

Abstract
Infective endocarditis (IE) caused by methicillin-resistant Staphylococcus aureus (MRSA) with reduced susceptibility to vancomycin and daptomycin has few adequate therapeutic options. Ceftaroline (CPT) is bactericidal against daptomycin (DAP)-nonsusceptible (DNS) and vancomycin-intermediate MRSA, but supporting data are limited for IE. This study evaluated the activities of ceftaroline, vancomycin, daptomycin, and the combination of ceftaroline plus daptomycin against DNS MRSA in a pharmacokinetic/pharmacodynamic (PK/PD) model of simulated endocardial vegetations (SEVs). Simulations of ceftaroline-fosamil (600 mg) every 8 h (q8h) (maximum concentration of drug in serum [Cmax], 21.3 mg/liter; half-life [t1/2], 2.66 h), daptomycin (10 mg/kg of body weight/day) (Cmax, 129.7 mg/liter; t1/2, 8 h), vancomycin (1 g) q8h (minimum concentration of drug in serum [Cmin], 20 mg/liter; t1/2, 5 h), and ceftaroline plus daptomycin were evaluated against 3 clinical DNS, vancomycin-intermediate MRSA in a two-compartment, in vitro, PK/PD SEV model over 96 h with a starting inoculum of ∼8 log10 CFU/g. Bactericidal activity was defined as a ≥3-log10 CFU/g reduction from the starting inoculum. Therapeutic enhancement of combinations was defined as ≥2-log10 CFU/g reduction over the most active agent alone. MIC values for daptomycin, vancomycin, and ceftaroline were 4 mg/liter, 4 to 8 mg/liter, and 0.5 to 1 mg/liter, respectively, for all strains. At simulated exposures, vancomycin was bacteriostatic, but daptomycin and ceftaroline were bactericidal. By 96 h, ceftaroline monotherapy offered significantly improved killing compared to other agents against one strain. The combination of DAP plus CPT demonstrated therapeutic enhancement, resulting in significantly improved killing versus either agent alone against 2/3 (67%) strains. CPT demonstrated bactericidal activity against DNS, vancomycin-intermediate MRSA at high bacterial densities. Ceftaroline plus daptomycin may offer more rapid and sustained activity against some MRSA in the setting of high-inoculum infections like IE and should also be considered.
INTRODUCTION
Daptomycin (DAP) has been the main alternative to vancomycin (VAN) for the management of serious infections, such as infective endocarditis (IE), caused by methicillin-resistant Staphylococcus aureus (MRSA) with reduced susceptibility to VAN, such as VAN-intermediate S. aureus (VISA) and heterogeneous VISA (hVISA) (1). Unfortunately, as the MIC for VAN increases, the DAP MIC may also increase (2–4). DAP-nonsusceptible (DNS) MRSA, defined as a DAP MIC of >1 mg/liter, have now been reported (5–9). While DNS MRSA infections remain relatively uncommon, the alternatives available are limited by bacteriostatic activity, inadequate pharmacokinetics, and/or toxicity (10).
Ceftaroline (CPT), the active metabolite of the prodrug CPT-fosamil (CPT-F), demonstrates in vitro bactericidal activity against MRSA, including hVISA, VISA, and DNS strains (11–14). Furthermore, some in vitro data suggest that CPT is more active against MRSA with reduced susceptibility to lipo- and glycopeptides than fully susceptible strains due to the phenomenon referred to as the “seesaw” effect (14–16). CPT-F represents a favorable alternative for the management of serious infections with VAN- and DAP-nonsusceptible MRSA not only because of its bactericidal activity against these strains but also because of its excellent safety profile (17). However, because the use of CPT-F for the management of infections such as infective endocarditis and osteomyelitis is not approved by the FDA, the data supporting the use of this drug in these settings comes from in vivo rabbit models and 2 recently published case series (18–21). While the observations of these studies support the notion that CPT-F is a viable treatment for patients with infective endocarditis, additional studies are still needed to further describe the role of CPT-F in this setting. Currently, the MRSA guidelines endorsed by the Infectious Disease Society of America recommend the addition of another agent to high-dose DAP (10 mg/kg of body weight/day), including a beta-lactam, for the treatment of persistent bacteremia (1). The addition of CPT-F to high-dose DAP has been used successfully for the treatment of infective endocarditis after the emergence of DAP nonsusceptibility and in vitro data, including pharmacokinetic/pharmacodynamic (PK/PD) models have demonstrated therapeutic enhancement of this combination (13, 22). The studies that have been conducted so far, however, have not evaluated CPT or the combination of CPT with DAP in the simulated endocardial vegetation (SEV) model which simulates barriers such as high bacterial inocula and tissue penetration into large vegetations that would be found in humans.
