

Abstract
Norovirus (NoV) is the leading cause of acute gastroenteritis worldwide, causing over 200,000 deaths a year. NoV is nonenveloped, with a single-stranded RNA genome, and is primarily transmitted person to person. The viral RNA-dependent RNA polymerase (RdRp) is critical for the production of genomic and subgenomic RNA and is therefore a prime target for antiviral therapies. Using high-throughput screening, nearly 20,000 “lead-like” compounds were tested for inhibitory activity against the NoV genogroup II, genotype 4 (GII.4) RdRp. The four most potent hits demonstrated half-maximal inhibitory concentrations (IC50s) between 5.0 μM and 9.8 μM against the target RdRp. Compounds NIC02 and NIC04 revealed a mixed mode of inhibition, while NIC10 and NIC12 were uncompetitive RdRp inhibitors. When examined using enzymes from related viruses, NIC02 demonstrated broad inhibitory activity while NIC04 was the most specific GII.4 RdRp inhibitor. The antiviral activity was examined using available NoV cell culture models; the GI.1 replicon and the infectious GV.1 murine norovirus (MNV). NIC02 and NIC04 inhibited the replication of the GI.1 replicon, with 50% effective concentrations (EC50s) of 30.1 μM and 71.1 μM, respectively, while NIC10 and NIC12 had no observable effect on the NoV GI.1 replicon. In the MNV model, NIC02 reduced plaque numbers, size, and viral RNA levels in a dose-dependent manner (EC50s between 2.3 μM and 4.8 μM). The remaining three compounds also reduced MNV replication, although with higher EC50s, ranging from 32 μM to 38 μM. In summary, we have identified novel nonnucleoside inhibitor scaffolds that will provide a starting framework for the development and future optimization of targeted antivirals against NoV.
INTRODUCTION
Noroviruses (NoVs) cause around 50% of all gastroenteritis cases worldwide (1) and are associated with the deaths of more than 200,000 people per year, mainly in developing countries (2). Of particular importance are NoVs that belong to genogroup II, genotype 4 (GII.4), which have been associated with all six major pandemics of acute gastroenteritis in the last 2 decades, and account for 80% of all human NoV infections (3). In addition, NoV is increasingly recognized as an important cause of chronic gastroenteritis in immunocompromised patients (4, 5). Apart from the human costs, NoV infections cause severe economic losses (6). The virus is highly transmissible, with a low infectious dose, and high numbers are excreted during acute illness: approximately 108 virions per gram of feces (7). Following an incubation period of 1 to 2 days, the clinical features of NoV infections include acute onset of nausea, vomiting, abdominal cramps, headaches, and diarrhea that generally last for 2 to 4 days (8). A member of the family Caliciviridae, NoV is nonenveloped and 27 to 35 nm in diameter with a single-stranded RNA genome of 7,400 to 7,700 nucleotides. The genome includes three open reading frames (ORFs). ORF1 encodes seven nonstructural (NS) proteins (NS1 to NS7) and includes a viral genome-linked protein (VPg) (NS5), a 3C-like protease (NS6), and an RNA-dependent RNA polymerase (RdRp) (NS7). ORF2 and ORF3 encode the major capsid protein (VP1) and minor basic protein (VP2), respectively (9).
Significant efforts have been made to identify a permissive cell line for human NoV; however, these attempts have largely failed (10). The lack of a cell culture system for human NoV has hindered both replication studies and the identification of NoV therapeutic agents. In 2003, murine NoV (MNV) was identified (11), which led to the development of the first cell culture system and small-animal model for NoV infection (12). Wobus et al. demonstrated that MNV replicates in cells of mononuclear origin, such as primary dendritic cells and macrophages (12). Building on this work with MNV, it has been recently shown that BALB/c Rag-γc-deficient mice could also support the replication of human GII.4 NoV (13). Another important breakthrough in the establishment of human NoV cell culture models was the development of a subgenomic replicon based on the prototype NoV GI strain, Norwalk virus (14). The replicon consists of the complete ORF1, encoding the viral replicative enzymes (NS1 to -7), while ORF2, which encodes the major structural protein, is disrupted by the insertion of a neomycin resistance gene. This allows replication of autonomous RNA in Huh-7 cells without the expression of viral structural genes, and therefore, no virions are produced (14). Replicons are invaluable for studying viral RNA replication and provide a very useful platform for antiviral development; for example, they have been instrumental in the recent advances in hepatitis C virus (HCV) antiviral development (reviewed in reference 15).
The highly infectious nature of NoV and its association with outbreaks in hospitals, senior care facilities, and cruise ships highlight the need for specific approaches to control NoV infections, through either vaccines or antivirals. This is of particular importance for individuals with a high risk of exposure, for immunocompromised patients with chronic NoV infections, and for those who are susceptible to complications and dehydration, including young children and the elderly. The NoV infectious cycle offers a number of potential targets for the development of direct-acting antivirals (DAAs). One key target for NoV antivirals is the viral polymerase (RdRp) because of its essential role in viral replication and the lack of homologous human enzymes. A number of studies have used recombinant NoV RdRp to characterize its biochemical properties in vitro, and the X-ray crystal structures of RdRps from human GI and GII and mouse GV NoVs have been solved (16–18).
There are only a few publications reporting NoV DAA development (reviewed in reference 19). The most advanced preclinical studies have so far focused on protease inhibitors (20, 21) or have repurposed available drugs that were originally developed to treat other viral (22–24) and nonviral (25) infections as NoV RdRp inhibitors. However, thus far, no novel molecules or scaffolds have been identified as specific inhibitors of the NoV RdRp.
In this study, we conducted a high-throughput screen (HTS) to identify small-molecule inhibitors of GII.4 NoV RdRp transcription that may provide a platform for the development of antivirals against this important clinical pathogen. Four scaffolds were identified, and their modes of RdRp inhibition were characterized. We further examined the specificities of these compounds across a range of calicivirus RdRps, and the antiviral activity was assessed using the human Norwalk GI.1 subgenomic replicon and the MNV infectious cell culture model.
MATERIALS AND METHODS
Recombinant RdRp expression, purification, and comparison.
Recombinant RdRps with a C-terminal hexahistidine tag were expressed in Escherichia coli and purified by nickel affinity chromatography, as described previously (26, 27). The RdRps of the following caliciviruses (shown with their corresponding GenBank accession numbers) were used in this study: NoV GII.4 Den Haag 2006b variant (EF684915), NoV GII.4 New Orleans 2009 variant (JQ613573), NoV GI.1 Norwalk (NC_001959), NoV GV.1 (MNV; DQ285629), NoV GII.7 (GQ849131), and sapovirus (SaV) GII (AY237420). Amino acid sequence analysis was performed using the MEGA5 software package (28), and a phylogenetic tree of protein sequences was produced using the neighbor-joining method.
