

Abstract
Although ampicillin is the most commonly used drug in neonates, developmental pharmacokinetic (PK) data to guide dosing are lacking. Ampicillin is primarily renally eliminated, and developmental changes are expected to influence PK. We conducted an open-label, multicenter, opportunistic, prospective PK study of ampicillin in neonates stratified by gestational age (GA) (≤34 or >34 weeks) and postnatal age (PNA) (≤7 or >7 days). Drug concentrations were measured by tandem mass spectrometry. PK data were analyzed using population nonlinear mixed-effects modeling in NONMEM 7.2. Monte Carlo simulations were conducted to determine the probability of target attainment for the time in which the total steady-state ampicillin concentrations remained above the MIC (T>MIC) for 50%, 75%, and 100% of the dosing interval. A total of 142 PK samples from 73 neonates were analyzed (median [range] GA, 36 [24 to 41] weeks; PNA, 5 [0 to 25] days). The median ampicillin dose was 200 (100 to 350) mg/kg/day. Postmenstrual age and serum creatinine were covariates for ampicillin clearance (CL). A simplified dosing regimen of 50 mg/kg every 12 h for GA of ≤34 weeks and PNA of ≤7 days, 75 mg/kg every 12 h for GA of ≤34 weeks and PNA of ≥8 and ≤28 days, and 50 mg/kg every 8 h for GA of >34 weeks and PNA of ≤28 days achieved the prespecified surrogate efficacy target in 90% of simulated subjects. Ampicillin CL was associated with neonatal development. A simplified dosing regimen stratified by GA and PNA achieves the desired surrogate therapeutic target in the vast majority of neonates.
INTRODUCTION
Ampicillin is the most commonly administered medication in the neonatal intensive care unit (NICU), yet its pharmacokinetics (PK) and safety in neonates are poorly described, and multiple dosing reference guides provide a variety of dosing recommendations based on limited PK data (see Table S1 in the supplemental material). The PK of ampicillin have been studied in children and adults (1–14), but data on dosing in the neonatal population are sparse. Current dosing regimens take into account the gestational age (GA)- and postmenstrual age (PMA)-related variation in renal drug clearance and recommend lower doses and less frequent dosing in the most premature neonates (1). However, the available data in the literature are insufficient to support dosing of ampicillin in the most extremely premature neonates (GA of ≤32 weeks at birth).
Given the inherent difficulties with multiple blood draws to acquire blood samples in neonates, capitalizing on standard-of-care procedures (such as biological sample collection from neonates already receiving drugs per routine medical care) has produced meaningful PK data, resulting in improved dosing recommendations for neonates (15). Opportunistic study designs offer an advantage over traditional PK studies given the low risk to participants and higher enrollment rates in a very difficult-to-study population such as neonates. However, opportunistic studies can be limited by the type and quantity of data collected, as well as by the reliability of standard-of-care procedures to generate data used for analysis. Exploring the utility of opportunistic PK study designs is imperative for neonates given recent legislation mandating studies in this population (16).
Using an opportunistic design, this study sought to characterize the developmental PK of ampicillin prescribed per standard of care to neonates across a wide age spectrum and to compare the pharmacodynamic (PD) target attainments of various dosing strategies.
MATERIALS AND METHODS
Study design.
This prospective, multicenter (n = 9) trial was conducted under the PK of Understudied Drugs Administered to Children per Standard of Care Study (POPS) (protocol NICHD-2011-POP01) in the Pediatric Trials Network (PTN). This protocol enrolled children <21 years of age receiving drugs per routine medical care. The data presented here were compiled from neonates receiving ampicillin. The protocol was reviewed and approved by the institutional review board of each participating institution. The guardian provided informed consent. Neonates were stratified by postnatal age (PNA) and GA (Table 1). The ampicillin dosing regimen, PK sampling, and demographic information (age, sex, race or ethnicity, GA at birth, birth weight, and actual weight) were recorded in an electronic database. Ampicillin dosing information for up to 8 doses prior to the first sampling dose was recorded. The following laboratory values were collected, if available, within 24 h before or after the dose of ampicillin closest to the time of biological sample collection: serum creatinine (SCR), plasma albumin, aspartate aminotransferase, alanine aminotransferase, and bilirubin (total and direct). Subjects were enrolled in the study for up to 90 days.
TABLE 1.
Demographic characteristicsa
| Parameter | Value for the indicated gestational age (wk) and PNA (days) |
Total | |||
|---|---|---|---|---|---|
| ≤34 |
>34 |
||||
| ≤7 | 8–28 | ≤7 | 8–28 | ||
| Group no. | 1 | 2 | 3 | 4 | |
| n | 21 | 7 | 27 | 18 | 73 |
| Postnatal age (days) at day of first plasma PK sample | |||||
| Mean (SD) | 2.6 (2.3) | 15.4 (4.0) | 2.9 (2.6) | 13.4 (5.4) | 6.6 (6.4) |
| Median (minimum, maximum) | 1.0 (0.0, 7.0) | 16.0 (9.0, 21.0) | 2.0 (0.0, 7.0) | 12.5 (8.0, 25.0) | 5.0 (0.0, 25.0) |
| Gestational age (wk) | |||||
| Mean (SD) | 30.3 (3.4) | 26.9 (2.5) | 38.2 (2.0) | 38.4 (1.8) | 34.9 (5.0) |
| Median (minimum, maximum) | 32.3 (24.0, 34.0) | 26.1 (25.0, 32.0) | 38.0 (34.0, 41.0) | 38.8 (35.0, 41.0) | 36.1 (24.0, 41.0) |
| No. (%) male | 9 (43) | 3 (43) | 18 (67) | 8 (44) | 38 (52) |
| Ethnicity, no. (%) | |||||
| Hispanic or Latino | 3 (14) | 1 (14) | 6 (22) | 3 (16) | 13 (18) |
| Not Hispanic or Latino | 18 (86) | 5 (71) | 19 (70) | 14 (78) | 56 (77) |
| Not reported | 0 | 1 (14) | 2 (7) | 1 (6) | 4 (6) |
| Race, no. (%) | |||||
| Black | 4 (19) | 3 (43) | 3 (11) | 2 (11) | 12 (16) |
| White | 16 (76) | 3 (43) | 23 (85) | 14 (78) | 56 (77) |
| Not reported | 0 | 0 | 0 | 1 (6) | 1 (1) |
| Other | 1 (5) | 1 (14) | 0 | 1 (6) | 3 (4) |
PK, pharmacokinetic; PNA, postnatal age.