The objective of the present study was to evaluate the activity of CPT alone or in combination with DAP against 3 strains of DNS MRSA with reduced susceptibility to VAN in an in vitro model of simulated endocardial vegetations.
MATERIALS AND METHODS
Bacterial strains.
Three clinical strains of DNS MRSA with VAN MICs of ≥4 mg/liter were evaluated (R6386, R6913, and R5995).
Antimicrobial agents.
DAP and VAN were purchased commercially from Cubist Pharmaceuticals (Lexington, MA) and Sigma Chemical Co. (St. Louis, MO), respectively. Analytical-grade CPT powder was provided by Forest Laboratories, Inc. (New York, NY).
Media.
Due to the calcium-dependent nature of DAP, Mueller-Hinton broth (MHB) (Difco, Detroit, MI) supplemented with 50 mg/liter of calcium and 12.5 mg/liter magnesium was used for susceptibility testing, and MHB containing 75 mg/liter of calcium was used for in vitro simulated endocardial vegetation (SEV) model experiments (due to binding of calcium by albumin in SEVs). Colony counts were determined using tryptic soy agar (TSA) (Difco, Detroit, MI) plates. Brain heart infusion agar (BHIA) (Difco, Detroit, MI) plates were used for VAN and CPT resistance screening, and Mueller-Hinton agar (MHA) (Difco, Detroit, MI) supplemented with 50 mg/liter of calcium was used for DAP resistance screening.
Susceptibility testing.
MICs were determined in duplicate by broth microdilution at ∼1 × 106 CFU/ml in MHB according to the Clinical Laboratory and Standards Institute guidelines (CLSI) for each study antimicrobial (23).
SEVs.
Simulated endocardial vegetations were prepared by mixing 0.05 ml of organism suspension (final inoculum, 1 × 108 CFU/g), 0.5 ml of human cryoprecipitate antihemolytic factor (AHF) from volunteer donors (American Red Cross, Detroit, MI), and 0.025 ml of platelet suspension (platelets mixed with normal saline with 250,000 to 500,000 platelets per clot) in 1.5-ml siliconized Eppendorf tubes. Bovine thrombin (5,000 units/ml) (0.05 ml) was added to each tube after insertion of a sterile monofilament line into the mixture. The resultant simulated vegetations were then removed from the Eppendorf tubes with a sterile plastic needle and introduced into the infection model. This methodology results in SEVs consisting of approximately 3 to 3.5 g/dl of albumin and 6.8 to 7.4 g/dl of total protein.
In vitro pharmacodynamic infection model.
An in vitro infection model consisting of a 250-ml one-compartment glass apparatus with ports, where the SEVs were suspended, was utilized for all simulations. The apparatus was prefilled with medium, and antibiotics were administered as boluses over a 96-h period into the central compartment via an injection port. The model apparatus was placed in a 37°C incubator throughout the procedure, and a magnetic stir bar was placed in the medium for thorough mixing of the drug in the model. Fresh medium was continuously supplied and removed from the compartment along with the drug via a peristaltic pump (Masterflex; Cole-Parmer Instrument Company, Chicago, IL) set to simulate the half-lives (t1/2s) of the antibiotics. Supplemental DAP was added at an appropriate rate to CPT combination models to compensate for the higher flow rate required to simulate CPT clearance (24). A total of 4 simulated regimens were evaluated on each isolate. Total drug concentrations were utilized due to the protein content in the SEVs. These regimens were CPT-F simulations of 600 mg every 8 h (q8h) (peak concentration, 21.3 mg/liter; average t1/2, 2.66 h) for 4 days, DAP simulations of 10 mg/kg every 24 h (q24h) (peak concentration, 129.7 mg/liter; average t1/2, 8 h) (25) for 4 days, VAN simulations of 1 g q8h (trough concentration, 20 mg/liter; average t1/2, 5 h) for 4 days, CPT-F simulations of 600 mg q8h plus DAP (10 mg/kg) q24h for 4 days, and a drug-free growth control for each isolate for 4 days.
