

Abstract
We report here the synthesis of 2-aminothiazolones along with their biological properties as novel anti-HIV agents. Such compounds have proven to act through the inhibition of the gp120-CD4 protein-protein interaction that occurs at the very early stage of the HIV-1 entry process. No cytotoxicity was found for these compounds, and broad antiviral activities against laboratory strains and pseudotyped viruses were documented. Docking simulations have also been applied to predict the mechanism, at the molecular level, by which the inhibitors were able to interact within the Phe43 cavity of HIV-1 gp120. Furthermore, a preliminary absorption, distribution, metabolism, and excretion (ADME) evaluation was performed. Overall, this study led the basis for the development of more potent HIV entry inhibitors.
INTRODUCTION
HIV-1 infection continues to be a major health threat worldwide. Approximately 40 million individuals are living with HIV-1, and new infections occur every year. Drug discovery and development have transformed HIV-1 infection into a chronic condition that can be controlled for many years through combination therapies with different classes of antiretroviral drugs, known as highly active antiretroviral therapy (HAART) (1). However, the need for lifelong use of HAART and the emergence of resistance to these drugs underscore the need to develop newer inhibitors with reduced toxicity and improved activity and resistance profiles (2–4). The attachment of HIV to host cells occurs via binding of the HIV envelope glycoprotein gp120 to the host CD4 receptor; thus, the inhibition of this protein-protein interaction offers an effective target in the development of new antiretroviral agents (5, 6). The crystal structure of gp120 bound to CD4 and the 17b Fab fragment antibody was solved in 1998 (7), revealing that the CD4 binding site of the HIV-1 gp120 envelope consists of a hydrophobic pocket capped by the CD4 Phe43, thus termed the Phe43 cavity. Research into gp120 inhibitors that are able to block the complex formation between gp120 and CD4 has received increasing attention in recent years and has led to the discovery of active small molecules characterized by a high degree of chemical diversity. BMS-378806 (BMS-806) and the related compounds 155 and BMS-488043, discovered through a cell-based screening assay, are nanomolar inhibitors that prevent the binding of gp120 to CD4 receptors (8–10). NBD-556 and NBD-557, first discovered by Zhao et al. (11) by using an HIV syncytium formation assay on a small library of 33,000 compounds, have been shown to compete with CD4 binding and possess low micromolar potency against several strains of HIV (11, 12). Interestingly, the crystal structure of NBD-556 in complex with gp120 was recently deposited in the Protein Data Bank (PDB code 3TGS), highlighting the binding mode of the compound within the Phe43 cavity of gp120 (13). NBD-556 analogues were then developed to study the structure-activity relationship (SAR) (14–17). Furthermore, molecular modeling techniques were successfully applied in the identification of new gp120-CD4 inhibitors (18). In this regard, our research group recently reported the successful application of different virtual screening approaches to the discovery of the hit compounds 1 to 6 (19, 20). These compounds showed micromolar inhibition of HIV-1 replication in cells infected by wild-type virus but were totally inactive toward the mutant Met475Ile, thus confirming that they target the CD4 binding site on HIV-1 gp120, as residue 475 belongs to the Phe43 cavity. On the other hand, the 2-aminothiazolone derivatives represent a versatile scaffold widely used in medicinal chemistry. Compounds containing the 2-aminothiazolone nucleus have been found to exhibit a broad spectrum of biological activities, such as antitumor (5-(2,4-dihydroxybenzylidene)-2-(phenylimino)-1,3-thiazolidin [DBPT]) (21), herbicidal (compound 7), and αvβ3 receptor antagonist activities (compound 8) (22). The 2-aminothiazolone derivatives with general formula 9 were recently assayed by us as HIV-1 integrase inhibitors, and they were found to be only moderately active (23).
Here, novel 2-aminothiazolones were synthesized and biologically tested in order to investigate their potential to inhibit HIV infection. The compounds were found to be able to inhibit the HIV replication at a micromolar/submicromolar concentration in vitro and were found to be nontoxic and endowed with a high genetic barrier to the development of resistance at least in vitro. Experiments demonstrated that the compounds acted as early inhibitors of the gp120-CD4 interaction. In line with these results, docking studies were performed next to elucidate the binding mode of the 2-aminothiazolones within the Phe43 cavity of gp120 and to identify amino acids involved in their mechanism of action. To the best of our knowledge, this is the first time that 2-aminothiazolone derivatives were investigated as antiretroviral agents targeting the gp120-CD4 interaction.
MATERIALS AND METHODS
Chemistry. (i) General information.
All commercially available reagents were used as purchased. Anhydrous reactions were run under a positive pressure of dry N2. Thin-layer chromatography (TLC) was carried out using Merck silica gel 60 F254 TLC plates. Chromatographic purifications were performed on columns packed with Merck 60 silica gel, 23-400 mesh, for the flash technique. 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded at 400 MHz on a Bruker Avance DPX 400 spectrometer. Chemical shifts are reported relative to (CH3)4Si at a δ value of 0.00 ppm (see supplemental material). Melting points were measured using a Gallenkamp melting point apparatus and are uncorrected. Elemental analyses were performed on a PerkinElmer PE 2004 elemental analyzer, and the data for C, H, and N are within 0.4% of the theoretical values. Enfuvirtide (ENF or T20) and azidothymidine (AZT) were obtained from the NIH AIDS reagents repository (https://www.aidsreagent.org), and maraviroc (MVC) and dolutegravir were obtained from ViiV Healthcare.
The final compounds 16a to 16l were obtained by starting from the commercially available methyl-4-iodosalycilate compound 10, which was used in a Suzuki coupling with the 5-formyl-2-furanylboronic acid compound 11 (Sigma-Aldrich SRL) (see the supplemental material). The coupled product 12 was then hydrolyzed with an aqueous solution of sodium hydroxide in MeOH-tetrahydrofuran (THF) to give, after 12 h, the free acid analogue 13. Next, to generate compounds with general structure 16 a-l, we adapted a recently published synthetic strategy that describes a one-pot multicomponent microwave-assisted reaction that allows the desired final compounds to be obtained directly. The methodology involves 4-(5-formylfuran-2-yl)-2-hydroxybenzoic acid, rhodanine, and primary amines that catalyze a Knoevenagel condensation between the aldehyde and rhodanine and then act as nucleophiles in the displacement of the thiocarbonyl sulfur. Using very short reaction times and an easy workup, this method allows one to obtain several final compounds by changing only the amines employed. We used 4-chloro-3-fluoro-benzylamine, 4-chloro-benzylamine, 3,4-dichloro-benzylamine, 3-fluoro-benzylamine, 4-fluoro-benzylamine, 3-chloro-4-fluoroaniline, 4-chloro-3-fluoroaniline, 4-fluorophenethylamine, 4-ethylphenethylamine, and phenethylamine. After dissolving all reagents in ethyl alcohol (EtOH), heating at 150°C for 20 min by microwave irradiation was required to complete the reaction. After acidic workup, the desired products were collected by filtration from ethanol (Fig. 1A). Compound 16m was obtained through a Suzuki reaction from commercial reagents 17 and 11. The coupled compound 18 reacts with rhodanine and 4-fluorophenethylamine, as described above (Fig. 1B).