A maximum of 10 research-only blood samples per subject was allowed in our study. A PK sampling scheme was employed such that no more than 2 ml/kg was obtained from each subject within a 90-day period. Participating sites were encouraged to collect biological samples opportunistically; thus, PK samples were obtained at the time of clinical laboratory evaluations per standard of care. However, the option to consent to sample collection for research purposes was given to the parent/guardian of the participating infant. PK sampling windows were provided to the sites as guidance for timing of PK sampling based on frequency of drug administration (see Table S2 in the supplemental material). Samples were stored frozen until ampicillin concentrations could be determined at the central laboratory. Plasma samples collected as part of this study were sent to a central laboratory for drug concentration measurements. A validated assay to measure ampicillin concentrations was performed at a central lab, OpAns Laboratory (Durham, NC), using a validated bioanalytical high-pressure liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) assay. The validated range of this method for ampicillin based on 10 μl of human plasma was 0.05 to 50 μg/ml. Quality controls were nominal concentrations of ampicillin in human plasma at 0.06, 4.0, and 40 μg/ml. The lower limit of quantification (LLOQ) was established using 5 quality control samples, independent of calibration standards. In addition, the lowest standard on the calibration curve was found to meet the following criteria for each validation run: the analyte response was at least 5 times the response compared with blank responses (i.e., signal-to-noise ratio of ≥5), and the analyte peak (response) was identifiable, discrete, and reproducible with a precision (coefficient of variation [CV]) of ≤20% and an accuracy of 80 to 120%. The LLOQ was defined by the lowest concentration on the standard curve and should not be confused with the limit of detection and/or the low quality control sample. The highest standard defined the upper limit of quantification.
PK analysis.
PK models for ampicillin concentration data were explored by nonlinear mixed-effects modeling using NONMEM 7.2 (Icon, Dublin, Ireland). Appropriate compartmental models were examined, and between-subject variability on model parameters was explored. Data were fitted to a 1-compartment model (ADVAN1 TRANS2) with the first-order conditional estimation method with interaction (FOCE-I). Proportional and additive residual error models were explored. Diagnostic plots were used to assess the appropriateness of this structure for the base and final models.
The investigation of the relationship between potential covariates and PK parameters proceeded by estimating the base population PK model, which included weight, with the generation of the Bayesian individual PK parameters (e.g., clearance [CL] and volume [V]). With these individual parameter estimates, their deviations from the typical population parameter values were also generated, i.e., individual subject ETAs (η). Next, graphical assessment of the relationships between PK parameters and potential covariates was performed by plotting ETAs versus potential clinically relevant covariates. Clinical variables were evaluated as potential covariates for PK parameters using a univariate screen in NONMEM followed by a multivariate assessment of the final population PK model. The following potential covariates were included in the univariate analysis: SCR, days of life (DOL) (which was PNA plus 1), GA, PMA, and birth weight. The construction of the final population PK model was based on NONMEM univariate and multivariate analyses with graphical exploration. During the model-building process, potential covariates that reduced the objective function by more than 3.84 (P < ∼0.05) were planned for inclusion in the subsequent multivariate analysis. A forward inclusion approach with backwards elimination was planned for the multivariate step, and a reduction of 7.88 (P < ∼0.005) was required for retention of a covariate in the final model.
Standard model diagnostic plots and procedures were used to evaluate model appropriateness. Empirical Bayesian estimates of individual subject PK parameters were generated from the final model. Model reliability was examined by visual predictive check (VPC) and 1,000-set bootstrapping procedures using WINGS for NONMEM. The model was considered reliable if the parameter estimates were within the 95% confidence intervals.
Analysis of surrogate pharmacodynamic endpoint using Monte Carlo simulations.
The PD target most correlated to maximal bactericidal activity for beta-lactams is the time in which free drug concentrations remain above the MIC (T>MIC) (17). The total, rather than free, drug concentration was used in this study due to low protein binding of ampicillin (∼10%) reported in neonates (18). Listeria monocytogenes and Escherichia coli (with ampicillin MIC90s of ≤2 μg/ml and ≤8 μg/ml, respectively, for susceptible isolates) can cause severe and fatal infections in neonates. Streptococci are also common pathogens in neonates but are very sensitive to ampicillin, with MIC90s of <0.5 μg/ml. Complete analyses were conducted to determine the probability of target attainment for achieving steady-state trough concentrations at 50%, 75%, and 100% T>MIC at MICs of 2 and 8 μg/ml.
Monte Carlo simulations were performed using the final population PK model to determine the distribution of steady-state ampicillin concentrations from the “typical” dose selected by clinicians for each age group in this study. The typical dose by age group (groups 1 to 4 as described in Table 1) was determined by dividing the average total daily dose for the group by the median dosing interval, rounded to the nearest 25 mg/kg. For groups 1 and 2, this was 100 mg/kg every 12 h; for group 3, 75 mg/kg every 8 h; and for group 4,100 mg/kg every 8 h.
