibeault D, Kukolj G. 2010. Preclinical characterization of BI 201335, a C-terminal carboxylic acid inhibitor of the hepatitis C virus NS3-NS4A protease. Antimicrob. Agents Chemother. 54:4611–4618. 10.1128/AAC.00787-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beaulieu PL, Bös M, Cordingley MG, Chabot C, Fazal G, Garneau M, Gillard JR, Jolicoeur E, LaPlante S, McKercher G, Poirier M, Poupart M-A, Tsantrizos YS, Duan J, Kukolj G. 2012. Discovery of the first thumb pocket 1 NS5B polymerase inhibitor (BILB 1941) with demonstrated antiviral activity in patients chronically infected with genotype 1 hepatitis C virus (HCV). J. Med. Chem. 55:7650–7666. 10.1021/jm3006788 [DOI] [PubMed] [Google Scholar]
- 42.Cotelle P. 2009. New hexahydrodiazocinonaphthyridine triones as HIV-1 integrase inhibitors: WO2008048538 A1. Expert Opin. Ther. Pat. 19:87–93. 10.1517/13543770802603577 [DOI] [PubMed] [Google Scholar]
- 43.Jurado KA, Wang H, Slaughter A, Feng L, Kessl JJ, Koh Y, Wang W, Ballandras-Colas A, Patel PA, Fuchs JR, Kvaratskhelia M, Engelman A. 2013. Allosteric integrase inhibitor potency is determined through the inhibition of HIV-1 particle maturation. Proc. Natl. Acad. Sci. U. S. A. 110:8690–8695. 10.1073/pnas.1300703110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Christ F, Shaw S, Demeulemeester J, Desimmie BA, Marchand A, Butler S, Smets W, Chaltin P, Westby M, Debyser Z, Pickford C. 2012. Small-molecule inhibitors of the LEDGF/p75 binding site of integrase block HIV replication and modulate integrase multimerization. Antimicrob. Agents Chemother. 56:4365–4374. 10.1128/AAC.00717-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsiang M, Jones GS, Niedziela-Majka A, Kan E, Lansdon EB, Huang W, Hung M, Samuel D, Novikov N, Xu Y, Mitchell M, Guo H, Babaoglu K, Liu X, Geleziunas R, Sakowicz R. 2012. New class of HIV-1 integrase (IN) inhibitors with a dual mode of action. J. Biol. Chem. 287:21189–21203. 10.1074/jbc.M112.347534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kessl JJ, Jena N, Koh Y, Taskent-Sezgin H, Slaughter A, Feng L, de Silva S, Wu L, Le Grice SF, Engelman A, Fuchs JR, Kvaratskhelia M. 2012. Multimode, cooperative mechanism of action of allosteric HIV-1 integrase inhibitors. J. Biol. Chem. 287:16801–16811. 10.1074/jbc.M112.354373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Engelman A, Kessl JJ, Kvaratskhelia M. 2013. Allosteric inhibition of HIV-1 integrase activity. Curr. Opin. Chem. Biol. 17:339–345. 10.1016/j.cbpa.2013.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Molla A, Vasavanonda S, Kumar G, Sham HL, Johnson M, Grabowski B, Denissen JF, Kohlbrenner W, Plattner JJ, Leonard JM, Norbeck DW, Kempf DJ. 1998. Human serum attenuates the activity of protease inhibitors toward wild-type and mutant human immunodeficiency virus. Virology 250:255–262. 10.1006/viro.1998.9383 [DOI] [PubMed] [Google Scholar]
- 49.Dudley MN, Blaser J, Gilbert D, Zinner SH. 1990. Significance of extravascular protein binding for antimicrobial pharmacodynamics in an in vitro capillary model of infection. Antimicrob. Agents Chemother. 34:98–101. 10.1128/AAC.34.1.98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Balakrishnan M, Yant SR, Tsai L, O'Sullivan C, Bam RA, Tsai A, Niedziela-Majka A, Stray KM, Sakowicz R, Cihlar T. 2013. Non-catalytic site HIV-1 integrase inhibitors disrupt core maturation and induce a reverse transcription block in target cells. PLoS One 8:e74163. 10.1371/journal.pone.0074163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bieniasz PD, Fridell RA, Aramori I, Ferguson SS, Caron MG, Cullen BR. 1997. HIV-1-induced cell fusion is mediated by multiple regions within both the viral envelope and the CCR-5 co-receptor. EMBO J. 16:2599–2609. 10.1093/emboj/16.10.2599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shen L, Peterson S, Sedaghat AR, McMahon MA, Callender M, Zhang H, Zhou Y, Pitt E, Anderson KS, Acosta EP, Siliciano RF. 2008. Dose-response curve slope sets class-specific limits on inhibitory potential of anti-HIV drugs. Nat. Med. 14:762–766. 10.1038/nm1777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Desimmie BA, Schrijvers R, Demeulemeester J, Borrenberghs D, Weydert C, Thys W, Vets S, Van Remoortel B, Hofkens J, De Rijck J, Hendrix J, Bannert N, Gijsbers R, Christ F, Debyser Z. 2013. LEDGINs inhibit late stage HIV-1 replication by modulating integrase multimerization in the virions. Retrovirology 10:57. 10.1186/1742-4690-10-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Trainor GL. 2007. The importance of plasma protein binding in drug discovery. Expert Opin. Drug Discov. 2:51–64. 10.1517/17460441.2.1.51 [DOI] [PubMed] [Google Scholar]
- 55.Moyle G, Gatell J, Perno CF, Ratanasuwan W, Schechter M, Tsoukas C. 2008. Potential for new antiretrovirals to address unmet needs in the management of HIV-1 infection. AIDS Patient Care STDS 22:459–471. 10.1089/apc.2007.0136 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.