FIG 1.

(A) Synthesis of aminothiazolones 16a to 16l. Step i, Pd(PPh3)2Cl2, Na2CO3, dimethylformamide (DMF)-EtOH, occurring at room temperature for 1 h; step ii, 1 N NaOH and MeOH-THF, with reflux, overnight; step iii, EtOH at 150°C in microwave (MW), 20 min. (B) Synthesis of the aminothiazolone 16m. Step i, Pd(PPh3)2Cl2, Na2CO3, DMF-EtOH at room temperature for 1 h; ii, EtOH at 150°C, MW, 20 min.
(ii) HPLC and MS analysis.
The purity of the tested compounds was assessed by reverse-phase liquid chromatography and a mass spectrometer (Agilent series 1100 liquid chromatogram/mass selective detector [LC/MSD]) equipped with a UV detector (λ, 254 nm) and an electrospray ionization (ESI) source. The LC elution method (using a Zorbax Eclipse XDB, 4.6 by 150 mm, 5-μm C8 column) involved the following for compounds 16a to 16m: the temperature was 25°C, in mobile phase, and was composed of 70% CH3CN and 30% H2O with 0.5% formic acid at a flow rate of 1.0 ml · min−1 (all solvents were high-performance liquid chromatography [HPLC] grade; Sigma-Aldrich). All analyzed compounds met ≥95% of the purity criteria. The mass spectrometry (MS) data were obtained using an Agilent 1100 LC/MSD VL system (G1946C), with a 0.4-ml · min−1 flow rate using a binary solvent system of a 95:5 ratio of CH3OH to H2O. UV detection was monitored at a λ of 254 nm. Mass spectra were acquired in negative mode scanning over the mass range of 50 to 1,500 Da. The following ion source parameters were used: drying gas flow, 9 ml · min−1; nebulizer pressure, 40 pound-force per square inch gauge (psig); and drying gas temperature, 350°C.
(iii) Microwave irradiation experiments.
Microwave reactions were conducted using a CEM Discover synthesis unit (CEM Corp., Matthews, NC, USA). The instrument consists of a continuous focused microwave power delivery system with operator-selectable power output from 0 to 300 W. The temperatures of the contents of the vessel were monitored with a calibrated infrared (IR) temperature control mounted under the reaction vessel. All experiments were performed using a stirring option, whereby the contents of the vessel were stirred by a rotating magnetic plate located below the floor of the microwave cavity and a Teflon-coated magnetic stir bar in the vessel.
Antiviral activity and cell toxicity. (i) Phenotypic analyses with fully replicating recombinant HIV-1 strains.
The human TZM-bl indicator cell line was obtained from the American Type Culture Collection (Manassas, VA) and maintained at 37°C and 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) medium containing 10% fetal bovine serum, 50 μg/ml penicillin, and 50 μg/ml streptomycin. The HIV-1 viruses NL(AD8) and NL4.3 were titrated as follows: serial 5-fold dilutions of each virus were made in quadruplicate wells in 96-well culture plates, in a total volume of 100 μl of growth medium, for a total of 8 dilution steps. Freshly trypsinized cells (20,000 cells in 100 μl of growth medium containing 75 μg/ml DEAE-dextran) were added to each well, and the plates were incubated at 37°C in a humidified 5% CO2-95% air environment. After 48 h of incubation, the medium was removed and viral infection was quantified using a β-galactosidase (CPRG) assay (Roche). Twenty thousand TZM-bl cells/well were seeded in 96-well plates in complete DMEM supplemented with 30 μg/ml DEAE-dextran (Sigma-Aldrich). Three hundred times the 50% tissue culture infective dose (TCID50)/ml of each strain was pretreated for 1 h at 37°C with six serial dilutions (range, 20.000 nM to 6.4 nM) of each compound and then added to the cells, as described previously (24, 25). Vehicle (0.1% dimethyl sulfoxide [DMSO])-treated cells served as a negative control. A CCR5 inhibitor (maraviroc) and an integrase inhibitor (dolutegravir) were used as positive-control drugs. After 2 days, viral infection was quantified using a CPRG assay (Roche). The inhibitory curves were fitted by nonlinear regression, allowing for the calculation of the 50% inhibitory concentration (IC50) using the Prism software. To evaluate the cell toxicity of the compounds, the metabolic XTT [2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide] test (Sigma-Aldrich) was performed according to the manufacturer's instructions.
(ii) Phenotypic analyses with pseudotyped viruses and compounds 16h and 16l.
Pseudoviruses were prepared by transfecting exponentially dividing HEK 293T cells (5 × 106 cells in 15 ml growth medium in a T-75 culture flask) with 5 μg of each env-expression plasmid (from the tier 2 panel, QH0692.42, SC422661.8, TRO.11, AC10.0.29, RHPA4259.7, REJO4541.67, WITO4160.33, and CAAN5342.A2; from the tier 3 panel, PVO.4 and TRJO4551.58) and 10 μg of an env-deficient HIV-1 backbone vector (pSG3ΔEnv) obtained from the AIDS reagents repository, using FuGENE 6 reagent (Roche), as previously described (26, 27). All the envelopes were from subtype B viruses and amplified from R5-tropic viruses during acute infection (28). Pseudovirus-containing culture supernatants were harvested 2 days after transfection, filtered (0.45 μm pore size), and stored at −80°C in 1-ml aliquots. After pseudovirus titration on TZM-bl cells, 40,000 TZM-bl cells/well were seeded in 96-well plates in complete DMEM supplemented with 30 μg/ml DEAE-dextran (Sigma-Aldrich). Three hundred times the TCID50/ml of each pseudovirus was pretreated for 1 h at 37°C with six serial dilutions (range, 20.000 nM to 6.4 nM) of compound 16l or 16h and then added to the cells. Vehicle (0.1% DMSO)-treated cells served as a negative control. A CCR5 inhibitor (maraviroc) was used as a positive-control drug. After 2 days, viral infection was quantified using a CPRG assay (Roche). The inhibitory curves were fitted by nonlinear regression, allowing for the calculation of the IC50 using the Prism software.
Time-of-addition assay.
A time-of-addition experiment was performed as previously described, with minor modifications (29), by using a single-cycle assay and the pseudotype virus REJO4541 clone 67. For the time-of-addition assay, 40.000 TZM-bl cells/well in a 96-well plate were infected with 1,500× TCID50/ml of the env-pseudotyped HIV-1 virus in complete medium supplemented with 30 μg/ml DEAE-dextran (Sigma-Aldrich). Virus was incubated with cells for 1 h at 4°C, and unbound virus was subsequently removed by extensive and repeated washing with phosphate-buffered saline (PBS) to synchronize the replication (29). To analyze the very early steps of infection, we performed a preincubation step with the virus and a representative compound (16l) (using IgGb12 obtained from the AIDS reagents repository as a comparison) for 1 h at 37°C, prior to cell infection. For the following 4 h, antiretroviral compounds inhibiting distinct viral replication steps (IgGb12, maraviroc, T20, AZT, and dolutegravir) and compound 16l were added at time zero and after 60, 75, 90, 120, 150, 180, 210, and 240 min. To ensure the complete inhibition of viral replication occurred, we used a 40-fold IC50, as previously evaluated for each compound on TZM-bl cells (7.5 μg/ml IgGb12, 0.7 μM maraviroc, 1.6 μM T20, 3.2 μM AZT, 1 μM dolutegravir, and 5 μM 16l). β-Galactosidase expression in cell lysates 48 h postinfection was used as a marker of HIV infection and was normalized to untreated control cells.
gp120-CD4-His ELISA binding assay.