Pharmacodynamic analysis.
Two SEVs were removed from each infection model (total of 36) at 0, 4, 8, 24, 32, 48, 56, 72, and 96 h. The SEVs were homogenized, diluted in cold saline, and plated onto TSA plates. The plates were incubated at 37°C for 24 h at which time colony counts were performed. The total reduction in log10 CFU/g over 96 h was determined by plotting time-kill curves based on the number of remaining organisms over the time period. Bactericidal (99.9% kill) and bacteriostatic activity were defined as reductions in colony count from the initial inocula of ≥3-log10 CFU/g and <3-log10CFU/g, respectively. Inactivity was defined as no observed reductions in initial inocula. Therapeutic enhancement of activity was defined as an increase in kill of ≥2-log10 CFU/g by a combination of antimicrobials versus the most active single agent of that combination. Combinations that resulted in a ≥1-log10 bacterial growth in comparison to the least-active single agent were considered antagonistic.
Pharmacokinetic analysis.
Pharmacokinetic samples were obtained through the injection port of each infection model at 0, 1, 2, 4, 8, 24, 32, 48, 56, 72, and 96 h for verification of target antibiotic concentrations. All samples were stored at −80°C until ready for analysis. CPT concentrations were determined by bioassay using Bacillus subtilis ATCC 6633. Blank 1/4-in. disks were spotted with 10 μl of standard concentrations or samples. Each standard was tested in duplicate by placing the disk on agar plates (antibiotic medium 11) inoculated with a 0.5 McFarland suspension of the test organism. This assay demonstrated an intraday coefficient of variance of less than 4.7% for high, medium, and low broth standards. The plates were incubated for 24 h at 37°C at which time the zone sizes were measured using a ProtoCOL plate reader (Microbiology International, Frederick, MD). DAP concentrations were determined using a validated high-performance liquid chromatography (HPLC) assay that conforms to the guidelines set forth by the College of American Pathologists and demonstrated an intraday coefficient of variance of less than 2% for high, medium, and low standards. Concentrations of VAN were determined using a fluorescence polarization immunoassay (TDx assay; Abbott Diagnostics). The VAN assay has a lower limit of detection of 2.0 mg/liter, with an interday coefficient of variance of less than 12% for low, medium, and high standards. The half-lives, peak concentrations, and area under the curve (AUC) (by trapezoidal method), or time spent above the MIC (T>MIC), were determined as appropriate for all antimicrobials utilizing PK Analyst software (version 1.10; MicroMath Scientific Software, Salt Lake City, UT).
Changes in susceptibility.
Development of resistance was evaluated at 96 h. Samples of 100 μl from each time point were plated on BHIA or calcium-supplemented MHA containing 3-fold the MIC of the respective antibiotic to assess for increases in MIC. The plates were examined for growth after 48 h of incubation at 37°C. Broth microdilution MICs following CLSI guidelines were performed on any isolate observed to grow on drug-containing agar plates used for resistance screening. If resistance was detected by 96 h, earlier time points were then screened to detect the first occurrence of MIC elevation.
Statistical analysis.
Changes in CFU/g at 24, 48, 72, and 96 h (days 1 to 4) were compared by one-way analysis of variance with Tukey's posthoc test. A P value of ≤0.05 was considered significant. All statistical analyses were performed using SPSS statistical software (release 21.0; SPSS, Inc., Chicago, IL).
RESULTS
Susceptibility testing.
MIC values for DNS VISA strains R6913, R6386, and R5995 were 4, 8, and 4 mg/liter to VAN, 4, 4, and 4 mg/liter to DAP, and 0.5, 1, and 0.5 mg/liter to CPT, respectively.
In vitro PK/PD model.