In addition, simulations were performed using the dose recommendations from references that are commonly used for neonatal doses: Neofax (1) and Harriet Lane (19). Based on the relatively high concentrations seen with the typical dose in the current study, a lower dosing strategy was also evaluated. This revised dosing regimen was as follows: group 1, 50 mg/kg every 12 h; group 2, 75 mg/kg every 12 h; group 3, 50 mg/kg every 8 h; and group 4, 75 mg/kg every 8 h. The derivation of this optimal dosing regimen was based on achieving steady-state trough concentrations in ≥90% of the simulated participants at an MIC of 8 μg/ml. Simulations were performed to encompass the full range of GA and PNA across all 4 groups.
A total of 1,920 virtual subjects, 480 in each age group, were included at GAs of 24, 26, 28, 30, 32, 34, 35, 36, 37, 38, 39, and 40 weeks and PNAs of 1, 3, 7, 10 14, 21, and 28 days. The current weight (kg) (WTKG) and SCR used for each cohort were from a prior trial in premature infants (20). An additional SCR variability of 30% (beyond fixed effects of GA and PNA) was included during the NONMEM simulation by including a random effect (ETA) on SCR with a variance (OMEGA) value of 0.09 (30% CV). Medians and 95% confidence intervals were generated for the steady-state total concentration-time profiles of each age group using the various dosing strategies.
Statistical analysis.
For continuous variables, descriptive statistics included number of observations, mean, standard deviation, median, minimum, and maximum value. Discrete-variable summaries included counts and proportions. Except for the PK modeling, data were analyzed using SAS software, version 9.2 or later (SAS, Cary, NC).
RESULTS
A total of 75 participants provided consent for the study, were enrolled based on PNA and GA, and had available PK samples. Of the 75 participants, the indications for ampicillin included presumed or confirmed infection (n = 38), sepsis (n = 31), necrotizing enterocolitis (n = 2), abdominal procedure (n = 2), meconium ileus with peritonitis (n = 1), and pneumonia (n = 1). PK samples were collected opportunistically at times when blood samples were collected for biochemical analysis, unless the guardian consented to allow extra blood draws for research purposes, which occurred for 28 (37%) of these participants. The first participant was enrolled in February 2012, and the last participant completed the study in August 2012. The demographic characteristics are summarized in Table 1. One hundred sixty available plasma PK samples were collected from the 75 participants, which is an average of 2.1 per participant. Seventeen (23%) participants had more than 2 samples collected.
Of the 75 participants, 2 were excluded for the following reasons: (i) one subject had only one concentration that was below the quantitative level (BQL), and PNA at the time of the first PK sample was beyond the 28-day threshold (specifically, 30 days), and (ii) another subject had a suspected error in dosing history (i.e., 10 mg for 1,290 g or 7.8 mg/kg) with a high observed concentration. Fourteen (9%) of 156 samples from the 73 remaining participants were excluded: 6 samples were BQL and were thought to be unreliable given the time after dose; 5 participants had levels drawn after 24 h, which, given the dosing interval, were deemed to not be reliable; 1 participant had a sample drawn during infusion or flush; 1 sample was thought to be contaminated; and 1 participant had a sample drawn after a one-time intramuscular administration.
A total of 73 participants with 142 observed drug concentrations were included in the PK analysis. The median (range) ampicillin dosing prescribed initially as standard of care for these 73 participants was 200 (100 to 350) mg/kg/day, administered every 6 to 12 h (Table 2). The median (range) time of PK sampling in neonates with useable ampicillin concentration data was 4.8 (0.2 to 15.2) hours after the most recent dose, and the median (range) observed concentration was 123 (0.85 to 464) μg/ml. Collection of blood for PK samples was relatively consistent throughout the dosing intervals. Of the 142 measured samples, SCR values were available within 2 days of the PK sample date for 109 (77%) of the samples. Of the 73 participants, 24 had at least 1 SCR drawn in the first 3 days of life.
TABLE 2.
Ampicillin as prescribed by primary physician
| Group | n | Daily dose (mg/kg/day)a | Amt per dose (mg/kg)a | Dosing interval | Typical POPSb dose |
|---|---|---|---|---|---|
| 1 | 21 | 200 (161–303) | 100 (81–109) | 19% every 8 h, 81% every 12 h | 100 mg/kg every 12 h |
| 2 | 7 | 185 (113–194) | 93 (57–97) | 100% every 12 h | 100 mg/kg every 12 h |
| 3 | 27 | 218 (100–307) | 100 (43–102) | 59% every 8 h, 41% every 12 h | 75 mg/kg every q 8 h |
| 4 | 18 | 282 (184–350) | 92 (46–100) | 44% every 6 h, 28% every 8 h, 28% every 12 h | 100 mg/kg every 8 h |
| Overall | 73 | 200 (100–350) | 98 (43–109) | 11% every 6 h, 34% every 8 h, 55% every 12 h | 100 mg/kg every 12 h |
Numbers represent median (range).
POPS, NIH-funded study supporting this work that focuses on pharmacokinetics of understudied drugs administered to children per standard of care.
The univariate screen using a 1-compartment population PK model identified SCR and PMA as potential covariates for CL and none for V (Table 3). The final model used the FOCE-I, and the explicit formulas for the typical values of V and CL were V (liters) = θ(1) · WTKG and CL (liters/h) = θ(2) · WTKG · (0.6/SCR)θ(3)(PMA/37)θ(4), where θ(1) = 0. 399, θ(2) = 0.078, θ(3) = 0.428, and θ(4) = 1.34. WTKG was weight (kg), SCR was serum creatinine (mg/dl), and PMA was postmenstrual age (weeks). The between-subject variability for CL was 23%, and the residual variability was 34%. The estimated values for the population PK parameters, covariate, and variances, along with the standard errors of these estimates, bootstrap medians, and 95% confidence intervals for these values, are listed in Table 4. The ETA shrinkage value for CL was 21%, while the EPS shrinkage value for CL was 13%. In an attempt to further refine our final PK model by replacing PMA for GA and PNA as covariates (along with WTKG and SCR), the objective function value did not improve, and hence our final PK model was retained.