Biochemical RdRp assays.
Polymerase activity was measured by monitoring the formation of double-stranded RNA (dsRNA) from a single-stranded homopolymeric template, poly(C), using the fluorescent dye PicoGreen (Life Technologies, Carlsbad, CA, USA), as described previously (29) with minor modifications. RdRp assays were performed in 384-well plates, and each reaction mixture contained 20 ng enzyme (13.3 nM), 5 μM GTP, 6 μg/ml poly(C) RNA, 2.5 mM MnCl2, 5 mM dithiothreitol (DTT), 0.01% bovine serum albumin (BSA), and 0.005% Tween 20 in 20 mM Tris-HCl, pH 7.5, with a final volume of 25 μl. Reactions were run for 10 min at 23°C and terminated with 10 mM EDTA, followed by PicoGreen staining and dsRNA quantitation. Alternatively, radioactive-GTP incorporation was measured on a scintillation counter, as described previously (27).
High-throughput screening.
An HTS was carried out to identify inhibitors of NoV using the RdRp of a representative GII.4 variant, Den Haag 2006b, which was associated with a global pandemic and was the predominant NoV in circulation between 2006 and 2008 (30). A random selection of 19,956 compounds from the Walter and Eliza lead-like compound library (The Walter and Eliza Hall Institute, Parkville, Australia) were screened at a final concentration of 10 μM, as outlined previously (29). Hits from the HTS were subjected to a confirmatory counterassay at 10 μM using radioactive-nucleotide incorporation to further exclude false positives that could have affected the fluorescence signal in the primary assay.
Mode of RdRp inhibition.
To examine the mode of enzyme inhibition by the lead hits, the kinetics of substrate (GTP) incorporation was examined in the presence or absence of inhibitor. Reactions were performed with increasing concentrations of GTP (from 0.2 to 66 μM) and 5, 10, 15, or 20 μM inhibitor. Kinetic parameters for each compound were determined by nonlinear regression and used to generate Lineweaver-Burk double-reciprocal plots.
Cell culture and cytotoxicity.
HG23 cells, a human hepatoma (Huh-7) cell line bearing the Norwalk virus (GI.1) subgenomic replicon (14), were kindly supplied by Kim Green (NIAID, NIH, Bethesda, MD, USA). MNV strain CW1 (12) was kindly provided by Herbert Virgin (Washington University, St. Louis, MO, USA). MNV stocks were prepared as described previously (31).
HG23 and RAW 264.7 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Life Technologies) supplemented with 10% fetal bovine serum (FBS) (Sigma-Aldrich, St. Louis, MO, USA), Glutamax (Life Technologies), and 100 U/ml penicillin-streptomycin (Life Technologies). G418 (Geneticin; Life Technologies) was also added to HG23 cells (750 μg/ml). To examine the cytotoxicity of compounds identified in this study, cell monolayers (1 × 104/well for HG23 or 2.0 × 104/well for RAW 264.7 cells) were treated with various concentrations of each compound for 72 h in 96-well plates. Cytotoxicity was measured by a resazurin-to-resorufin conversion assay (CellTiter-Blue; Promega, Madison, WI, USA), and fluorescence was measured on a Fluostar Optima microplate reader (BMG Labtech, Ortenberg, Germany).
Inhibition of the GI.1 Norwalk replicon.
The effects of the identified compounds on the replication of the Norwalk subgenomic replicon (14) were examined. The nucleoside inhibitor (NI) 2′-C-methylcytidine (2CM) (Sigma-Aldrich), which has previously been shown to inhibit the replication of the Norwalk replicon (23, 32), was used as a positive control in all cell culture experiments, and dimethyl sulfoxide (DMSO) (0.5% [vol/vol]) was added to untreated cells. HG23 cells were seeded in 96-well plates at a density of 1 × 104 cells/well in antibiotic-free medium and treated with test compounds as described for cytotoxicity analysis. Total RNA was isolated using the RNeasy kit (Qiagen) 72 h later for the quantitation of replicon RNA levels.
Inhibition of murine norovirus replication.
Inhibition of MNV replication in RAW 264.7 cells was determined by plaque reduction assays, as described previously (33) with modifications. Briefly, 6-well plates were seeded with 1 × 106 RAW 264.7 cells/well and incubated overnight at 37°C. The monolayers were then inoculated with 80 PFU of MNV in DMEM. After a 1-h adsorption at 37°C, the medium was removed, and the wells were overlaid with 0.75% (wt/vol) low-melting-point agarose in minimum essential medium (MEM) (Life Technologies) containing test compounds. Cells were incubated for 48 h at 37°C and then fixed with formaldehyde (4% [vol/vol]) and stained with crystal violet (0.2% [wt/vol]). Inhibition of MNV was measured by quantitation of the total plaque surface area using the image-processing program ImageJ (34). To measure MNV RNA replication, RAW 264.7 cells were seeded in 96-well plates at a density of 2.0 × 104 cells/well. Test compounds were added on the following day, and the cells were incubated for 1 h at 37°C. The cells were then infected with MNV (multiplicity of infection [MOI] = 0.1) and incubated for 48 h. Total RNA was extracted for quantitation using a QIAamp Viral RNA Mini kit (Qiagen).
qRT-PCR.
Viral RNA was quantitated from either HG23 cells or MNV-infected RAW 264.7 cells by quantitative reverse transcriptase (qRT) PCR (7). In brief, cDNA was synthesized using a SuperScript Vilo cDNA Synthesis Kit (Life Technologies). Replicon (GI.1) and MNV (GV.1) RNAs were measured using an iTaq Universal SYBR green Supermix (Bio-Rad, CA) as described previously (7). NS7-specific primers were used for both GI-based replicon and MNV quantitation and included Replicon Fwd (5′-CCAACTGAAACCCTTTGCGG-3′), Replicon Rev (5′-AGGCATCAGCGTAAGACCAC-3′), MNV Fwd (5′-TGGACGTCGGCGACTATAAG-3′), and MNV Rev (5′-ACCACCTCGTCATCACCATA-3′).
Structure-activity relationship analysis.
To identify molecules from the remaining 90,000 compounds in the library with structural similarity to leading HTS hits, all functionality was removed from the hit structures, and the library was screened using ActivityBase SARview software (IDBS, Guilford, United Kingdom), as described previously (35). All identified molecules were screened in triplicate at 10 μM for the ability to inhibit the NoV GII.4 RdRp, and the results were compared to the primary HTS hits.
RESULTS
Identification of RdRp inhibitors by high-throughput screening.