Pharmacodynamic responses to simulated antimicrobial regimens for each strain are summarized in Fig. 1A to C. VAN was bacteriostatic against all strains, but DAP and CPT were bactericidal at the simulated exposures. By 96 h, DAP monotherapy resulted in significantly improved killing compared to VAN against strain R6913 but was similar to that of CPT (P = 0.38); however, the combination of CPT plus DAP resulted in significantly improved killing compared to the effect of DAP against this strain (P = 0.037), although it was statistically similar to that of CPT. The combination of DAP plus CPT demonstrated therapeutic enhancement, resulting in significantly improved killing versus either agent alone against strains R6386 and R5995 (P < 0.001). DAP-containing regimens achieved bactericidal activity significantly faster than CPT monotherapy for all strains (4.5 ± 2.1 h versus 35.4 ± 17.5 h; P = 0.0025), but the combination of DAP plus CPT resulted in significantly faster time to bactericidal activity than DAP monotherapy (4.2 ± 1.6 h versus 6.5 ± 0.5 h; P = 0.0023).
FIG 1.
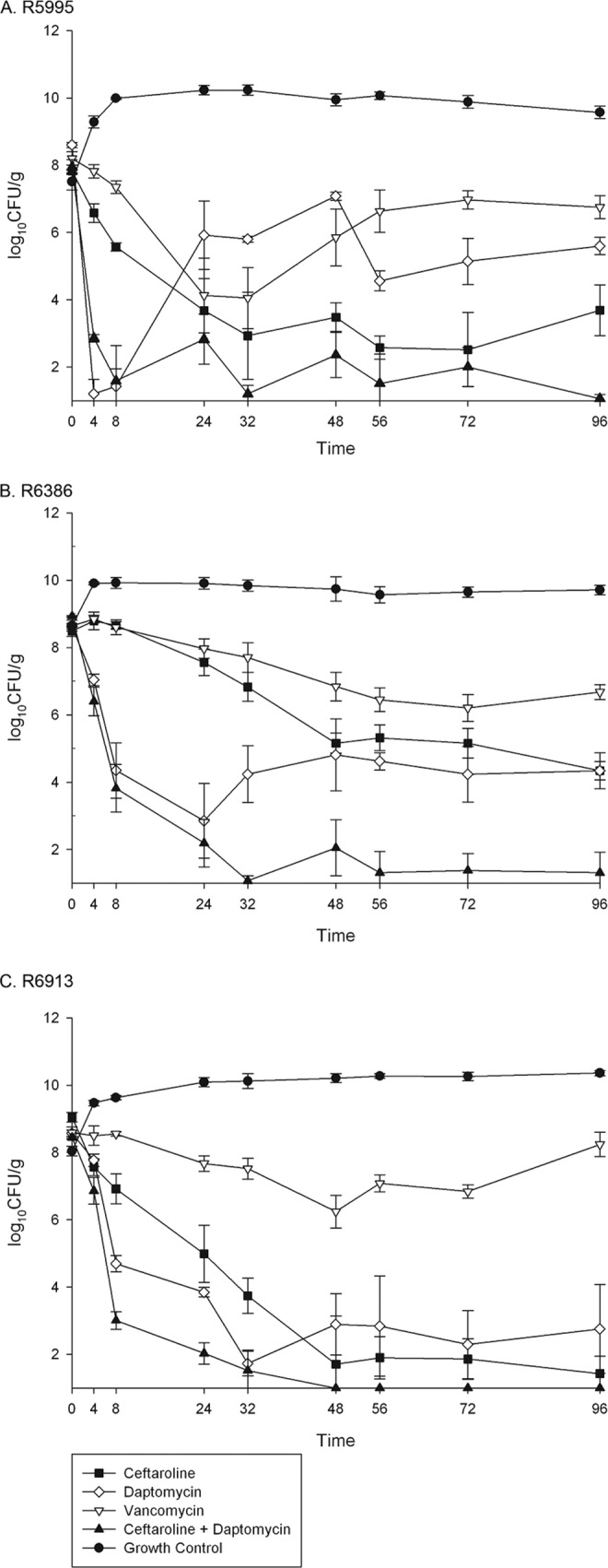
Pharmacodynamics of simulated antimicrobial regimens over 96 h against 3 strains of daptomycin-nonsusceptible, vancomycin-intermediate MRSA in a pharmacokinetic/pharmacodynamic model of simulated endocardial vegetations. Time is shown in hours on the x axes.
Pharmacokinetics.