Competition between 2-aminothiazolone derivatives and CD4 was determined through a gp120-CD4-6His tag binding enzyme-linked immunosorbent assay (ELISA), as previously described, with minor modifications (9, 11). Briefly, 96-well plates (Corning) were coated overnight at room temperature with 100 μl of sheep anti-gp120 antibody D7324 (Aalto Bio Reagents, Dublin, Ireland) at 5 μg/ml in 100 mM NaHCO3. The wells were blocked with 1% nonfat milk in PBS at 37°C for 1 h. The coated plates were incubated with 0.5 μg/ml recombinant gp120 (Abcam) in PBS at 37°C for 1 h and washed three times with 0.1% PBS-Tween (PBS-T). Soluble CD4 6His-tagged (Life Technologies) at 0.20 μg/ml in PBS and equal volumes of the tested compound at six different concentrations (50 μM to 1.5 μM) were added to the wells and incubated at 37°C for 1 h. Vehicle (DMSO) was used as a negative control, and IgGb12 was used as positive control. The wells were washed five times with 0.1% PBS-T and incubated for an hour at 37°C with a goat anti-His antibody (Roche) at a dilution of 1:1,000 in PBS-0.5% milk. After five washes with 0.1% PBS-T, 3,3′,5,5′-tetramethylbenzidine (TMB) chromogenic substrate for peroxidase (Pierce) was added, and the absorbance at 450 nm was read.
Computational studies. (i) Protein preparation.
The HIV-1 gp120 three-dimensional coordinates were extracted from the crystallographic complex (PDB code 1G9M) (7) and were energy minimized to remove unfavorable contacts through the all-atom optimized potentials for liquid simulations (OPLS) force field and the Polak-Ribière conjugate gradient method. A continuum solvation method, with water as the solvent, was also applied. Extended cutoffs were used, and convergence was set to 0.05 kJ/mol · Å. The homology models of HIV-1 gp120 for NL4.3 (CXCR4-tropic strain) and AD8 (CCR5-tropic strain) were performed with the software PRIME, using two crystallographic complexes (PDB codes 2B4C [30] and 1G9M) as a template and the primary sequences of the two strains as queries. The obtained models were energy minimized by the software MacroModel from the Schrödinger suite, using the Polak-Ribière conjugate gradient algorithm and OPLS_2005 force field (Schrödinger).
Docking studies.
Compounds were built using the Maestro 9.0 graphical interface and modeled using the Merck molecular force field (MMFF) in Gibbs born-surface area (GB/SA) water, as implemented in MacroModel (Schrödinger). Docking studies were performed using the program GOLD (version 5.1) (31). The ChemScore was chosen as the fitness function. The genetic algorithm (GA) parameter settings of GOLD were employed with the search efficiency set to 100%. Finally, results differing <1.5 Å in ligand all-atom root mean square deviation (RMSD) were clustered together. For each inhibitor, the first ranked solutions and the lowest energy conformation of the most populated cluster were analyzed. Pictures of the modeled ligand-enzyme complexes, together with graphic manipulations, were rendered with the PyMOL package (version 1.2r3pre [http://www.pymol.org/]).
(ii) Molecular dynamics.
Molecular dynamic (MD) simulations were performed using the AMBER 12 suite of programs (http://ambermd.org/) and the ff03.r1 force field. An appropriate number of counterions (7 Cl− ions for NL4.3 and 3 Cl− ions for AD8 complex) were added to neutralize the system, and the complexes were placed in an octagonal box of TIP3P water molecules. The distance between the box walls and the protein was set to 10 Å. MD runs were carried out with a previously validated protocol (32). Before MD simulation, two stages of energy minimization were performed to remove bad contacts. In first stage, we kept the protein fixed with a constraint of 500 kcal/mol and we minimized the positions of the water molecules. In the second stage, we minimized the entire system, applying a constraint of 10 kcal/mol on the alpha carbons. MD trajectories were run using the minimized structure as the starting input. Constant-volume simulations were performed for 50 ps, during which time the temperature was raised from 0 to 300 K using the Langevin dynamics method. Next, 150 ps of constant-pressure MD simulations were performed at 300 K in three steps of 50 ps each. During the three periods of this second stage, the alpha carbons were blocked with harmonic force constants of 10, 5, and 1 kcal/mol · Å for steps 1, 2, and 3, respectively. Finally, a 15-ns MD simulation without restraint was run at a constant temperature of 300 K and a constant pressure of 1 atm.
ADME assay. (i) Chemicals.
All solvents and reagents were from Sigma-Aldrich SRL (Milan, Italy). Dodecane was purchased from Fluka (Milan, Italy). Pooled 20-mg/ml human liver microsomes (HLM) from male donors were from BD Gentest-Biosciences (San Jose, CA). Milli-Q quality water (Millipore, Milford, MA, USA) was used. Hydrophobic filter plates (MultiScreen-IP clear plates, 0.45-μm-diameter pore size), 96-well microplates, and 96-well UV-transparent microplates were obtained from Millipore (Bedford, MA, USA).
(ii) Parallel artificial membrane permeability assay.
Donor solution (0.5 mM) was prepared by diluting 1 mM dimethyl sulfoxide (DMSO) compound stock solution using 0.025 M phosphate buffer (pH 7.4). The filters were coated with 5 μl of a 1% (wt/vol) dodecane solution of phosphatidylcholine. Donor solution (150 μl) was added to each well of the filter plate. Three hundred microliters of solution (50% DMSO in phosphate buffer) was added to each well of the acceptor plate. All compounds were tested in three different plates on different days. The sandwich was incubated for 5 h at room temperature with gentle shaking. After the incubation time, the plates were separated and samples were taken from both the receiver and donor sides and analyzed using LC with UV detection at 280 nm. LC analysis was performed with a PerkinElmer (series 200) instrument equipped with an UV detector (PerkinElmer 785A UV/visible detector). Chromatographic separation was conducted using a Polaris C18 column (150 to 4.6 mm, 5-μm particle size) at a flow rate of 0.8 ml · min−1, with a mobile phase composed of 50% acetonitrile (ACN), 50% H2O, and 0.1% formic acid for all compounds. Permeability (Papp) for the parallel artificial membrane permeability assay (PAMPA) was calculated according to the following equation (equation 1), obtained from the Wohnsland and Faller and Sugano et al. equations (33, 34), with some modifications in order to obtain permeability values in cm · s−1,
| (1) |
where VA is the volume in the acceptor well, VD is the volume in the donor well (cm3), A is the effective area of the membrane (cm2), t is the incubation time(s), and r is the ratio between the drug concentration in the acceptor and the equilibrium concentration of the drug in the total volume (VD + VA). The drug concentration was estimated by using the peak area integration. The percent membrane retention was calculated according to equation 2,
| (2) |
where r is the ratio between drug concentration in the acceptor and equilibrium concentration, and D, A, and Eq represent the drug concentrations in the donor, acceptor and equilibrium solution, respectively.