TABLE 3.
Ampicillin model-building processa
| Model | Population model | Objective function value | Change in objective function from base model |
|---|---|---|---|
| V (base model) | V = θV · WTKG | 1,284 | |
| CL (base model) | CL = θCL · WTKG | 1,284 | |
| Birth wt | CL = θCL · WTKG · (BW/2,500)θ(2),BW | 1,284 | 0 |
| DOL | CL = θCL · WTKG · (DOL/7)θ(2),DOL | 1,278 | −6.108 |
| GA | CL = θCL · WTKG · (GA/36)θ(2).GA | 1,257 | −26.81 |
| PMA | CL = θCL · WTKG · (PMA/37)θ(2),PMA | 1,251 | −33.34 |
| SCR | CL = θCL · WTKG · (0.6/SCR)θ(2),SCR | 1,249 | −34.85 |
| SCR, PMA (final) | CL = θCL · WTKG · (0.6/SCR)θ(2),SCR · (PMA/37)θ(2),PMA | 1,229 | −55.19 |
V, volume; WTKG, = weight in kg; CL, clearance; BW, birth weight; DOL, days of life (defined as postnatal age + 1 day); GA, gestational age; PMA, postmenstrual age; SCR, serum creatinine.
TABLE 4.
Final pharmacokinetic model parametersa
| Parameter | Symbol | Point estimate | % RSE | 95% Bootstrap confidence interval |
||
|---|---|---|---|---|---|---|
| 2.5% | Median | 97.5% | ||||
| CL | θ(2) | 0.078 | 4.37 | 0.071 | 0.077 | 0.084 |
| V | θ(1) | 0.399 | 6.34 | 0.350 | 0.398 | 0.452 |
| CL, SCR | θ(2),SCR | 0.428 | 21.40 | 0.235 | 0.433 | 0.639 |
| CL, PMA | θ(2),PMA | 1.34 | 23.73 | 0.651 | 1.31 | 1.96 |
| CL interindividual variability (CV, %) | ω2CL | 22.8 | 0.07 | 12.1 | 21.9 | 28.7 |
| Residual variability (CV, %) | σ2 | 33.9 | 0.08 | 26.6 | 33.5 | 41.4 |
CL, clearance (liters/h); V, volume (liters); SCR, serum creatinine (mg/dl); PMA, postmenstrual age (weeks); CV, coefficient of variation; RSE, relative standard error.
Individual subject post hoc CL estimates using the final model appeared to increase with GA and PNA, as reflected by increasing CL with each group (i.e., group 1 had the lowest CL and group 4 the highest) (Table 5). The half-life decreased with increasing both components of PMA, GA and PNA, as would be expected with the increasing CL when V was constant (Fig. 1). The goodness-of-fit plots demonstrated that the model generally fit the observed concentrations for both the population and the individual (Fig. 2). The post hoc Bayesian population estimates for V and CL were similar to the median bootstrap analysis values and were within the 95% confidence intervals obtained from the bootstrap analysis (Table 4). One hundred percent of the bootstrap data sets converged to ≥3 significant digits. The medians of bootstrap fixed-effects parameter estimates were within 1.5% of population estimates from the original data set for all parameters. The VPC using individual time points indicated that the model adequately described the data; 29% of the observations fell outside the 90% prediction interval (Fig. 3).
TABLE 5.
Individual empirical Bayesian post hoc parameter estimatesa
| Group | n | Clearance (liters/h/kg) | Volume (liters/kg) | Half-life (h) | Steady-state concn (μg/ml) |
|
|---|---|---|---|---|---|---|
| Minimum | Maximum | |||||
| 1 | 21 | 0.055 (0.03–0.07) | 0.40 (0.40–0.40) | 5.0 (3.9–9.4) | 77 (36–320) | 318 (244–563) |
| 2 | 7 | 0.070 (0.03–0.07) | 0.40 (0.40–0.41) | 4.0 (3.8–8.3) | 33 (21–145) | 266 (159–368) |
| 3 | 27 | 0.086 (0.04–0.13) | 0.40 (0.40–0.40) | 3.2 (2.2–6.2) | 48 (5–173) | 274 (127–413) |
| 4 | 18 | 0.11 (0.06–0.13) | 0.40 (0.40–0.41) | 2.4 (2.1–4.7) | 28 (5–129) | 246 (138–203) |
| Overall | 73 | 0.072 (0.03–0.13) | 0.40 (0.40–0.41) | 3.3 (2.1–9.4) | 47 (5–320) | 281 (127–563) |
All values are medians and ranges.
FIG 1.
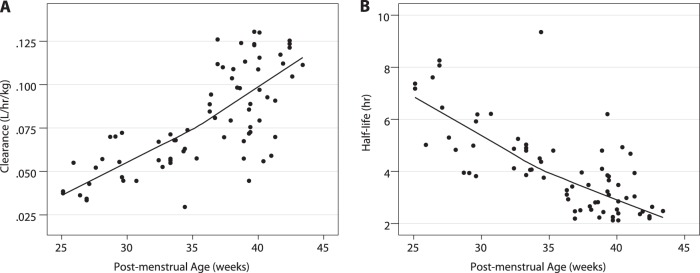
Clearance (A) and half-life (B) versus postmenstrual age.
FIG 2.