The PK parameters for CPT achieved were a maximum concentration of drug in serum (Cmax) of 21.39 ± 1.12 mg/liter (target Cmax of 21.3 mg/liter) and half-life (t1/2) of 2.64 ± 0.23 h (target t1/2 of 2.66 h) for the central compartment. The average time above the MIC was 100% of the dosing interval for all 3 strains. The PK parameters for DAP achieved in the model were a Cmax of 131.9 ± 4.24 mg/liter (target, 129.7 mg/liter) and t1/2 of 8.41 ± 0.41 h (target, 8 h) with an average area under the curve from 0 to 24 h (AUC0–24) of 1,375.1 ± 0.79 mg · h/liter. The PK parameters for VAN achieved in the model were a minimum concentration of drug in serum (Cmin) of 20.3 ± 0.1 mg/liter (target, 20 mg/liter) and t1/2 of 4.5 ± 0.01 h (target, 5 h) with an AUC0–24 of 1,360 ± 6.8 mg · h/liter correlating with a VAN AUC0–24/MIC ratio of 340 for all strains.
Changes in susceptibility.
Isolates with MICs that were higher than the baseline MICs to CPT, DAP, and VAN were not detected on resistance screening plates.
DISCUSSION
The prevalence of DNS- and VISA-related MRSA infections remains relatively low; however, these organisms tend to arise from difficult-to-treat, high-inoculum infections such as infective endocarditis and osteomyelitis (10). Only limited data exist to support alternatives to VAN and DAP in the setting of serious infections with DNS and VISA strains of MRSA, but cases of successful management of these infections with CPT-F continue to be reported (26). To address the lack of data in this area, we evaluated the pharmacodynamics of humanized CPT exposures in a PK/PD model of simulated endocardial vegetations. CPT demonstrated bactericidal activity against DNS VISA strains of MRSA even at the high bacterial densities present in the simulated vegetations. At the simulated exposure of VAN, with a target trough concentration of 20 mg/liter, VAN demonstrated modest, bacteriostatic activity against the DNS VISA strains tested. This is unsurprising, as the AUC/MIC ratios (areas under the curve over 24 h in the steady state divided by the MICs) achieved against these strains were not optimal, since optimization would be clinically unrealistic. DAP demonstrated bactericidal activity against these strains; however, this was at exposures typically achieved with a 10-mg/kg/day dose. Even at this high-dose simulation, bacterial regrowth was observed. The synergistic combination of DAP plus CPT appears to offer more rapid activity against DNS MRSA strains than CPT alone and provides more sustained bacterial suppression with less regrowth than DAP alone. Against strain R6913, CPT, DAP, and CPT plus DAP had similar amounts of activity, and final colony counts at 96 h were not significantly different. Although all three strains had a daptomycin MIC of 4 mg/liter, we observed a surprising degree of heterogeneity in the response to the simulated DAP exposures. R6913 was killed effectively by DAP monotherapy in the model, while R6386 and R5995 responded initially but ultimately regrew. Interestingly, DAP monotherapy against R5995 resulted in >7-log10 CFU/g reduction in colony counts by just 4 h but regrew to ∼6 log10 CFU/g by 24 h. This response seems to suggest heterogeneous resistance to DAP, where initial exposure eradicates the more susceptible population and selects for the resistant subpopulations capable of growing in high concentrations of DAP. DAP nonsusceptibility is known to be a complex phenotype that may arise from a variety of possible genetic abnormalities (27). This variability in genetic pathways leading to the vancomycin-intermediate phenotype as well as the DNS phenotype may explain why responses to DAP were varied among strains with the same MICs.
The combination of CPT plus DAP is known to enhance DAP binding by ∼7-fold and to increase the activity of human cationic antimicrobial peptides produced by the immune system (13). This enhancement of activity appears to be related to an alteration of cell surface charge that makes DAP or cationic peptide binding more favorable (13, 28). Given that there were no immune cells or antimicrobial peptides in our model, it is possible that CPT may be more active in vivo due to synergy with peptides such as cathelicidins, defensins, and platelet-derived microbicidal proteins (29). Furthermore, the synergy with DAP may be potent enough to allow for reduced doses of CPT and/or DAP to achieve similar pharmacodynamics, which may offer cost savings in the clinic and reduce risk of toxicity without sacrificing efficacy (30). Further research should focus on evaluating less frequent dosing of CPT and lower doses of DAP to determine the minimum exposures necessary to prevent resistance and achieve pharmacodynamics similar to the pharmacodynamics of higher doses.