(iii) Water solubility assay.
One milligram of each solid compound was added to 1 ml of water. The samples were shaken in a shaker bath at room temperature for 24 to 36 h. The suspensions were filtered through a 0.45-μm nylon filter (Acrodisc), and the solubilized compound was determined by LC-MS/MS assay. For each compound, the determination was performed in triplicate. For the quantification, we used an LC-MS system consisted of a Varian apparatus (Varian, Inc.), including a vacuum solvent degassing unit, two pumps (212-LC), a triple quadrupole MSD (model 320-LC) mass spectrometer with electrospray (ES) interface, and Varian MS workstation system control version 6.9 software. Chromatographic separation was obtained using a Pursuit C18 column (50 by 2.0 mm) (Varian) with 3-μm particle size and gradient elution, with eluent A being ACN and eluent B consisting of an aqueous solution of 0.1% formic acid. The analysis started with 0% of eluent A, which was linearly increased to 70% in 10 min and then slowly increased to 98% in 15 min. The flow rate was 0.3 ml/min and the injection volume was 5 μl. The instrument operated in negative mode with the following parameters: detector, 1,850 V; drying gas pressure, 25.0 lb/in2; desolvation temperature, 300.0°C; nebulizing gas, 45.0 lb/in2; needle, 5,000 V; and shield, 600 V. Air and nitrogen were used as the nebulizer and drying gases, respectively. Collision-induced dissociation was performed using argon as the collision gas at a pressure of 1.8 mtorr in the collision cell. The transitions, as well as the capillary voltage and the collision energy used for compounds 16h and 16l, are summarized in Table 1. Quantification of the single compound was made by a comparison with apposite calibration curves realized with standard solutions in methanol.
TABLE 1.
Chromatographic and MS parameters of selected compounds
| Compound | Monitored transition (m/z) | Capillary voltage (V) | Collision energy (eV) | tRa (min) |
|---|---|---|---|---|
| 16h | 214.6 | 23.5 | −100 | 12.1 |
| 433.2 | 21.0 | |||
| 16l | 214.6 | 24.5 | −40 | 12.8 |
| 417.2 | 20.5 |
tR, retention time.
(iv) Microsomal stability assay.
Each compound was incubated in the DMSO solution at 37°C for 60 min in 125 mM phosphate buffer (pH 7.4) and 5 μl of human liver microsomal protein (0.2 mg · ml−1), in the presence of a NADPH-generating system, at a final volume of 0.5 ml (final compound concentration, 50 μM); DMSO did not exceed 2% concentration in the final solution. The reaction was stopped by cooling in ice and adding 1.0 ml of acetonitrile. The reaction mixtures were then centrifuged, and the parent drug and metabolites were subsequently determined by LC-UV-MS. Chromatographic analysis was performed with an Agilent 1100 LC/MSD VL system (G1946C) (Agilent Technologies, Palo Alto, CA), constituted by a vacuum solvent degassing unit, a binary high-pressure gradient pump, an 1100 series UV detector, and an 1100 MSD model VL benchtop mass spectrometer. Chromatographic separation was obtained using a Varian Polaris C18-A column (150 to 4.6 mm, 5 μm particle size) and gradient elution, with eluent A being ACN and eluent B consisting of an aqueous solution of 0.1% formic acid. The analysis started with 2% of eluent A, which was rapidly increased to 70% in 12 min and then slowly increased to 98% in 20 min. The flow rate was 0.8 ml · min−1 and the injection volume was 20 μl. The Agilent 1100 series mass spectra detection (MSD) single quadrupole instrument was equipped with the orthogonal spray API-ES (Agilent Technologies, Palo Alto, CA). Nitrogen was used as nebulizing and drying gas. The pressure of the nebulizing gas, the flow of the drying gas, the capillary voltage, the fragmentor voltage, and the vaporization temperature were set at 40 lb/in2, 9 liter · min−1, 3,000 V, 70 V, and 350°C, respectively. UV detection was monitored at 280 nm. The LC-ESI-MS determination was performed by operating the MSD in the negative ion mode. Spectra were acquired over the scan range m/z 100 to 1,500 using a step size of 0.1 atomic mass units (u). The percentage of unmetabolized compound was calculated by a comparison with reference solutions.
RESULTS
Inhibition of HIV-1 in cell culture.
All the synthesized compounds were tested in vitro to evaluate their ability to inhibit HIV replication in human TZM-bl cells infected with HIV-1 NL4.3 (a CXCR4-tropic strain) or AD8 (a CCR5-tropic strain), and the biological results are listed in Table 2 together with the toxicity data. Remarkably, all compounds showed low micromolar/submicromolar activities, with the exception of compound 16m, which was inactive. With 16m being the only compound in this series that does not contain salicylic moiety, we can argue that this group of compounds is fundamental for the anti-HIV activity of the 2-aminothiazolone derivatives under study. Furthermore, a substituent-dependent effect was observed, as the best results were obtained when phenylethylamines were introduced on the thiazolone ring (16h to 16l), while the substitution with shorter chains, such as benzylamines (16a to 16e) or phenylamines (16f and 16g), determined a small reduction of the inhibitory potency against HIV-1.
TABLE 2.