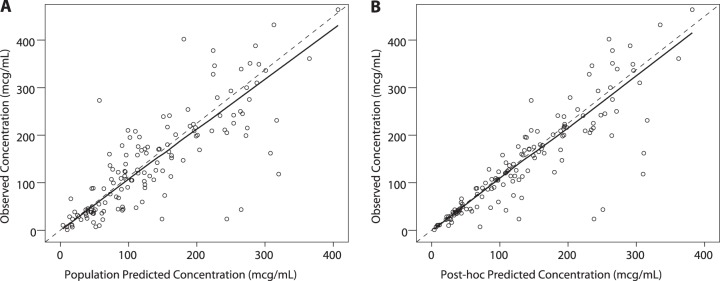
Observed versus population (A) and individual (B) predictions, final model.
FIG 3.
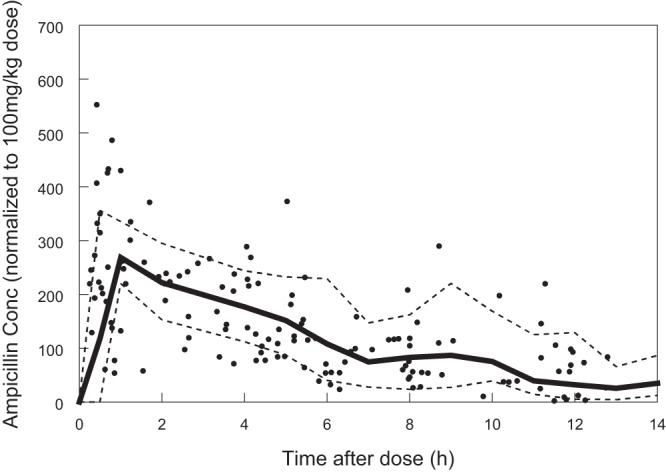
Visual predictive check. Circles, observed concentrations (Conc); solid line, predicted median concentration; dashed lines: 90% confidence intervals.
Using the PD surrogate of 100% T>MIC, all standard-of-care ampicillin doses used in this study achieved predicted trough concentrations at steady state of >2 μg/ml in 100% of participants; 100% of participants in groups 1 and 2 and 89% in groups 3 and 4 had predicted trough concentrations of ≥8 μg/ml. All participants in groups 3 and 4 with trough concentrations of <8 μg/ml were dosed every 12 h compared to every 8 h.
Because of variability in the primary caregiver's dose selection, we evaluated standardized dosing using Monte Carlo simulations. Based on the Monte Carlo simulations, >97% of virtual participants in all 4 age groups (at typical POPS doses listed in Table 2) achieved 100% T>MIC at an MIC of 8 μg/ml (Table 6). In contrast, 10% of simulated subjects in at least one group failed to meet the surrogate PD target of 100% T>MIC at an MIC of 8 μg/ml when dosing recommendations found in pediatric guidelines were used (Table 6). With the goal of achieving trough concentrations of ≥8 μg/ml in ≥90% of the simulated subjects, we simplified the dosing regimens from several references and devised an optimal GA- and PNA-based dosing regimen for ampicillin (Table 7). Although PMA was the covariate in our final PK model, we replaced it with GA and PNA in streamlining the dosing regimens, as these variables are easier to integrate into practical dosing guidelines for clinical practice. Analysis of PD target attainment using PMA-based dosing demonstrated results similar to those for GA- and PNA-based dosing.
TABLE 6.
Probability of target attainment from Monte Carlo simulations using the final pharmacokinetic model
| Groupa | % of subjects meeting MIC target of: |
|||||
|---|---|---|---|---|---|---|
| 2 μg/ml |
8 μg/ml |
|||||
| 50% T>MICb | 75% T>MIC | 100% T>MIC | 50% T>MIC | 75% T>MIC | 100% T>MIC | |
| Harriet Lane | ||||||
| 1 | 100 | 100 | 100 | 100 | 100 | 99.8 |
| 2 | 100 | 100 | 100 | 100 | 100 | 99.8 |
| 3 | 100 | 100 | 98.8 | 100 | 100 | 90.2 |
| 4 | 100 | 100 | 100 | 100 | 100 | 99.2 |
| Neofax | ||||||
| 1 | 100 | 100 | 1 | 100 | 100 | 98.1 |
| 2 | 100 | 100 | 99.8 | 100 | 100 | 96.9 |
| 3 | 100 | 100 | 98.8 | 100 | 100 | 90.2 |
| 4 | 100 | 100 | 99.2 | 100 | 100 | 90.2 |
| Typical POPS doses | ||||||
| 1 | 100 | 100 | 100 | 100 | 100 | 99.2 |
| 2 | 100 | 100 | 100 | 100 | 100 | 97.1 |
| 3 | 100 | 100 | 99.6 | 100 | 100 | 98.1 |
| 4 | 100 | 100 | 100 | 100 | 100 | 98.3 |
Group numbers refer to the age group categories defined in Table 1.
T>MIC, time above MIC.
TABLE 7.
Optimal dosing regimen based on Monte Carlo simulations using the final pharmacokinetic model
| Gestational age (wk) | Postnatal age (days) | Maintenance dose (mg/kg) | Dosing interval (h) |
|---|---|---|---|
| ≤34 | ≤7 | 50 | 12 |
| ≤34 | ≥8 and ≤28 | 75 | 12 |
| >34 | ≤28 | 50 | 8 |
DISCUSSION
Ampicillin is a commonly used drug in neonates. However, the insufficient PK studies (especially in very premature neonates), coupled with the lack of uniformity in dosing regimens (which are based on GA, PNA, weight, and PMA in pediatric dosing compendiums [see Table S1 in the supplemental material]), have led to confusion on optimal drug dosing in this vulnerable population. Early pharmacological studies of serum and cerebrospinal fluid drug concentrations after intramuscular injection of ampicillin in term and preterm (<2,500 g) neonates were performed between 1967 and 1974. These studies included a combined total of 156 neonates (GA not specified) and showed that the serum half-life of ampicillin decreases rapidly in the first 2 weeks of life as a result of increasing drug clearance. The doses studied ranged from 25 to 150 mg/kg administered every 8 to 12 h according to PNA (2, 4, 8). In a recent study comparing clinical efficacies of ampicillin and penicillin in combination with gentamicin therapy for early-onset sepsis in infants (including GA of <26 weeks), both were well tolerated, with no difference in adverse events or laboratory abnormalities. However, the focus of this study was clinical effectiveness comparison and not safety profiling (9).