Given the antibacterial efficacy of CPT observed in our PK/PD model of SEVs and the emerging clinical and animal data, CPT-F appears to be a suitable alternative to manage patients with IE secondary to DNS or VISA MRSA. Although clinical data are still lacking, the combination of DAP plus CPT-F may also be a suitable choice for these infections and may offer faster clearance of bacteremia and more sustained suppression than either agent alone. Further clinical evaluation of this combination is warranted.
ACKNOWLEDGMENTS
This work was funded by an investigator-initiated grant from Forest Laboratories. M.J.R. is funded in part by NIH R21A1092055-01.
M.J.R. has received grant support, consulted for, or provided lectures for Cubist, Durata, Forest, Novartis, and Sunovion. B.J.W., K.E.B., and C.E.I. have no potential conflicts of interest to disclose.
We thank Abbott Laboratories for the use of the fluorescence polarization immunoassay analyzer for determination of vancomycin concentrations.
Footnotes
Published ahead of print 24 March 2014
REFERENCES
- 1.Liu C, Bayer A, Cosgrove SE, Daum RS, Fridkin SK, Gorwitz RJ, Kaplan SL, Karchmer AW, Levine DP, Murray BE, Rybak MJ, Talan DA, Chambers HF. 2011. Clinical practice guidelines by the Infectious Diseases Society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children: executive summary. Clin. Infect. Dis. 52:285–292. 10.1093/cid/cir034 [DOI] [PubMed] [Google Scholar]
- 2.Sakoulas G, Alder J, Thauvin-Eliopoulos C, Moellering RC, Jr, Eliopoulos GM. 2006. Induction of daptomycin heterogeneous susceptibility in Staphylococcus aureus by exposure to vancomycin. Antimicrob. Agents Chemother. 50:1581–1585. 10.1128/AAC.50.4.1581-1585.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Werth BJ, Vidaillac C, Murray KP, Newton KL, Sakoulas G, Nonejuie P, Pogliano J, Rybak MJ. 2013. Novel combinations of vancomycin plus ceftaroline or oxacillin against methicillin-resistant vancomycin-intermediate Staphylococcus aureus (VISA) and heterogeneous VISA. Antimicrob. Agents Chemother. 57:2376–2379. 10.1128/AAC.02354-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cui L, Tominaga E, Neoh HM, Hiramatsu K. 2006. Correlation between reduced daptomycin susceptibility and vancomycin resistance in vancomycin-intermediate Staphylococcus aureus. Antimicrob. Agents Chemother. 50:1079–1082. 10.1128/AAC.50.3.1079-1082.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kelley PG, Gao W, Ward PB, Howden BP. 2011. Daptomycin non-susceptibility in vancomycin-intermediate Staphylococcus aureus (VISA) and heterogeneous-VISA (hVISA): implications for therapy after vancomycin treatment failure. J. Antimicrob. Chemother. 66:1057–1060. 10.1093/jac/dkr066 [DOI] [PubMed] [Google Scholar]
- 6.Tenover FC, Sinner SW, Segal RE, Huang V, Alexandre SS, McGowan JE, Jr, Weinstein MP. 2009. Characterisation of a Staphylococcus aureus strain with progressive loss of susceptibility to vancomycin and daptomycin during therapy. Int. J. Antimicrob. Agents 33:564–568. 10.1016/j.ijantimicag.2008.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Avery LM, Steed ME, Woodruff AE, Hasan M, Rybak MJ. 2012. Daptomycin-nonsusceptible vancomycin-intermediate Staphylococcus aureus vertebral osteomyelitis cases complicated by bacteremia treated with high-dose daptomycin and trimethoprim-sulfamethoxazole. Antimicrob. Agents Chemother. 56:5990–5993. 10.1128/AAC.01046-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mariani PG, Sader HS, Jones RN. 2006. Development of decreased susceptibility to daptomycin and vancomycin in a Staphylococcus aureus strain during prolonged therapy. J. Antimicrob. Chemother. 58:481–483. 10.1093/jac/dkl256 [DOI] [PubMed] [Google Scholar]