Structure, anti-HIV activity, and ADME properties of compounds 16a to 16m
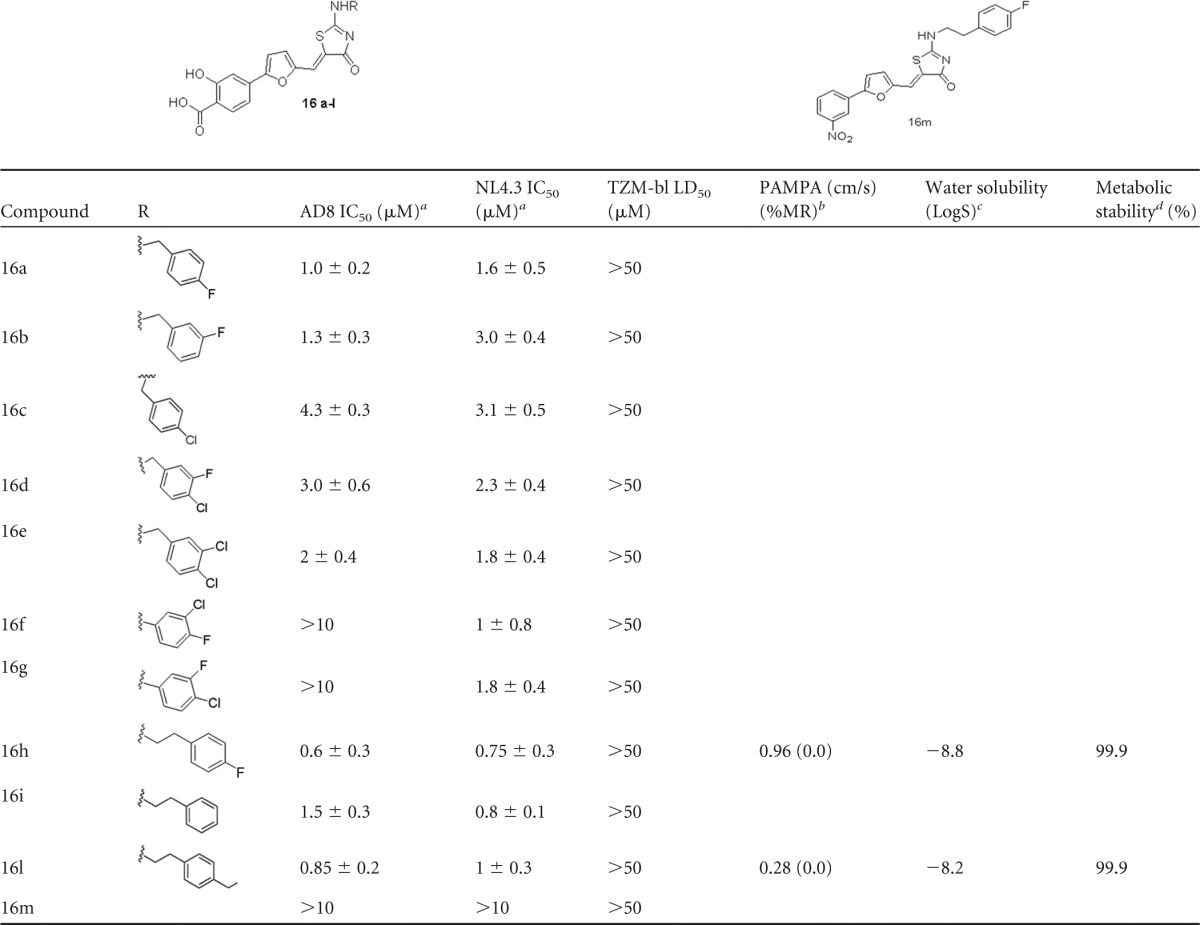
Values are expressed as mean ± SD. The experiments were repeated three times in triplicate.
PAMPA calculated as Papp × 10−6 cm/s, see Materials and Methods for details; %MR, percent membrane retention.
LogS = log mol · liter−1.
Expressed as percentage of unmodified parent.
When compounds 16h and 16l were tested against tier 2 and tier 3 pseudotyped viruses (all subtype B strains), both compounds showed a similar but not fully overlapping spectrum of neutralization (Table 3). A vesicular stomatitis virus protein G (VSV-G) control pseudovirus was also used in parallel, but none of the compounds showed any inhibitory effect against it. The reference compound maraviroc showed an IC50 of 19 ± 5 nM (mean ± standard deviation [SD]) with all R5 strains (data not shown). Noteworthy, no toxicity was observed for the tested compounds (i.e., 50% lethal dose [LD50] > 50 μM). Further modifications aiming at improving these results for the most active compounds, 16h to 16l, are in progress.
TABLE 3.
Activities of compounds 16h and 16l on HIV-1 laboratory strains (AD8 and NL4.3) and on tier 2 and 3 pseudovirusesa
| Compound | IC50 (mean ± SD) (μM) for: |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AD8 | NL4.3 | Pseudoviruses |
||||||||||
| QH0692.42 | SC422661.8 | PVO.4 | TRO.11 | AC10.0.29 | RHPA4259.7 | REJO4541.67 | TRJO4551.58 | WITO4160.33 | CAAN5342.A2 | |||
| 16h | 0.6 ± 0.3 | 0.75 ± 0.3 | 2.9 ± 0.3 | 1.5 ± 0.2 | 1.5 ± 0.4 | 7.7 ± 1.3 | 1 ± 0.3 | >10 | 1.5 ± 0.4 | 3.9 ± 0.2 | 3.9 ± 0.6 | 5.6 ± 0.3 |
| 16l | 0.8 ± 0.2 | 1 ± 0.3 | 4.1 ± 1.3 | 4.1 ± 1.3 | 1.3 ± 0.3 | 8.7 ± 1.4 | 1 ± 0.4 | >10 | 1.5 ± 0.5 | 3.3 ± 0.2 | 5 ± 0.1 | 5.6 ± 1.3 |
The IC50 values in μM (±SD) of dolutegravir on laboratory strains was 4 ± 3 nM; the IC50 of maraviroc on pseudoviruses was 19 ± 5 nM IC50. The experiments were repeated three times in triplicate.
Mechanism-of-action studies and time of intervention and gp120-CD4 binding assay.
A time-of-addition experiment was carried out to determine the exact inhibition target of the 2-aminothiazolones. This experiment determines how long the addition of an anti-HIV compound can be postponed within the viral replication cycle before losing its antiviral activity. Reference compounds with a known mode of action were included. IgGb12 is an anti-gp120 antibody, maraviroc is a CCR5-receptor antagonist, and T20 is a fusion inhibitor that acts by binding to the envelope glycoprotein gp41. The nucleoside analogue AZT inhibits the reverse transcription process, while dolutegravir is an inhibitor of the integration process. The profile of IgGb12 was the same as that seen with 16l (Fig. 2). These data confirmed gp120 to be a target of interaction of 16l.
FIG 2.

Time-of-addition assay. The target of the antiviral compound 16l was identified by comparing its activity in the time scale to those of reference drugs. In the assay, a panel of reference drugs sequentially targeting distinct replication steps of HIV-1 from entry to the integration into cell chromosome was used (shown as bars at the bottom): IgGb12 (an anti-gp120 antibody that binds the CD4-binding site), maraviroc (a CCR5 coreceptor inhibitor), enfuvirtide (T20, a fusion inhibitor), azidothymidine (AZT) (a reverse transcriptase inhibitor), and dolutegravir (DTG) (an integrase inhibitor). For compound 16l, the efficacy was at its maximum only when the virus was pretreated as for IgGb12, and it was ineffective once the virus attached the CD4 cell surface receptor.
The gp120-CD4 binding assay demonstrated a direct and dose-dependent inhibition by compounds 16h and 16l (IC50 for both, 20 μM) of the interaction between a dual tropic gp120 that was used in the assay and soluble CD4 (Fig. 3).
FIG 3.