A strength of the present study was its opportunistic design, which capitalized on obtaining samples drawn at the same time as when blood samples were collected for biochemical analysis in neonates on drugs per routine medical care. Preliminary data obtained through opportunistic studies have served in the design of phase I to III trials in children. For example, through the infrastructure of the Pediatric Pharmacology Research Unit funded by the National Institutes of Health, Wade et al. obtained timed and scavenged plasma samples to characterize the PK of fluconazole in premature neonates (15). This PK analysis led to the dose selection for a phase III randomized, placebo-controlled trial of fluconazole prophylaxis in premature neonates (clinicaltrials.gov, NCT00734539). In our large opportunistic study, we evaluated the population PK of ampicillin in 73 neonates as young as 24 weeks of gestation and up to 28 days PNA. This broad age spectrum enabled the construction of a population PK model that incorporated developmental changes, including age-related alterations in body composition and acquisition of renal function, which occurs during the first month of life. Notably, some developmental processes that affect drug PK may not occur linearly with age. In fact, the most striking changes in a drug's PK transpire within the first year of life, including a rapid surge in renal function due to an increase in renal blood flow within the first 2 weeks of life (21, 22). Furthermore, drug PK may not be solely dependent on PNA, particularly in the extremely premature neonates, because nephrogenesis, which contributes to an increasing glomerular filtration rate in utero, requires completion by 36 weeks of gestation (21, 22).
In the final population PK model, WTKG, a marker for growth, was assumed to be a significant covariate for CL and V. In addition to WTKG, PMA and SCR were identified as important covariates for CL. A maturational change in ampicillin CL was evident by the inclusion of both PMA and SCR as covariates. The need to account for developmental maturation using PMA (which is composed of PNA and GA) is consistent with prior investigations of ampicillin PK in neonates that correlated drug elimination to PNA (2, 4, 8). Furthermore, SCR was expected to be strongly correlated to CL given the predominantly renal elimination of this drug.
The population PK parameter estimate for elimination half-life in the present study was comparable to prior reports on premature neonates, with inverse proportionality to PNA (2, 4, 8). In our cohort of premature neonates, the median half-life ranged from 3.2 to 5.0 h for neonates with PNAs of ≤7 days but decreased to 2.4 to 4.0 h for those with PNAs of ≥8 and ≤28 days. In addition, ampicillin CL increased rapidly by 27% after the first week of life (i.e., group 1 versus 2 with a GA of ≤34 weeks and group 3 versus 4 with a GA of >34 weeks). Furthermore, ampicillin CL increased by 56% from the younger (GA of ≤34 weeks) to the older (GA of >34 weeks) cohorts. Previous investigations did not specifically provide CL and V estimates. A 1-compartment model appropriately described our data, similar to the case in a prior study that reported no substantial improvement in fit with a 2-compartment model compared to a 1-compartment model after intramuscular injection of ampicillin (23). Our model was precise as evidenced by population CL and V point estimates nearly identical to the median bootstrap values and narrow 95% confidence intervals. The percentage of observations outside the prediction interval was higher than expected (i.e., >10%) in the VPC. In close examination of the VPC, bias was absent, but the difference between observed and expected concentrations was likely due to the inability to characterize the ETA for V. Furthermore, our final PK model supported a single ETA only for CL (not V). The small sample size in our study likely contributed to this outcome.
The Monte Carlo simulation demonstrated that the higher doses of ampicillin currently being prescribed by most physicians, as evident by the average daily dose of ampicillin ordered for the neonates in this study, achieved the surrogate PD endpoint of trough concentrations at steady state of ≥8 μg/ml in >97% of virtual subjects compared to 90% of virtual subjects with the current dosing references. Compared to the dosing regimens recommended in pediatric references, our proposed optimal dosing strategy for ampicillin provides fewer dosing groups, accounts for renal function that dramatically changes within the first few weeks of life (including preterm infants ≤29 weeks), and incorporates more convenient, less frequent dosing (every 8 h rather than every 6 h).
This study had several strengths and limitations. We assessed the PK of ampicillin over a broader range of GA and PNA than in previous studies; thus, we were better able to delineate the developmental aspects associated with ampicillin. While the final population PK model allowed us to characterize the CL and V of ampicillin in these preterm and term neonates, we were limited in evaluating intrapatient variability because the average number of samples per subject was only 2. Similarly, we did not assess the role of drug transporters in contributing to the CL and V of ampicillin. The expression and function of drug transporters responsible for drug distribution and those residing in the kidney (such as the organic anion transporters) vary with age (24). Approximately 90% of ampicillin is excreted as unchanged drug in the kidney (18). While not examined in our study, these age-related differences in drug transporters may have contributed to ampicillin CL. As this was an opportunistic study, we did not control for consistent dosing. Doses ranged from 100 to 350 mg/kg/day and generally exceeded the recommended doses. Notably, our Monte Carlo simulations did not account for ampicillin penetration into the cerebrospinal fluid, which has been reported at a range of 11 to 65% based on doses of 120 to 200 mg/kg/day administered in infants <1 month old with meningitis (8). Further studies are needed to explore optimal dosing for the treatment of meningitis, especially in premature infants <1 month old, for whom only one study is currently available (8).
Conclusions.