- 9.Fowler VG, Jr, Boucher HW, Corey GR, Abrutyn E, Karchmer AW, Rupp ME, Levine DP, Chambers HF, Tally FP, Vigliani GA, Cabell CH, Link AS, DeMeyer I, Filler SG, Zervos M, Cook P, Parsonnet J, Bernstein JM, Price CS, Forrest GN, Fatkenheuer G, Gareca M, Rehm SJ, Brodt HR, Tice A, Cosgrove SE, S. aureus Endocarditis and Bacteremia Study Group 2006. Daptomycin versus standard therapy for bacteremia and endocarditis caused by Staphylococcus aureus. N. Engl. J. Med. 355:653–665. 10.1056/NEJMoa053783 [DOI] [PubMed] [Google Scholar]
- 10.Boucher HW, Sakoulas G. 2007. Perspectives on daptomycin resistance, with emphasis on resistance in Staphylococcus aureus. Clin. Infect. Dis. 45:601–608. 10.1086/520655 [DOI] [PubMed] [Google Scholar]
- 11.Villegas-Estrada A, Lee M, Hesek D, Vakulenko SB, Mobashery S. 2008. Co-opting the cell wall in fighting methicillin-resistant Staphylococcus aureus: potent inhibition of PBP 2a by two anti-MRSA beta-lactam antibiotics. J. Am. Chem. Soc. 130:9212–9213. 10.1021/ja8029448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steed ME, Rybak MJ. 2010. Ceftaroline: a new cephalosporin with activity against resistant gram-positive pathogens. Pharmacotherapy 30:375–389. 10.1592/phco.30.4.375 [DOI] [PubMed] [Google Scholar]
- 13.Werth BJ, Sakoulas G, Rose WE, Pogliano J, Tewhey R, Rybak MJ. 2013. Ceftaroline increases membrane binding and enhances the activity of daptomycin against daptomycin-nonsusceptible vancomycin-intermediate Staphylococcus aureus in a pharmacokinetic/pharmacodynamic model. Antimicrob. Agents Chemother. 57:66–73. 10.1128/AAC.01586-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Werth BJ, Steed ME, Kaatz GW, Rybak MJ. 2013. Evaluation of ceftaroline activity against heteroresistant vancomycin-intermediate Staphylococcus aureus and vancomycin-intermediate methicillin-resistant S. aureus strains in an in vitro pharmacokinetic/pharmacodynamic model: exploring the “seesaw effect”. Antimicrob. Agents Chemother. 57:2664–2668. 10.1128/AAC.02308-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Steed M, Vidaillac C, Rybak MJ. 2011. Evaluation of ceftaroline activity versus daptomycin (DAP) against DAP-nonsusceptible methicillin-resistant Staphylococcus aureus strains in an in vitro pharmacokinetic/pharmacodynamic model. Antimicrob. Agents Chemother. 55:3522–3526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vidaillac C, Leonard SN, Rybak MJ. 2009. In vitro activity of ceftaroline against methicillin-resistant Staphylococcus aureus and heterogeneous vancomycin-intermediate S. aureus in a hollow fiber model. Antimicrob. Agents Chemother. 53:4712–4717. 10.1128/AAC.00636-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.File TM, Jr, Wilcox MH, Stein GE. 2012. Summary of ceftaroline fosamil clinical trial studies and clinical safety. Clin. Infect. Dis. 55(Suppl 3):S173–S180. 10.1093/cid/cis559 [DOI] [PubMed] [Google Scholar]
- 18.Lin JC, Aung G, Thomas A, Jahng M, Johns S, Fierer J. 2013. The use of ceftaroline fosamil in methicillin-resistant Staphylococcus aureus endocarditis and deep-seated MRSA infections: a retrospective case series of 10 patients. J. Infect. Chemother. 19:42–49. 10.1007/s10156-012-0449-9 [DOI] [PubMed] [Google Scholar]
- 19.Ho TT, Cadena J, Childs LM, Gonzalez-Velez M, Lewis JS., Jr 2012. Methicillin-resistant Staphylococcus aureus bacteraemia and endocarditis treated with ceftaroline salvage therapy. J. Antimicrob. Chemother. 67:1267–1270. 10.1093/jac/dks006 [DOI] [PubMed] [Google Scholar]