Using an in vitro HTS, nonnucleoside inhibitors (NNI) of the NoV GII.4 RdRp (Den Haag 2006b variant) were identified from 19,956 compounds randomly selected from a larger, 110,000-compound library. Figure 1 shows a summary of the pathway to the identification of the four most potent RdRp inhibitors in this study. Overall, test plates demonstrated suitable measures for HTS quality quantitated by Z′ and Z factors, as described in reference 36 (Fig. 2A). Both measures were above the acceptable limit of 0.5 (average Z′ = 0.77 ± 0.06; Z = 0.72 ± 0.07), with the exception of 2 out of 57 plates, which had Z factor scores of −0.64 and 0.31. The apparent drop in quality was attributed to two highly fluorescent compounds, which were subsequently omitted from the analysis. Of the 19,956 compounds screened, 35 hits demonstrated greater than 3 times the standard deviation (SD) of the average inhibition relative to control reactions, or >15.5% inhibition of RdRp activity (Fig. 2B). All 35 hits were subsequently counterscreened using a radioactive-nucleotide incorporation RdRp assay, and 12 compounds demonstrated >40% inhibition of RdRp activity, while 14 had no inhibitory activity and were most likely fluorescence quenchers. Four other compounds that appeared to increase the activity of NoV RdRp by ∼2-fold (Fig. 2B) also had no effect when retested at 10 μM using the fluorescence and radioactivity assays (data not shown). Dose-response curves for RdRp inhibition were generated for the 12 most potent hits, and the half-maximal inhibitory concentrations (IC50s) ranged from 5 to 30 μM. The most potent of these compounds, a phenylthiazole carboxamide (NIC02) and a pyrazole acetamide (NIC04), inhibited the GII.4 NoV RdRp with IC50s of 5.0 and 5.5 μM, respectively (Fig. 3 and Table 1). NIC10, a triazole, and NIC12, a pyrazolidinedione, demonstrated slightly higher IC50s of 9.2 and 9.8 μM, respectively (Fig. 3 and Table 1).
FIG 1.
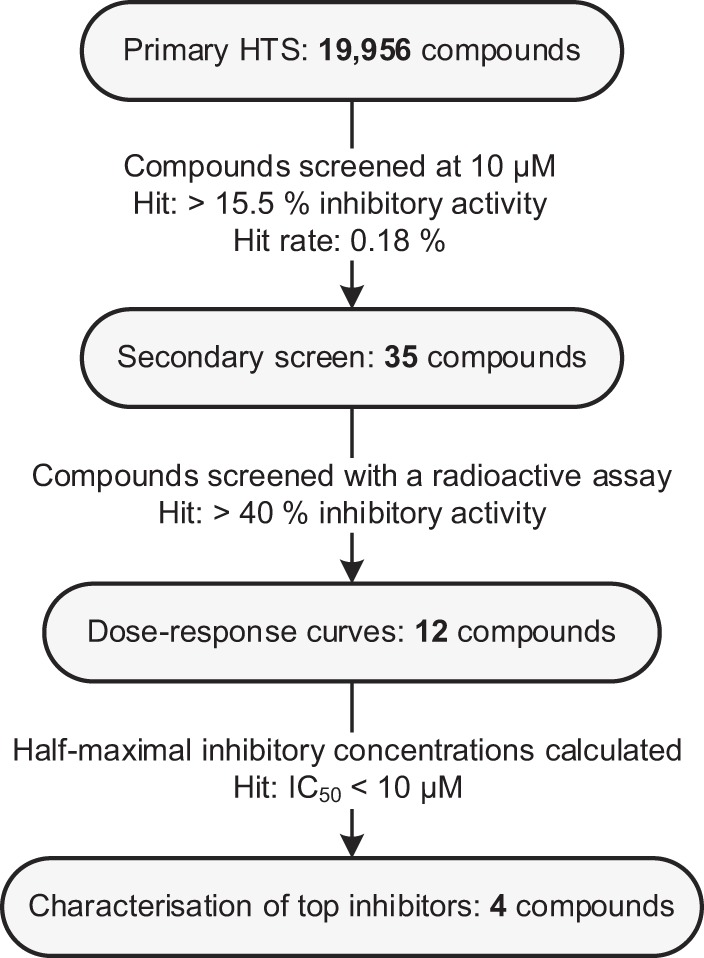
Outline of the pathway to identify NoV RdRp inhibitors in this study.
FIG 2.
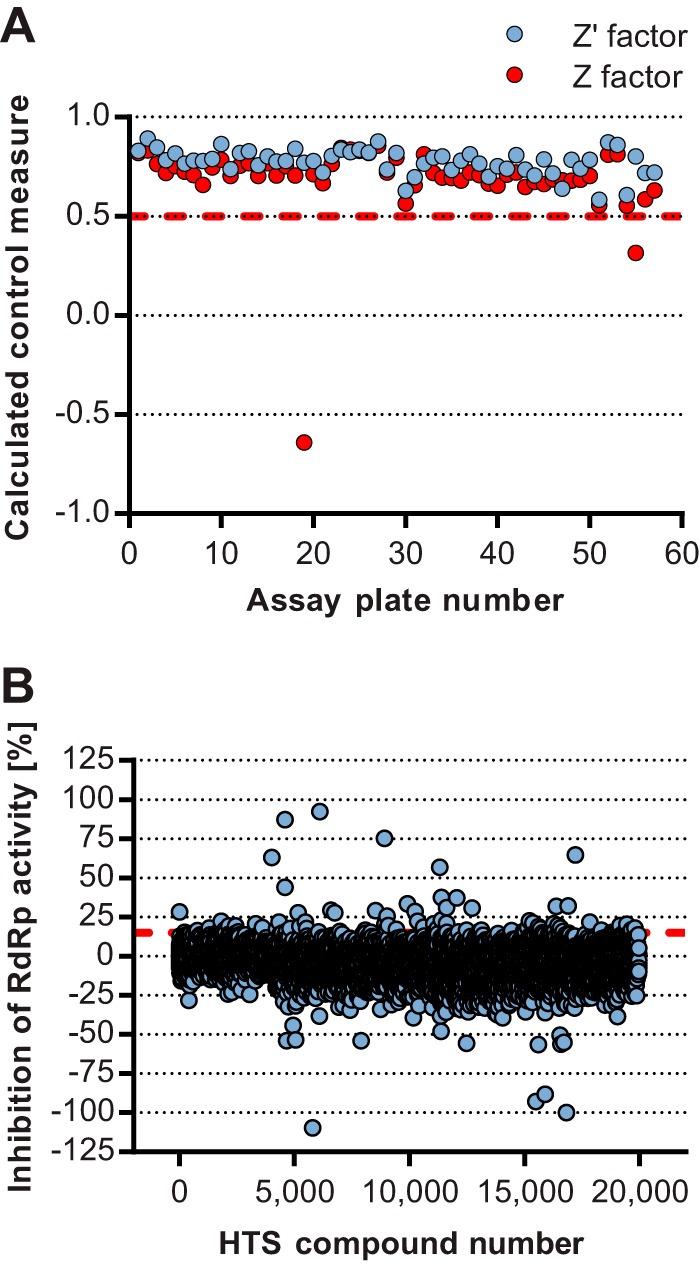
HTS for inhibitors of the NoV GII.4 RdRp. Compounds were screened for inhibitory activity against the NoV RdRp. (A) Control measurement for test plates in chronological order. The Z′ factor and Z factor were determined for each plate, and a value of >0.5 was considered acceptable, which is highlighted with a dashed red line. (B) Inhibition results of the HTS. RdRp inhibition was calculated as a percentage of internal plate controls. The hit selection cutoff, which represented inhibition exceeding 3 times the SD of the mean distribution, is shown as a dashed red line.