This large opportunistic population PK study of ampicillin in neonates demonstrated the importance of PMA in drug CL. We simplified the dosing regimens from several references and devised an ampicillin dosing regimen of 50 to 75 mg/kg every 8 to 12 h, based on GA and PNA (Table 7). This revised dosing regimen achieved trough concentrations of ≥8 μg/ml in 90% of subjects. Furthermore, although several references suggest dosing every 6 h for some PNA and GA groups, adjusting the total dose to allow for dosing every 8 h decreases the frequency of ampicillin administration yet maintains therapeutic drug exposure.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded under NICHD contract HHSN2752010000031 for the Pediatric Trials Network (principal investigator, D.K.B.) and by grant U54D071600 from the National Institutes of Health (principal investigator, E.V.C.). Research reported in this publication was supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under award number UL1TR001117. The assay measuring ampicillin concentrations was performed at OpAns Laboratory (Durham, NC).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
M.L. receives support from the U.S. government for his work in pediatric and neonatal clinical pharmacology (government contract HHSN267200700051C [principal investigator, D.K.B.] under the Best Pharmaceuticals for Children Act) and from NICHD (K23HD068497). D.K.B. receives support from the U.S. government for his work in pediatric and neonatal clinical pharmacology (1R01HD057956-05, 1K24HD058735-05, UL1TR001117, and NICHD contract HHSN275201000003I) and the nonprofit organization Thrasher Research Fund for his work in neonatal candidiasis (www.thrasherresearch.org); he also receives research support from industry for neonatal and pediatric drug development (www.dcri.duke.edu/research/coi.jsp). E.V.C. receives salary support from the U.S. government (U54 HD071600-01) and research support from Trius, Cerexa Pharmaceuticals, Abbott, and Theravance. M.C.-W. receives support for research from the National Institutes of Health (NIH) (1K23HD064814), the National Center for Advancing Translational Sciences of the NIH (UL1TR001117), the Food and Drug Administration (1U01FD004858-01), the Biomedical Advanced Research and Development Authority (BARDA) (HHSO100201300009C), and the nonprofit organization Thrasher Research Fund (www.thrasherresearch.org) and from industry for drug development in adults and children (www.dcri.duke.edu/research/coi.jsp). J.E.S. received support from the U.S. government for her work in pediatric pharmacology (U10 HD045934 05). The other authors have no potential conflicts of interest.
Members of the PTN Administrative Core Committee are as follows: Katherine Berezny, Duke Clinical Research Institute, Durham, NC; Jeffrey Barrett, Children's Hospital of Philadelphia, Philadelphia, PA; Gregory L. Kearns, Children's Mercy Hospital, Kansas City, MO; Andre Muelenaer, Virginia Tech Carilion School of Medicine, Roanoke, VA; T. Michael O'Shea, Wake Forest Baptist Medical Center, Winston Salem, NC; Ian M. Paul, Penn State College of Medicine, Hershey, PA; P. Brian Smith, Duke Clinical Research Institute, Durham, NC; John van den Anker, George Washington University School of Medicine and Health, Washington, DC; Kelly Wade, Children's Hospital of Philadelphia, Philadelphia, PA; Thomas J. Walsh, Weill Cornell Medical College of Cornell University, New York, NY; David Siegel, Perdita Taylor-Zapata, Anne Zajicek, Zhaoxia Ren, and Alice Pagan, The Eunice Kennedy Shriver National Institute of Child Health and Human Development; and Ravinder Anand, Traci Clemons, and Gina Simone, The EMMES Corporation (Data Coordinating Center). Members of the PTN study team, principal investigators (PIs), and study coordinators (SCs) are as follows: Kristi Prather (statistician) and Barrie Harper (project leader), Duke Clinical Research Institute, Durham, NC; Leslie Wilson (SC), Riley Hospital for Children at Indiana University, Indianapolis; Janice Bernhardt (SC), University of North Carolina at Chapel Hill, NC; Kelli Brown and Tressa Bratton (SCs), University of Louisville and Kosair Children's Hospital, Louisville, KY; Wade Rich and Nancy Tang (SCs), University of California, San Diego, CA; Kathryn Smith (SC), Rady Children's Hospital, San Diego, CA; Kathleen Neville (PI) and Rose Thompson (SC), Children's Mercy Hospital and Clinics, Kansas City, MO; Laura James (PI) and Dawn Hansberry (SC), Arkansas Children's Hospital, Little Rock; Michael Reed (PI) and Tonia Polanski (SC), Akron Children's Hospital, OH; and Ram Yogev (PI) and Molly Hartrich (SC), Children's Memorial Hospital, Chicago, IL.
Footnotes
Published ahead of print 10 March 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02374-13.