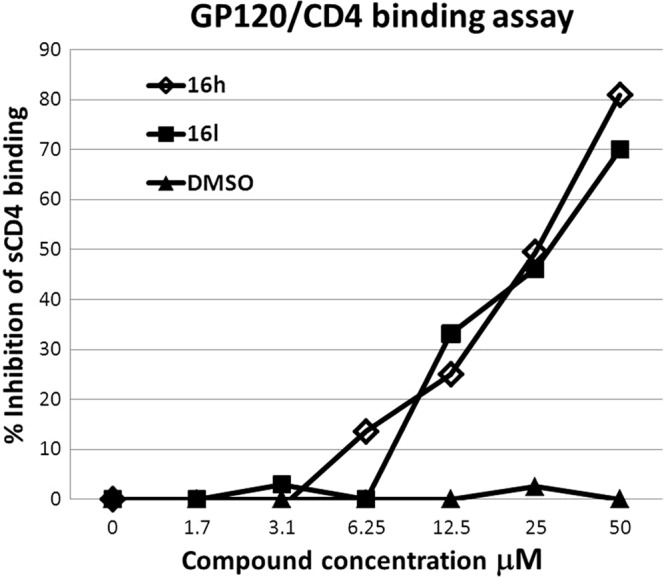
gp120-CD4-His tag binding assay. Two representative 2-aminothiazolone derivative molecules, 16h and 16l, were tested in a competitive ELISA to demonstrate that gp120 binding to CD4 was the target of this class of compounds. Both compounds were able to displace soluble CD4 from a dual tropic gp120 in a dose-dependent manner (IC50 for both compounds, 20 μM). DMSO was also tested at the same concentrations present at each compound dilution. The experiments were repeated twice in triplicate.
Molecular modeling.
Compounds 16a to 16m were subjected to docking analysis into a refined gp120 core structure (PDB code 1G9M) (7) by focusing calculations on the Phe43 cavity and its surrounding residues. An analysis of the docked complexes suggested that 2-aminothiazolone derivatives made some of the interactions previously identified as crucial for the activity of gp120-CD4 small-molecule inhibitors. In detail, the aromatic side chains of the amines at the 2 position were deeply embedded at the bottom of the cavity where established hydrophobic interactions with residues Phe382, Met475, Val255, Trp427, Trp112, Ile424, and Tyr384, and a hydrogen bond was present between the NH and the backbone carbonyl oxygen of Gly473. The thiazolone ring interacts with Ile371, Gly473, and Glu370 by filling the region occupied by the CD4 residue Phe43 in the gp120-CD4 complex. Finally, the salicylic ring was involved in polar contacts with an arginine residue at position 476 of gp120 in a solvent-exposed area. As an example, Fig. 4A shows the predicted binding mode of compound 16h. The alignment between the pose predicted for compound 16h and the binding mode of the NBD-based derivative DMJ-II-121 within the Phe43 cavity (PDB code 4I54) highlights common interactions between the two inhibitors, such as the hydrogen bond with Gly473 and the hydrophobic contacts with Trp112, Phe382, Tyr384, and Ile424. Conversely, the interaction with Arg476 is exclusively observed for compound 16h (Fig. 4B). A noteworthy finding is that compounds 16f and 16g were active against HIV-1 NL4.3 but not against AD8-infected cells. In order to investigate this peculiar behavior, two homology models were built using the primary sequence of the NL4.3 and AD8 strains. An initial analysis of the two three-dimensional (3D) structures showed no differences in the Phe43 binding sites. HIV-1 NL4.3 and AD8 strains differ in their ability to utilize either CCR5 or CXCR4 as a coreceptor, and this specificity is largely determined by the sequence of the V3 loop of the viral envelope protein gp120 (35). Accordingly, most of the mutations found in the primary sequence are located in the V3 and V4 loops, >5 Å away from the Phe43 binding site residues in the 3D structures. To evaluate the effects of distal amino acid mutations on the binding profile of 16f, the compound was docked into both homology models, and the two complexes were submitted to 15-ns molecular dynamic (MD) simulations. In agreement with the biological results, compound 16f was found to be more stable in complex with NL4.3 than AD8, and this might explain its different activity profile against the two strains. Figure 5 shows the root mean square deviation calculated on ligand heavy atoms during the time of simulation with respect to the starting pose in complexes AD8 (blue) and NL4.3 (red).
FIG 4.

(A) Binding mode of compound 16h (blue) as predicted by computational studies. (B) Phe43 cavity occupied at the same time by 16h (blue) and DMJ-II-121 (green) (PDB code 4I54).
FIG 5.

Time evolution of root mean square deviation (RMSD) (Å) calculated on heavy atoms of ligand 16f in AD8 (blue lines) and NL4.3 (red lines).
In vitro ADME.
We initiated early preclinical in vitro adsorption, distribution, metabolism, and excretion (ADME) studies with compounds 16h and 16l to determine their aqueous solubility, parallel artificial membrane permeability (PAMPA), and human liver microsomes (HLM) stability in order to early assess the absorption/stability profiles of these drug candidates (Table 2). Passive membrane permeability was evaluated with the PAMPA assay, while compound solubility was evaluated according to the method developed by Avdeef et al. (36), and the results were expressed as LogS (log mol · liter−1). Metabolic stability was finally evaluated by incubating the abovementioned compounds with 5 μl of human pooled HLM for 1 h at 37°C in order to simulate phase I metabolism. Compounds 16h and 16l showed low values of membrane permeation (permeability classifications: high, >20 × 10−6 cm/s; medium, 10 to 20 × 10−6 cm/s; low, <10 × 10−6 cm/s). However, it has been reported that salicylic acid derivatives might pass through cellular membranes by active transport; thus, further experiments in this direction are necessary to better characterize the permeability profiles of these compounds (37). Furthermore, the ADME investigation highlighted good metabolic stability (>99%) and low aqueous solubility (less than −8, when the desired LogS for a drug candidate should be between −4 and −6).
DISCUSSION
Compared to other steps in the replication of HIV-1 that have been successfully inhibited by many available antiretroviral agents, the entry process remains an elusive but extremely interesting target for both therapeutic and preventive approaches against HIV-1 infection (38). Moreover, clinically available entry inhibitors still have several limitations in terms of neutralization spectra or pharmacokinetic (PK) properties. In fact, maraviroc (MVC), which targets the interaction of gp120 with the CCR5 coreceptor (39), can inhibit only CCR5-using HIV strains, and tropism prediction by genotypic or phenotypic determination is currently mandatory before starting MVC treatment (40); also, enfuvirtide (ENF or T20), which prevents gp41-mediated fusion of the viral and host cell membranes is an injectable peptide associated with still completely unsolved side effects, mainly due to its mode of administration and antigenicity (41, 42). Only two small-molecule compounds and their derivatives have recently entered the preclinical and clinical development process (BMS-626529 and NBD-556) and show promising antiretroviral activity (8–12).
In this paper, we show that a novel class of small-molecule compounds can inhibit the earliest CD4-gp120 protein-protein interactions at the host cell viral interface without any cell toxicity in vitro (Table 2). Moreover, we show that these compounds were able to inhibit CXCR4- and/or CCR5-using laboratory strains and that at least two of these compounds were active against some tier 1 and tier 2 pseudotyped viruses, suggesting a wider spectrum of neutralization (27). The tested viruses are very different and have distinct coreceptor usage; however, neutralization assays with more tier 1 and tier 2 pseudovirus panels, with envelopes coming from non-B subtypes, or antiviral assays on clinical isolates, will be needed in order to further extend this observation.