- 20.Jacqueline C, Amador G, Batard E, Le Mabecque V, Miegeville AF, Biek D, Caillon J, Potel G. 2011. Comparison of ceftaroline fosamil, daptomycin and tigecycline in an experimental rabbit endocarditis model caused by methicillin-susceptible, methicillin-resistant and glycopeptide-intermediate Staphylococcus aureus. J. Antimicrob. Chemother. 66:863–866. 10.1093/jac/dkr019 [DOI] [PubMed] [Google Scholar]
- 21.Jacqueline C, Caillon J, Le Mabecque V, Miegeville AF, Hamel A, Bugnon D, Ge JY, Potel G. 2007. In vivo efficacy of ceftaroline (PPI-0903), a new broad-spectrum cephalosporin, compared with linezolid and vancomycin against methicillin-resistant and vancomycin-intermediate Staphylococcus aureus in a rabbit endocarditis model. Antimicrob. Agents Chemother. 51:3397–3400. 10.1128/AAC.01242-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rose WE, Schulz LT, Andes D, Striker R, Berti AD, Hutson PR, Shukla SK. 2012. Addition of ceftaroline to daptomycin after emergence of daptomycin-nonsusceptible Staphylococcus aureus during therapy improves antibacterial activity. Antimicrob. Agents Chemother. 56:5296–5302. 10.1128/AAC.00797-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clinical and Laboratory Standards Institute. 2011. Performance standards for antimicrobial susceptibility testing; twenty-first informational supplement. CLSI document M100–S21. Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 24.Blaser J. 1985. In-vitro model for simultaneous simulation of the serum kinetics of two drugs with different half-lives. J. Antimicrob. Chemother. 15(Suppl A):125–130. 10.1093/jac/15.suppl_A.125 [DOI] [PubMed] [Google Scholar]
- 25.Benvenuto M, Benziger DP, Yankelev S, Vigliani G. 2006. Pharmacokinetics and tolerability of daptomycin at doses up to 12 milligrams per kilogram of body weight once daily in healthy volunteers. Antimicrob. Agents Chemother. 50:3245–3249. 10.1128/AAC.00247-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jongsma K, Joson J, Heidari A. 2013. Ceftaroline in the treatment of concomitant methicillin-resistant and daptomycin-non-susceptible Staphylococcus aureus infective endocarditis and osteomyelitis: case report. J. Antimicrob. Chemother. 68:1444–1445. 10.1093/jac/dkt009 [DOI] [PubMed] [Google Scholar]
- 27.Bayer AS, Schneider T, Sahl HG. 2013. Mechanisms of daptomycin resistance in Staphylococcus aureus: role of the cell membrane and cell wall. Ann. N. Y. Acad. Sci. 1277:139–158. 10.1111/j.1749-6632.2012.06819.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mehta S, Singh C, Plata KB, Chanda PK, Paul A, Riosa S, Rosato RR, Rosato AE. 2012. Beta-lactams increase the antibacterial activity of daptomycin against clinical methicillin-resistant Staphylococcus aureus strains and prevent selection of daptomycin-resistant derivatives. Antimicrob. Agents Chemother. 56:6192–6200. 10.1128/AAC.01525-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sakoulas G, Bayer AS, Pogliano J, Tsuji BT, Yang SJ, Mishra NN, Nizet V, Yeaman MR, Moise PA. 2012. Ampicillin enhances daptomycin- and cationic host defense peptide-mediated killing of ampicillin- and vancomycin-resistant Enterococcus faecium. Antimicrob. Agents Chemother. 56:838–844. 10.1128/AAC.05551-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barber KE, Werth BJ, Rybak MJ. 2013. Evaluation of therapeutic de-escalation of the novel combination of ceftaroline (CPT) plus daptomycin (DAP) against DAP non-susceptible (DNS) methicillin-resistant Staphylococcus aureus (MRSA) in an in vitrohollow-fiber pharmacokinetic/pharmacodynamic model, abstr A-467 Abstr. 53rd Intersci. Conf. Antimicrob. Agents Chemother., Denver, CO [Google Scholar]