FIG 3.
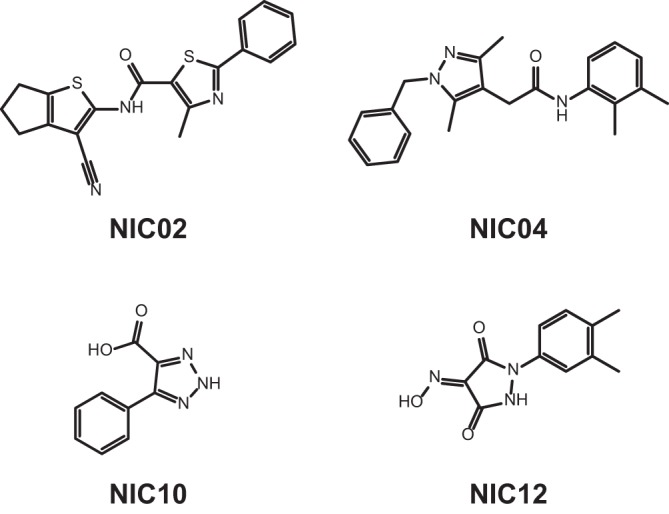
Chemical structures of the NoV RdRp inhibitors in this study.
TABLE 1.
Activities of the identified inhibitors using the NoV RdRp and cell culture models
| Compound | CLogPa | GII.4 RdRp IC50 (μM)b | GI.1 replicon EC50 (μM)c | GV.1 MNV EC50 (μM)c |
|---|---|---|---|---|
| NIC02 | 3.6 | 5.0 (3.6–6.9) | 30.1 (19.6–45.9) | 4.8 (1.7–13.3) |
| NIC04 | 3.5 | 5.5 (4.5–6.7) | 71.1 (56.7–89.1) | 32.8 (22.0–48.9) |
| NIC10 | 0.8 | 9.2 (7.4–11.3) | >100 | 34.5 (22.6–52.5) |
| NIC12 | 0.4 | 9.8 (7.4–13.0) | >100 | 38.1 (17.7–82.4) |
CLogP, calculated partition coefficient.
IC50s were determined by in vitro radioactive-GTP incorporation RdRp assays (95% CI in parentheses).
EC50s were determined using cell-based replicon and infectious-NoV model systems (95% CI in parentheses).
Mode of inhibition.
To characterize the mode of NoV GII.4 RdRp inhibition by each of the four lead hits, the kinetics of GTP incorporation was examined in the absence or presence of increasing inhibitor concentrations (see Fig. S1 in the supplemental material). Double-reciprocal Lineweaver-Burk plots indicated a mixed mode of inhibition for NIC02 and NIC04, where both the substrate affinity and the reaction velocity decreased with higher inhibitor concentrations (Fig. 4A and B). In contrast, the Lineweaver-Burk plots for NIC10 and NIC12 were representative of an uncompetitive mechanism of RdRp inhibition (Fig. 4C and D). For these two compounds, the apparent substrate affinity increased at higher compound concentrations, as indicated by a decrease in Km values, while the reaction velocity decreased.
FIG 4.

Differential mechanisms of RdRp inhibition by the four NIC compounds. In order to determine the mode of inhibition, RdRp activity was measured at different concentrations of substrate (0.2 to 66 μM) in the presence of 0 to 20 μM inhibitors. Double-reciprocal plots were generated for NIC02 (A), NIC04 (B), NIC10 (C), and NIC12 (D). RFU, relative fluorescence units.
Activities of identified NNIs across related calicivirus RdRps.
To determine the breadth of the inhibitory activities for the four identified compounds, purified recombinant RdRps from five caliciviruses of varying relatedness to the target GII.4 Den Haag 2006b were used (Fig. 5A). These RdRps represented a different variant within GII.4 (GII.4 New Orleans 2009 variant), a different genotype within the same genogroup (GII.7), and different genogroups within the genus (GI.1 Norwalk and GV.1 MNV). Additionally, an RdRp from sapovirus (GII), which represents another genus within the family Caliciviridae, was examined. A phylogenetic analysis of the protein sequences revealed the relationship of the enzymes examined in this study (Fig. 5A), with amino acid identities ranging from 32.6% (sapovirus) to 97.1% (New Orleans 2009) compared to the target GII.4 Den Haag 2006b RdRp sequence (Fig. 5B).
FIG 5.

Inhibitory profiles of lead NoV inhibitors against RdRps from related caliciviruses. (A) Unrooted neighbor-joining tree of RdRp amino acid sequences used in this study. The evolutionary distances were computed using the number-of-differences method and represent the number substitutions per sequence, indicated by the scale bar. (B) Amino acid percentage identities between the analyzed enzymes. (C) Inhibitory activities of the identified compounds against enzymes from related viruses. The compounds were assayed in triplicate at increasing concentrations, and average changes in IC50s are shown with standard deviations for each enzyme relative to the target, GII.4 Den Haag 2006b RdRp.
All four lead hits demonstrated inhibitory activity against both GII.4 enzymes (New Orleans 2009 and Den Haag 2006b variants; 97% identity), with similar IC50s for each NIC (Fig. 5C), as assessed by in vitro RdRp assays. Of the four lead hits, NIC02 showed the broadest inhibitory activity, with similar levels of inhibition across RdRps from all six caliciviruses (±0.19-fold change in the IC50) (Fig. 5C). Given its inhibitory activity against distantly related RdRps, we examined the inhibitory activities of NIC02 against different classes of polymerases. NIC02 did not demonstrate any inhibition of enzyme activity, even at concentrations up to 100 μM, against an RNA-dependent DNA polymerase (RdDp) (avian myeloblastosis virus RdDp) or a DNA-dependent DNA polymerase (DdDp) (Taq DdDp) (data not shown).