We used the time-of-addition assay to indirectly demonstrate that the very early entry step is inhibited by these novel molecules, and indeed, the addition of our representative compound could not be postponed to virus seeding before completely losing its antiviral activity in cell culture. These results were in agreement with the gp120-CD4-His competition assay that directly demonstrated the interaction and inhibition of CD4 binding to gp120. These results suggest that these novel compounds may be used either sequentially or concurrently with currently available HIV-1 entry inhibitors. Although it is theoretically possible that resistance to these novel compounds may also occur through the selection of CD4-independent viruses, these variants are rarely selected in vivo and have an increased sensitivity to host immune control compared to CD4-dependent viruses (43), thus, this may not be a clinically relevant hurdle to their usage. Therefore, the novel small molecules with broad neutralization profiles, reduced toxicity, and favorable PK profiles are still needed to improve current lifelong antiretroviral regimens. In fact, novel combinations of antiretroviral drugs with a distinct mechanism of action and nonoverlapping resistance profiles will offer novel strategies to treat or prevent HIV-1 infection.
The next steps for continuing drug development of these compounds will be oriented to improve their aqueous solubility. A series of chemical modifications will be performed to introduce polar groups in the part of the molecule exposed to the solvent as well as to increase the binding affinity toward the receptor in agreement with molecular modeling predictions. Furthermore, other strategies to overcome the solubility issue can be pursued, such as the development of prodrugs or liposome encapsulation (44).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the European Union collaborative project CHAARM (grant HEALTH-F3-2009-242135) and by the Italian Ministero dell'Istruzione, dell'Università e della Ricerca, Prin 2010 research project (grant 2010W2KM5L).
Footnotes
Published ahead of print 10 March 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02739-13.
REFERENCES
- 1.Antonelli G, Turriziani O. 2012. Antiviral therapy: old and current issues. Int. J. Antimicrob. Agents 40:95–102. 10.1016/j.ijantimicag.2012.04.005 [DOI] [PubMed] [Google Scholar]
- 2.Esté JA, Cihlar T. 2010. Current status and challenges of antiretroviral research and therapy. Antiviral Res. 85:25–33. 10.1016/j.antiviral.2009.10.007 [DOI] [PubMed] [Google Scholar]
- 3.Asahchop EL, Wainberg MA, Sloan RD, Tremblay CL. 2012. Antiviral drug resistance and the need for development of new HIV-1 reverse transcriptase inhibitors. Antimicrob. Agents Chemother. 56:5000–5008. 10.1128/AAC.00591-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Luca A, Dunn D, Zazzi M, Camacho R, Torti C, Fanti I, Kaiser R, Sonnerborg A, Codoñer FM, Van Laethem K, Vandamme AM, Bansi L, Ghisetti V, van de Vijver DA, Asboe D, Prosperi MC, Di Giambenedetto S, SEHERE Collaboration in Chain 2013. Declining prevalence of HIV-1 drug resistance in antiretroviral treatment-exposed individuals in Western Europe. J. Infect. Dis. 207:1216–1220. 10.1093/infdis/jit017 [DOI] [PubMed] [Google Scholar]
- 5.Myszka DG, Sweet RW, Hensley P, Brigham-Burke M, Kwong PD, Hendrickson WA, Wyatt R, Sodroski J, Doyle ML. 2000. Energetics of the HIV gp120-CD4 binding reaction. Proc. Natl. Acad. Sci. U. S. A. 97:9026–9031. 10.1073/pnas.97.16.9026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wyatt R, Sodroski J. 1998. The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens. Science 280:1884–1888. 10.1126/science.280.5371.1884 [DOI] [PubMed] [Google Scholar]
- 7.Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. 1998. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393:648–659. 10.1038/31405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ho HT, Fan L, Nowicka-Sans B, McAuliffe B, Li CB, Yamanaka G, Zhou N, Fang H, Dicker I, Dalterio R, Gong YF, Wang T, Yin Z, Ueda Y, Matiskella J, Kadow J, Clapham P, Robinson J, Colonno R, Lin PF. 2006. Envelope conformational changes induced by human immunodeficiency virus type 1 attachment inhibitors prevent CD4 binding and downstream entry events. J. Virol. 80:4017–4025. 10.1128/JVI.80.8.4017-4025.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin PF, Blair W, Wang T, Spicer T, Guo Q, Zhou N, Gong YF, Wang HG, Rose R, Yamanaka G, Robinson B, Li CB, Fridell R, Deminie C, Demers G, Yang Z, Zadjura L, Meanwell N, Colonno R. 2003. A small molecule HIV-1 inhibitor that targets the HIV-1 envelope and inhibits CD4 receptor binding. Proc. Natl. Acad. Sci. U. S. A. 100:11013–11018. 10.1073/pnas.1832214100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Si Z, Madani N, Cox JM, Chruma JJ, Klein JC, Schön A, Phan N, Wang L, Biorn AC, Cocklin S, Chaiken I, Freire E, Smith AB, III, Sodroski JG. 2004. Small-molecule inhibitors of HIV-1 entry block receptor-induced conformational changes in the viral envelope glycoproteins. Proc. Natl. Acad. Sci. U. S. A. 101:5036–5041. 10.1073/pnas.0307953101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao Q, Ma L, Jiang S, Lu H, Liu S, He Y, Strick N, Neamati N, Debnath AK. 2005. Identification of N-phenyl-N′-(2,2,6,6-tetramethyl-piperidin-4-yl)-oxalamides as a new class of HIV-1 entry inhibitors that prevent gp120 binding to CD4. Virology 339:213–225. 10.1016/j.virol.2005.06.008 [DOI] [PubMed] [Google Scholar]