REFERENCES
- 1.Thomson Reuters Clinical Editorial Staff. 2011. Neofax®, 24th ed. Thomson Reuters, Hoboken, NJ [Google Scholar]
- 2.Axline SG, Yaffe SJ, Simon HJ. 1967. Clinical pharmacology of antimicrobials in premature infants. II. Ampicillin, methicillin, oxacillin, neomycin, and colistin. Pediatrics 39:97–107 [PubMed] [Google Scholar]
- 3.Barrons RW, Murray KM, Richey RM. 1992. Populations at risk for penicillin-induced seizures. Ann. Pharmacother. 26:26–29 [DOI] [PubMed] [Google Scholar]
- 4.Boe RW, Williams CP, Bennett JV, Oliver TK., Jr 1967. Serum levels of methicillin and ampicillin in newborn and premature infants in relation to postnatal age. Pediatrics 39:194–201 [PubMed] [Google Scholar]
- 5.Chmel H, Emmanuel G, Lie T, Anderson L, Ireland J. 1990. A prospective, double-blind, randomized study comparing the efficacy and safety of low-dose ciprofloxacin with ampicillin in the treatment of bronchitis. Diagn. Microbiol. Infect. Dis. 13:149–151. 10.1016/0732-8893(90)90098-G [DOI] [PubMed] [Google Scholar]
- 6.Eshelman FN, Spyker DA. 1978. Pharmacokinetics of amoxicillin and ampicillin: crossover study of the effect of food. Antimicrob. Agents Chemother. 14:539–543. 10.1128/AAC.14.4.539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ginsburg CM, McCracken GH, Jr, Thomas ML, Clahsen J. 1979. Comparative pharmacokinetics of amoxicillin and ampicillin in infants and children. Pediatrics 64:627–631 [PubMed] [Google Scholar]
- 8.Kaplan JM, McCracken GH, Jr, Horton LJ, Thomas ML, Davis N. 1974. Pharmacologic studies in neonates given large dosages of ampicillin. J. Pediatr. 84:571–577. 10.1016/S0022-3476(74)80684-0 [DOI] [PubMed] [Google Scholar]
- 9.Metsvaht T, Ilmoja ML, Parm U, Maipuu L, Merila M, Lutsar I. 2010. Comparison of ampicillin plus gentamicin vs. penicillin plus gentamicin in empiric treatment of neonates at risk of early onset sepsis. Acta Paediatr. 99:665–672. 10.1111/j.1651-2227.2010.01687.x [DOI] [PubMed] [Google Scholar]
- 10.Nahata MC, Vashi VI, Swanson RN, Messig MA, Chung M. 1999. Pharmacokinetics of ampicillin and sulbactam in pediatric patients. Antimicrob. Agents Chemother. 43:1225–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ripa S, Ferrante L, Prenna M. 1990. Pharmacokinetics of sulbactam/ampicillin in humans after intravenous and intramuscular injection. Chemotherapy 36:185–192. 10.1159/000238765 [DOI] [PubMed] [Google Scholar]
- 12.Shaffer CL, Davey AM, Ransom JL, Brown YL, Gal P. 1998. Ampicillin-induced neurotoxicity in very-low-birth-weight neonates. Ann. Pharmacother. 32:482–484 [DOI] [PubMed] [Google Scholar]
- 13.Sheffield MJ, Lambert DK, Henry E, Christensen RD. 2010. Effect of ampicillin on the bleeding time of neonatal intensive care unit patients. J. Perinatol. 30:527–530. 10.1038/jp.2009.192 [DOI] [PubMed] [Google Scholar]
- 14.Sjovall J, Alvan G, Huitfeldt B. 1986. Intra- and inter-individual variation in pharmacokinetics of intravenously infused amoxycillin and ampicillin to elderly volunteers. Br. J. Clin. Pharmacol. 21:171–181. 10.1111/j.1365-2125.1986.tb05172.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wade KC, Wu D, Kaufman DA, Ward RM, Benjamin DK, Jr, Sullivan JE, Ramey N, Jayaraman B, Hoppu K, Adamson PC, Gastonguay MR, Barrett JS, National Institute of Child Health and Human Development Pediatric Pharmacology Research Unit 2008. Population pharmacokinetics of fluconazole in young infants. Antimicrob. Agents Chemother. 52:4043–4049. 10.1128/AAC.00569-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Food and Drug Administration Safety and Innovation Act (FDASIA). 2012. Public law 112-144. http://www.gpo.gov/fdsys/pkg/PLAW-112publ144/pdf/PLAW-112publ144.pdf
- 17.Lodise TP, Butterfield J. 2011. Use of pharmacodynamic principles to inform beta-lactam dosing: “S” does not always mean success. J. Hosp. Med. 6(Suppl 1):S16–S23. 10.1002/jhm.869 [DOI] [PubMed] [Google Scholar]
- 18.Taketomo CK, Hodding JH, Kraus DM. 2013. Pediatric & neonatal dosage handbook, 19th ed. LexiComp, Hudson, OH [Google Scholar]
- 19.Tschudy MM, Arcara KM. 2012. The Harriet Lane handbook: a manual for pediatric house officers, 19th ed. Elsevier Mosby, Philadelphia, PA [Google Scholar]
- 20.Smith PB, Cohen-Wolkowiez M, Castro LM, Poindexter B, Bidegain M, Weitkamp JH, Schelonka RL, Ward RM, Wade K, Valencia G, Burchfield D, Arrieta A, Bhatt-Mehta V, Walsh M, Kantak A, Rasmussen M, Sullivan JE, Finer N, Brozanski BS, Sanchez P, van den Anker J, Blumer J, Kearns GL, Capparelli EV, Anand R, Benjamin DK., Jr 2011. Population pharmacokinetics of meropenem in plasma and cerebrospinal fluid of infants with suspected or complicated intra-abdominal infections. Pediatr. Infect. Dis. J. 30:844–849. 10.1097/INF.0b013e31822e8b0b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van den Anker JN, Schwab M, Kearns GL. 2011. Developmental pharmacokinetics. Handb. Exp. Pharmacol. 205:51–75. 10.1007/978-3-642-20195-0_2 [DOI] [PubMed] [Google Scholar]
- 22.Kearns GL, Abdel-Rahman SM, Alander SW, Blowey DL, Leeder JS, Kauffman RE. 2003. Developmental pharmacology—drug disposition, action, and therapy in infants and children. N. Engl. J. Med. 349:1157–1167. 10.1056/NEJMra035092 [DOI] [PubMed] [Google Scholar]
- 23.Hermans J, Driessen O, Sorgedrager N, Spoor M. 1975. Pharmacokinetic analysis of ampicillin concentration in neonates: comparison of two pharmacokinetic models and of two numerical methods. Arzneimittelforschung 25:947–949 [PubMed] [Google Scholar]
- 24.Nigam SK, Bhatnagar V. 2013. How much do we know about drug handling by SLC and ABC drug transporters in children? Clin. Pharmacol. Ther. 94:27–29. 10.1038/clpt.2013.82 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.