NIC10 had a narrower spectrum of inhibitory activity. When compared to the GII.4 RdRp, NIC10 demonstrated 11-fold and 56-fold reductions in the IC50 values against SaV and GI.1 RdRps, respectively, and a 1.9-fold reduction against MNV but had comparable activity against the GII.7 RdRp (Fig. 5C). NIC12 showed a modest loss of potency against the GI.1 (2.3-fold) and GII.7 (4.6-fold) RdRps compared to GII.4 and had little inhibitory activity against the SaV RdRp, with 33.9% ± 14.1% inhibition of RdRp activity at 100 μM (Fig. 5C). Interestingly, however, NIC12 was 4-fold more active against the MNV RdRp than the GII.4 enzymes. Finally, NIC04 appeared to be the most specific scaffold for the GII.4 enzymes, with significant losses of inhibitory activity against GII.7 (4.6-fold), GI.1 (3.1-fold), and SaV RdRp (8.7-fold), while no inhibition was observed against the MNV RdRp (Fig. 5C).
Inhibition of the NoV replicon.
The antiviral activities of the identified compounds were assessed by monitoring the replication of the GI.1 Norwalk virus replicon in Huh-7 cells (14). Cells were treated with increasing concentrations of each compound (1 to 100 μM), and replicon RNA levels were quantitated 72 h later by qRT-PCR. NIC02 and NIC04 inhibited replicon replication in a dose-dependent manner, with 50% effective concentrations (EC50s) of 30.1 μM (95% confidence interval [CI], 19.6 to 45.9 μM) and 71.1 μM (95% CI, 56.7 to 89.1 μM), respectively (Fig. 6A and Table 1). NIC10 and NIC12 had no effect on the replication of the GI.1 replicon when tested at concentrations up to 100 μM. The compound cytotoxicity was assessed simultaneously and compared to that of vehicle (0.5% DMSO)-treated cells. NIC02 was toxic to the Huh-7 cells (Fig. 6B and Table 1) at concentrations greater than 10 μM, with a half-maximal cytotoxic concentration (CC50) of 134 μM. In contrast, NIC04, NIC10, and NIC12 had no cytotoxic effects at concentrations up to 100 μM. The nucleoside analogue 2CM, which was used as a positive control, reduced RNA replicon levels by 84.9% ± 1.4% relative to untreated cells with no observable cytotoxicity when examined at 10 µM (data not shown).
FIG 6.
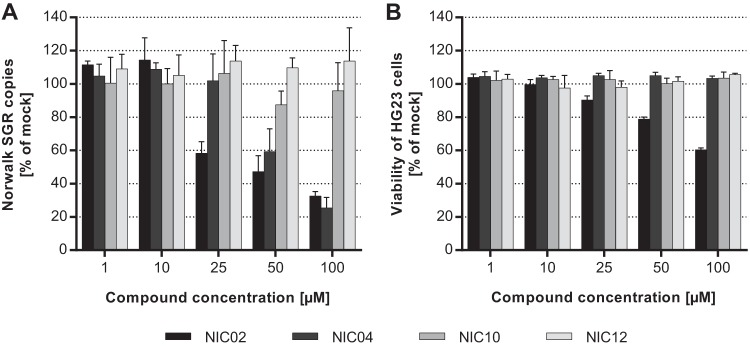
Inhibition of the Norwalk subgenomic replicon (SGR) by lead norovirus NNIs. (A) The inhibitory activities of the top four compounds were assessed using Huh-7 cells harboring the Norwalk replicon (HG23 cells). NIC02 and NIC04 inhibited the replicon with EC50s of 30.1 μM and 71.1 μM, respectively. NIC10 and NIC12 had limited effect on the replication of the replicon. (B) Cytotoxicities of the NIC compounds were examined at concentrations ranging from 1 μM to 100 μM. Only NIC02 demonstrated cytotoxic effect at high concentrations (CC50 = 134 μM). All results are the averages of triplicate experiments plotted with standard deviations.
Inhibition of infectious murine norovirus GV.1.
To examine the antiviral activities of the four lead hits (NIC02, NIC04, NIC10, and NIC12) in an infectious norovirus cell culture system, the GV.1 murine norovirus (MNV strain CW1) was used. Monolayers of RAW 264.7 cells were treated with test compounds and infected with MNV for 48 h. MNV replication was assessed either by plaque reduction assays or by viral RNA genome quantitation by qRT-PCR (NIC02 only). Inhibitory activity was demonstrated by a reduction in both plaque size and numbers with all four compounds compared to DMSO-treated MNV-infected cells (see Fig. S2 in the supplemental material). Quantitation of inhibitory activity, based on reduction in plaque area, revealed an EC50 of 4.8 μM for NIC02 (95% CI, 1.7 to 13.3), the most potent of the four inhibitors (Fig. 7A). The quantitation of MNV RNA genomes from infected cells also revealed an EC50 of 2.3 μM for NIC02 (Fig. 7C). Higher concentrations were required to inhibit the replication of MNV with NIC04, NIC10, and NIC12, with EC50s of 32.8 (95% CI, 22.0 to 48.9), 34.5 (95% CI, 22.6 to 52.5), and 38.1 (95% CI, 17.7 to 82.4) μM, respectively (Table 1).
FIG 7.

Effects of NIC compounds on the replication of murine norovirus. The antiviral activities of the NIC compounds were examined using infectious MNV. (A) Mean plaque areas for the different treatments shown as percentages of mock (DMSO)-treated cells. NIC02 demonstrated the highest inhibition of MNV replication (EC50 = 4.8 μM). NIC04, NIC10, and NIC12 had similar efficacies, with EC50s of 32.8, 34.5, and 38.1 μM, respectively. (B) Cytotoxicity profiles of the NIC compounds with uninfected RAW 264.7 cells, shown as percentages of viable cells compared to untreated wells. (C) Inhibition of MNV by NIC02 was assessed by quantitation of viral RNA levels where the antiviral activity was similar to that observed with the plaque reduction assay, with an EC50 of 2.3 μM. The results are the averages of triplicate experiments plotted with standard deviations.
The reduction in plaque size for NIC04, NIC10, and NIC12 was not due to cytotoxic effects of the test compounds, as no cell death was observed with concentrations up to 100 μM (Fig. 7B). However, toxicity to RAW 264.7 cells was observed with NIC02, with a CC50 of 57.1 μM, which was more than 10-fold higher than the EC50 of NIC02. The nucleoside analogue 2CM was also cytotoxic to RAW 264.7 cells at concentrations above 1 μM (CC50 value of 12.2 μM); however, at 1 μM, the MNV plaque area was reduced by 93.5% ± 0.3% relative to untreated cells (data not shown).
Preliminary structure-activity analysis.
In order to gain initial insights into the structure-activity relationships (SARs) for the four molecules, the compound library used in the primary HTS was searched to identify relevant analogues to NIC02, NIC04, NIC10, and NIC12. A total of 182 molecules were identified, which were examined for inhibitory activity at 10 μM against the NoV GII.4 RdRp (see Table S1 in the supplemental material). Three analogues of NIC02 were found, and only one retained some inhibitory activity at 10 μM: 17% compared to 63% enzyme inhibition for the original hit (see Table S1 in the supplemental material). None of the analogues for NIC04 (112 compounds) or NIC10 (33 compounds) demonstrated increased activity compared to the original hit molecules (see Table S1 in the supplemental material). Of 34 NIC12 analogues identified, three were more potent than the original hit (48% inhibition at 10 μM), with 55%, 62%, and 76% inhibition of RdRp activity at 10 μM, respectively (see Table S1 in the supplemental material).
DISCUSSION
There is a clear unmet need for safe, effective antiviral therapies to combat norovirus infections, both for prophylactic use to prevent NoV transmission in an outbreak setting and to treat immunocompromised patients with chronic NoV infections. Four small “drug-like” molecules were identified as inhibitors of NoV RdRp activity and represent new scaffolds for the development of therapeutic molecules (Fig. 3). The compounds demonstrated inhibitory activity in the low micromolar range and comprised a phenylthiazole-5-carboxamide (NIC02), a pyrazole-4-acetamide (NIC04), a triazole (NIC10), and a pyrazolidine-3,5-dione (NIC12). Three of these compounds were novel scaffolds that have not been previously reported as viral NNIs; however, NIC12 has been reported in a separate HTS as a “weak” inhibitor of the poliovirus RdRp activity, with approximately 22% inhibition when tested at 86 μM (37), suggesting NIC12 is more potent against NoV than against poliovirus RdRp.
The mechanism by which each of these compounds inhibited the NoV RdRp was examined using double-reciprocal Lineweaver-Burk plots (Fig. 4). Interestingly, the two larger compounds, NIC02 and NIC04 (365 and 347 Da, respectively), inhibited the NoV RdRp by a mixed mechanism, whereas NIC10 and NIC12 (189 and 233 Da, respectively) had a mode of action that represented uncompetitive inhibition. This suggests that NIC02 and NIC04 bind to both free enzyme and enzyme-substrate complexes, while NIC10 and NIC12 bind only to the enzyme-substrate complex and therefore likely bind to an allosteric pocket that is distinct from the substrate-binding site. Taken together, these findings indicate that NIC02 and NIC04 occupy a different binding pocket(s) of the NoV RdRp than NIC10 and NIC12.
The four compounds demonstrated variable inhibition profiles when examined across a panel of related RdRps in vitro. A broad spectrum of inhibitory activity was observed for NIC02, with similar IC50s across RdRps from different species within the family Caliciviridae (Fig. 5). These results suggest that NIC02 could inhibit the enzymes in a nonspecific manner, e.g., protein reactivity (38). However, the lack of NIC02 inhibitory activity against an RdDp and a DdDp, as well as our subsequent validation of its inhibitory activity in available cell culture models of NoV, demonstrated that it was an inhibitor of the viral RdRp. An explanation for its broad inhibitory activity, therefore, could be that NIC02 binds to a highly conserved RdRp motif. Using the infectious GV.1 MNV system, the antiviral activity of NIC02 was demonstrated both by reduction of plaque size (EC50 = 4.8 μM) (Fig. 7A) and by a reduction in viral genome replication in treated cells (EC50 = 2.3 μM) (Table 1 and Fig. 7C). NIC02 also inhibited the replication of the GI.1 NoV replicon, although it was less potent than MNV, with an EC50 of 30.1 μM (Fig. 6A). It should be noted, however, that NIC02 was the only compound of the four lead hits to display cytotoxicity against Huh-7 cells (CC50 = 134 μM) and RAW 264.7 cells (CC50 = 57.1 μM).
The inhibitory profiles of the remaining three hits were more restricted. NIC10 was significantly less potent against the RdRps of NoV GI.1 and SaV (56- and 11-fold, respectively) than against the GII and MNV RdRps. These findings were consistent with the cell culture observations; NIC10 did not inhibit the replication of the GI.1 replicon in Huh-7 cells but did inhibit MNV in cell culture, with an EC50 of 34.5 μM (Table 1). Interestingly, in the RdRp assays, NIC12 was more potent against GV.1 MNV than all three GII enzymes; however, it demonstrated only modest activity against MNV in cell culture (EC50 = 38.1 μM) (Table 1 and Fig. 7). When examined for inhibitory activity against NoV GI.1, NIC12 was only 2.3-fold less potent than the GII RdRps (Fig. 5) but had no effect on the replication of the GI.1 replicon. The lack of activity of NIC12 in the viral culture systems could be explained by a lack of cell internalization or metabolic instability; these parameters were not measured in the current study. This is further supported by the large difference in lipophilicity of NIC12 (calculated partition coefficient [CLogP] = 0.4) compared to NIC02 and NIC04 (CLogP = 3.6 and 3.5, respectively), which were both active in the viral cell culture models. Finally, NIC04 demonstrated the highest specificity toward GII enzymes, which is consistent with the high EC50 observed with the GI.1 replicon (71.1 μM). However, NIC04 was not active against GV.1 MNV RdRp in the in vitro assays but weakly inhibited MNV replication (EC50 = 32.8 μM).
Only a few studies have reported the development of small compound inhibitors as potential NoV DAAs, targeting two of the viral nonstructural proteins, the NS6 protease and the NS7 RdRp. Using structure-based design approaches, Tiew et al. synthesized a series of peptidyl protease inhibitors (21). Derivatives were later developed from these scaffolds with inhibitory activities (EC50s) between 0.2 and 2.8 μM, as assessed using the NoV GI.1 replicon model. These compounds also had broad antiviral activity, inhibiting homologous proteases of coronaviruses and picornaviruses (20). Two classes of NIs, originally developed for the treatment of other viral infections (22–24), have also demonstrated activity against the NoV RdRp. The nucleoside analogue 2CM, developed as an inhibitor of viruses within the family Flaviviridae, including HCV, has been shown to inhibit NoV replication in vitro (replicon and MNV) with EC50s between 1.3 μM and 18 μM (23, 24). Recently, the antiviral activity of 2CM has also been demonstrated in mice infected with MNV (32). The use of 2CM in humans, however, remains in some doubt, as clinical trials of valopicitabine, an oral prodrug of 2CM, were halted due to gastrointestinal adverse effects in HCV-infected patients. T-705 (favipiravir) is another nucleoside analogue with broad inhibitory activity against viral RdRps, although it was originally developed as an influenza virus inhibitor (39). T-705 was found to inhibit MNV replication (22), but only at high concentrations (EC50 = 250 μM compared to 0.2 μM for influenza virus).
Using available crystal structures, Mastrangelo et al. (25) targeted the active site of the NoV RdRp with an in silico approach to identify inhibitors from a panel of commercially available compounds. Two molecules were identified as potential NoV NNIs: suramin, a drug used in the treatment of sleeping sickness caused by the protozoan Trypanosoma, and its analogue, NF023. These relatively large compounds inhibited the NoV RdRp at nanomolar concentrations, with IC50s of 24.6 nM and 71.5 nM for suramin and NF023, respectively (25). The studies described above have so far been limited to the use of known viral polymerase inhibitors repositioned against a new viral target, the NoV RdRp, or alternative uses for existing drugs. Therefore, no new compounds have yet been explored as potential inhibitors of the NoV RdRp. The identification of the new scaffolds in this study provides a platform for NoV-specific antiviral development. However, current limitations for NoV drug development associated with the lack of a human GII.4 NoV culture system indicate a more difficult pathway to clinical use than that for other viruses that can be easily cultured in vitro. Further screening of the compound library revealed three NIC12-like molecules with increased potency against the NoV GII.4 RdRp compared to the original hit (see Table S1 in the supplemental material). This indicates that NIC12 could represent an attractive scaffold for medicinal chemistry optimization in the search for viral polymerase inhibitors. The examination of the three remaining scaffolds, in contrast, did not reveal any compounds with increased RdRp-inhibitory activity. However, the analysis was limited either by large structural modifications of the hit molecules (NIC04 and NIC10) or by the limited number of chemical analogues in the library (NIC02) (see Table S1 in the supplemental material), and further SAR would be useful to identify more potent derivatives.
In summary, through an HTS approach, we have identified four inhibitors of the NoV polymerase with low micromolar activity. They can be explored for further design efforts to yield potent NNIs with minimal undesirable biological effects. We next intend to determine the RdRp binding sites through mutational and crystallography studies, in order to guide structure-activity relationship analyses for the development of effective NoV therapeutics.
Supplementary Material
ACKNOWLEDGMENTS
We thank Kim Green and Skip Virgin for reagents and Rowena Bull and Chris Marquis for technical assistance.
This work was partially funded by a National Health and Medical Research Council project grant (APP1010327) and an Australian Research Council discovery project grant (DP120104073).
We have no conflicts of interest to disclose.
A.A.E. and P.A.W. conceived and designed the experiments. A.A.E., A.G.K., J.-S.E., and K.L.L. performed the experiments. A.A.E., K.L.L., J.M.M., and P.A.W. analyzed the data. J.M.M. contributed reagents/materials/analysis tools. P.A.W. and J.M.M. obtained funding. A.A.E. and P.A.W. wrote the paper.
Footnotes
Published ahead of print 17 March 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02799-13.
REFERENCES
- 1.Estes MK, Prasad BV, Atmar RL. 2006. Noroviruses everywhere: has something changed? Curr. Opin. Infect. Dis. 19:467–474. 10.1097/01.qco.0000244053.69253.3d [DOI] [PubMed] [Google Scholar]
- 2.Patel MM, Widdowson MA, Glass RI, Akazawa K, Vinje J, Parashar UD. 2008. Systematic literature review of role of noroviruses in sporadic gastroenteritis. Emerg. Infect. Dis. 14:1224–1231. 10.3201/eid1408.071114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tu ET, Bull RA, Greening GE, Hewitt J, Lyon MJ, Marshall JA, McIver CJ, Rawlinson WD, White PA. 2008. Epidemics of gastroenteritis during 2006 were associated with the spread of norovirus GII.4 variants 2006a and 2006b. Clin. Infect. Dis. 46:413–420. 10.1086/525259 [DOI] [PubMed] [Google Scholar]
- 4.Bok K, Green KY. 2012. Norovirus gastroenteritis in immunocompromised patients. N. Engl. J. Med. 367:2126–2132. 10.1056/NEJMra1207742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bull RA, Eden JS, Luciani F, McElroy K, Rawlinson WD, White PA. 2012. Contribution of intra- and interhost dynamics to norovirus evolution. J. Virol. 86:3219–3229. 10.1128/JVI.06712-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoffmann S, Batz MB, Morris JG., Jr 2012. Annual cost of illness and quality-adjusted life year losses in the United States due to 14 foodborne pathogens. J. Food Prot. 75:1292–1302. 10.4315/0362-028X.JFP-11-417 [DOI] [PubMed] [Google Scholar]
- 7.Tu ET, Bull RA, Kim MJ, McIver CJ, Heron L, Rawlinson WD, White PA. 2008. Norovirus excretion in an aged-care setting. J. Clin. Microbiol. 46:2119–2121. 10.1128/JCM.02198-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Green KY, Chanock RM, Kapikan AZ. 2001. Human calicivirus, p 841–874 In Knipe DM, Howley PM. (ed), Fields virology, 4th ed, vol 1 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 9.Jiang X, Wang M, Wang K, Estes MK. 1993. Sequence and genomic organization of Norwalk virus. Virology 195:51–61. 10.1006/viro.1993.1345 [DOI] [PubMed] [Google Scholar]
- 10.Duizer E, Schwab KJ, Neill FH, Atmar RL, Koopmans MP, Estes MK. 2004. Laboratory efforts to cultivate noroviruses. J. Gen. Virol. 85:79–87. 10.1099/vir.0.19478-0 [DOI] [PubMed] [Google Scholar]
- 11.Karst SM, Wobus CE, Lay M, Davidson J, Virgin HW. 2003. STAT1-dependent innate immunity to a Norwalk-like virus. Science 299:1575–1578. 10.1126/science.1077905 [DOI] [PubMed] [Google Scholar]
- 12.Wobus CE, Karst SM, Thackray LB, Chang KO, Sosnovtsev SV, Belliot G, Krug A, Mackenzie JM, Green KY, Virgin HW. 2004. Replication of Norovirus in cell culture reveals a tropism for dendritic cells and macrophages. PLoS Biol. 2:e432. 10.1371/journal.pbio.0020432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taube S, Kolawole AO, Hohne M, Wilkinson JE, Handley SA, Perry JW, Thackray LB, Akkina R, Wobus CE. 2013. A mouse model for human norovirus. MBio 4:e00450–13. 10.1128/mBio.00450-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang K-O, Sosnovtsev SV, Belliot G, King AD, Green KY. 2006. Stable expression of a Norwalk virus RNA replicon in a human hepatoma cell line. Virology 353:463–473. 10.1016/j.virol.2006.06.006 [DOI] [PubMed] [Google Scholar]
- 15.Aghemo A, De Francesco R. 2013. New horizons in hepatitis C antiviral therapy with direct-acting antivirals. Hepatology 58:428–438. 10.1002/hep.26371 [DOI] [PubMed] [Google Scholar]
- 16.Högbom M, Jäger K, Robel I, Unge T, Rohayem J. 2009. The active form of the norovirus RNA-dependent RNA polymerase is a homodimer with cooperative activity. J. Gen. Virol. 90:281–291. 10.1099/vir.0.005629-0 [DOI] [PubMed] [Google Scholar]
- 17.Lee J-H, Alam I, Han KR, Cho S, Shin S, Kang S, Yang JM, Kim KH. 2011. Crystal structures of murine norovirus-1 RNA-dependent RNA polymerase. J. Gen. Virol. 92:1607–1616. 10.1099/vir.0.031104-0 [DOI] [PubMed] [Google Scholar]
- 18.Ng KK-S, Pendás-Franco N, Rojo J, Boga JA, Machín À, Alonso JMM, Parra F. 2004. Crystal structure of Norwalk virus polymerase reveals the carboxyl terminus in the active site cleft. J. Biol. Chem. 279:16638–16645. 10.1074/jbc.M400584200 [DOI] [PubMed] [Google Scholar]
- 19.Rohayem J, Bergmann M, Gebhardt J, Gould E, Tucker P, Mattevi A, Unge T, Hilgenfeld R, Neyts J. 2010. Antiviral strategies to control calicivirus infections. Antiviral Res. 87:162–178. 10.1016/j.antiviral.2010.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim Y, Lovell S, Tiew K-C, Mandadapu SR, Alliston KR, Battaile KP, Groutas WC, Chang K-O. 2012. Broad-spectrum antivirals against 3C or 3C-like proteases of picornaviruses, noroviruses, and coronaviruses. J. Virol. 86:11754–11762. 10.1128/JVI.01348-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tiew K-C, He G, Aravapalli S, Mandadapu SR, Gunnam MR, Alliston KR, Lushington GH, Kim Y, Chang K-O, Groutas WC. 2011. Design, synthesis, and evaluation of inhibitors of Norwalk virus 3C protease. Bioorg. Med. Chem. Lett. 21:5315–5319. 10.1016/j.bmcl.2011.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rocha-Pereira J, Jochmans D, Dallmeier K, Leyssen P, Nascimento MSJ, Neyts J. 2012. Favipiravir (T-705) inhibits in vitro norovirus replication. Biochem. Biophys. Res. Commun. 424:777–780. 10.1016/j.bbrc.2012.07.034 [DOI] [PubMed] [Google Scholar]
- 23.Costantini VP, Whitaker T, Barclay L, Lee D, McBrayer TR, Schinazi RF, Vinje J. 2012. Antiviral activity of nucleoside analogues against norovirus. Antivir. Ther. 17:981–991. 10.3851/IMP2229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rocha-Pereira J, Jochmans D, Dallmeier K, Leyssen P, Cunha R, Costa I, Nascimento MSJ, Neyts J. 2012. Inhibition of norovirus replication by the nucleoside analogue 2′-C-methylcytidine. Biochem. Biophys. Res. Commun. 427:796–800. 10.1016/j.bbrc.2012.10.003 [DOI] [PubMed] [Google Scholar]
- 25.Mastrangelo E, Pezzullo M, Tarantino D, Petazzi R, Germani F, Kramer D, Robel I, Rohayem J, Bolognesi M, Milani M. 2012. Structure-based inhibition of Norovirus RNA-dependent RNA polymerases. J. Mol. Biol. 419:198–210. 10.1016/j.jmb.2012.03.008 [DOI] [PubMed] [Google Scholar]
- 26.Bull RA, Hyde J, Mackenzie JM, Hansman GS, Oka T, Takeda N, White PA. 2011. Comparison of the replication properties of murine and human calicivirus RNA-dependent RNA polymerases. Virus Genes 42:16–27. 10.1007/s11262-010-0535-y [DOI] [PubMed] [Google Scholar]
- 27.Jones LA, Clancy LE, Rawlinson WD, White PA. 2006. High-affinity aptamers to subtype 3a hepatitis C virus polymerase display genotypic specificity. Antimicrob. Agents Chemother. 50:3019–3027. 10.1128/AAC.01603-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739. 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eltahla AA, Lackovic K, Marquis C, Eden JS, White PA. 2013. A fluorescence-based high-throughput screen to identify small compound inhibitors of the genotype 3a hepatitis C virus RNA polymerase. J. Biomol. Screen. 18:1027–1034. 10.1177/1087057113489883 [DOI] [PubMed] [Google Scholar]
- 30.Eden JS, Bull RA, Tu E, McIver CJ, Lyon MJ, Marshall JA, Smith DW, Musto J, Rawlinson WD, White PA. 2010. Norovirus GII.4 variant 2006b caused epidemics of acute gastroenteritis in Australia during 2007 and 2008. J. Clin. Virol. 49:265–271. 10.1016/j.jcv.2010.09.001 [DOI] [PubMed] [Google Scholar]
- 31.McCartney SA, Thackray LB, Gitlin L, Gilfillan S, Virgin HW, Colonna M. 2008. MDA-5 recognition of a murine norovirus. PLoS Pathog. 4:e1000108. 10.1371/journal.ppat.1000108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rocha-Pereira J, Jochmans D, Debing Y, Verbeken E, Nascimento MS, Neyts J. 2013. The viral polymerase inhibitor 2′-C-methylcytidine inhibits Norwalk virus replication and protects against norovirus-induced diarrhea and mortality in a mouse model. J. Virol. 87:11798–11805. 10.1128/JVI.02064-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rocha-Pereira J, Cunha R, Pinto DC, Silva AM, Nascimento MS. 2010. (E)-2-styrylchromones as potential anti-norovirus agents. Bioorg. Med. Chem. 18:4195–4201. 10.1016/j.bmc.2010.05.006 [DOI] [PubMed] [Google Scholar]
- 34.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9:671–675. 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sykes ML, Baell JB, Kaiser M, Chatelain E, Moawad SR, Ganame D, Ioset J-R, Avery VM. 2012. Identification of compounds with anti-proliferative activity against Trypanosoma brucei brucei strain 427 by a whole cell viability based HTS campaign. PLoS Negl. Trop. Dis. 6:e1896. 10.1371/journal.pntd.0001896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang JH, Chung TDY, Oldenburg KR. 1999. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 4:67–73. 10.1177/108705719900400206 [DOI] [PubMed] [Google Scholar]
- 37.Campagnola G, Gong P, Peersen OB. 2011. High-throughput screening identification of poliovirus RNA-dependent RNA polymerase inhibitors. Antiviral Res. 91:241–251. 10.1016/j.antiviral.2011.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baell JB, Holloway GA. 2010. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 53:2719–2740. 10.1021/jm901137j [DOI] [PubMed] [Google Scholar]
- 39.Furuta Y, Takahashi K, Fukuda Y, Kuno M, Kamiyama T, Kozaki K, Nomura N, Egawa H, Minami S, Watanabe Y, Narita H, Shiraki K. 2002. In vitro and in vivo activities of anti-influenza virus compound T-705. Antimicrob. Agents Chemother. 46:977–981. 10.1128/AAC.46.4.977-981.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.