- 12.Madani N, Schön A, Princiotto AM, Lalonde JM, Courter JR, Soeta T, Ng D, Wang L, Brower ET, Xiang SH, Kwon YD, Huang CC, Wyatt R, Kwong PD, Freire E, Smith AB, III, Sodroski J. 2008. Small-molecule CD4 mimics interact with a highly conserved pocket on HIV-1 gp120. Structure 16:1689–1701. 10.1016/j.str.2008.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kwon YD, Finzi A, Wu X, Dogo-Isonagie C, Lee LK, Moore LR, Schmidt SD, Stuckey J, Yang Y, Zhou T, Zhu J, Vicic DA, Debnath AK, Shapiro L, Bewley CA, Mascola JR, Sodroski JG, Kwong PD. 2012. Unliganded HIV-1 gp120 core structures assume the CD4-bound conformation with regulation by quaternary interactions and variable loops. Proc. Natl. Acad. Sci. U. S. A. 109:5663–5668. 10.1073/pnas.1112391109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Curreli F, Choudhury S, Pyatkin I, Zagorodnikov VP, Bulay AK, Altieri A, Kwon YD, Kwong PD, Debnath AK. 2012. Design, synthesis, and antiviral activity of entry inhibitors that target the CD4-binding site of HIV-1. J. Med. Chem. 55:4764–4775. 10.1021/jm3002247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lalonde JM, Elban MA, Courter JR, Sugawara A, Soeta T, Madani N, Princiotto AM, Kwon YD, Kwong PD, Schön A, Freire E, Sodroski J, Smith AB., III 2011. Design, synthesis and biological evaluation of small molecule inhibitors of CD4-gp120 binding based on virtual screening. Bioorg. Med. Chem. 19:91–101. 10.1016/j.bmc.2010.11.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.LaLonde JM, Kwon YD, Jones DM, Sun AW, Courter JR, Soeta T, Kobayashi T, Princiotto AM, Wu X, Schön A, Freire E, Kwong PD, Mascola JR, Sodroski J, Madani N, Smith AB., III 2012. Structure-based design, synthesis, and characterization of dual hotspot small-molecule HIV-1 entry inhibitors. J. Med. Chem. 55:4382–4396. 10.1021/jm300265j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lalonde JM, Le-Khac M, Jones DM, Courter JR, Park J, Schön A, Princiotto AM, Wu X, Mascola JR, Freire E, Sodroski J, Madani N, Hendrickson WA, Smith AB., III 2013. Structure-based design and synthesis of an HIV-1 entry inhibitor exploiting X-ray and thermodynamic characterization. ACS Med. Chem. Lett. 4:338–343. 10.1021/ml300407y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Debnath AK. 2013. Rational design of HIV-1 entry inhibitors. Methods Mol. Biol. 993:185–204. 10.1007/978-1-62703-342-8_13 [DOI] [PubMed] [Google Scholar]
- 19.Caporuscio F, Tafi A, González E, Manetti F, Esté JA, Botta M. 2009. A dynamic target-based pharmacophoric model mapping the CD4 binding site on HIV-1 gp120 to identify new inhibitors of gp120-CD4 protein-protein interactions. Bioorg. Med. Chem. Lett. 19:6087–6091. 10.1016/j.bmcl.2009.09.029 [DOI] [PubMed] [Google Scholar]
- 20.Tintori C, Selvaraj M, Badia R, Clotet B, Esté JA, Botta M. 2013. Computational studies identifying entry inhibitor scaffolds targeting the Phe43 cavity of HIV-1 gp120. ChemMedChem 8:475–483. 10.1002/cmdc.201200584 [DOI] [PubMed] [Google Scholar]
- 21.Teraishi F, Wu S, Zhang L, Guo W, Davis JJ, Dong F, Fang B. 2005. Identification of a novel synthetic thiazolidin compound capable of inducing c-Jun NH2-terminal kinase-dependent apoptosis in human colon cancer cells. Cancer Res. 65:6380–6387. 10.1158/0008-5472.CAN-05-0575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dayam R, Aiello F, Deng J, Wu Y, Garofalo A, Chen X, Neamati N. 2006. Discovery of small molecule integrin alphavbeta3 antagonists as novel anticancer agents. J. Med. Chem. 49:4526–4534. 10.1021/jm051296s [DOI] [PubMed] [Google Scholar]
- 23.Rinaldi M, Tintori C, Franchi L, Vignaroli G, Innitzer A, Massa S, Esté JA, Gonzalo E, Christ F, Debyser Z, Botta M. 2011. A versatile and practical synthesis toward the development of novel HIV-1 integrase inhibitors. ChemMedChem 6:343–352. 10.1002/cmdc.201000510 [DOI] [PubMed] [Google Scholar]
- 24.Canducci F, Ceresola ER, Boeri E, Spagnuolo V, Cossarini F, Castagna A, Lazzarin A, Clementi M. 2011. Cross-resistance profile of the novel integrase inhibitor Dolutegravir (S/GSK1349572) using clonal viral variants selected in patients failing raltegravir. J. Infect. Dis. 204:1811–1815. 10.1093/infdis/jir636 [DOI] [PubMed] [Google Scholar]
- 25.Canducci F, Marinozzi MC, Sampaolo M, Boeri E, Spagnuolo V, Gianotti N, Castagna A, Paolucci S, Baldanti F, Lazzarin A, Clementi M. 2010. Genotypic/phenotypic patterns of HIV-1 integrase resistance to raltegravir. J. Antimicrob. Chemother. 65:425–433. 10.1093/jac/dkp477 [DOI] [PubMed] [Google Scholar]
- 26.Burioni R, Mancini N, De Marco D, Clementi N, Perotti M, Nitti G, Sassi M, Canducci F, Shvela K, Bagnarelli P, Mascola JR, Clementi M. 2008. Anti-HIV-1 response elicited in rabbits by anti-idiotype monoclonal antibodies mimicking the CD4-binding site. PLoS One 3:e3423. 10.1371/journal.pone.0003423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mascola JR, D'Souza P, Gilbert P, Hahn BH, Haigwood NL, Morris L, Petropoulos CJ, Polonis VR, Sarzotti M, Montefiori DC. 2005. Recommendations for the design and use of standard virus panels to assess neutralizing antibody responses elicited by candidate human immunodeficiency virus type 1 vaccines. J. Virol. 79:10103–10107. 10.1128/JVI.79.16.10103-10107.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li M, Gao F, Mascola JR, Stamatatos L, Polonis VR, Koutsoukos M, Voss G, Goepfert P, Gilbert P, Greene KM, Bilska M, Kothe DL, Salazar-Gonzalez JF, Wei X, Decker JM, Hahn BH, Montefiori DC. 2005. Human immunodeficiency virus type 1 env clones from acute and early subtype B infections for standardized assessments of vaccine-elicited neutralizing antibodies. J. Virol. 79:10108–10125. 10.1128/JVI.79.16.10108-10125.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Daelemans D, Pauwels R, De Clercq E, Pannecouque C. 2011. A time-of-drug addition approach to target identification of antiviral compounds. Nat. Protoc. 6:925–933. 10.1038/nprot.2011.330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang CC, Tang M, Zhang MY, Majeed S, Montabana E, Stanfield RL, Dimitrov DS, Korber B, Sodroski J, Wilson IA, Wyatt R, Kwong PD. 2005. Structure of a V3-containing HIV-1 gp120 core. Science 310:1025–1028. 10.1126/science.1118398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. 2003. Improved protein-ligand docking using GOLD. Proteins 52:609–623. 10.1002/prot.10465 [DOI] [PubMed] [Google Scholar]
- 32.Tintori C, Veljkovic N, Veljkovic V, Botta M. 2010. Computational studies of the interaction between the HIV-1 integrase tetramer and the cofactor LEDGF/p75: insights from molecular dynamics simulations and the informational spectrum method. Proteins 78:3396–3408. 10.1002/prot.22847 [DOI] [PubMed] [Google Scholar]
- 33.Wohnsland F, Faller B. 2001. High-throughput permeability pH profile and high-throughput alkane/water log P with artificial membranes. J. Med. Chem. 44:923–930. 10.1021/jm001020e [DOI] [PubMed] [Google Scholar]
- 34.Sugano K, Hamada H, Machida M, Ushio H. 2001. High throughput prediction of oral absorption: improvement of the composition of the lipid solution used in parallel artificial membrane permeation assay. J. Biomol. Screen. 6:189–196. 10.1177/108705710100600309 [DOI] [PubMed] [Google Scholar]
- 35.Moore JP, Kitchen SG, Pugach P, Zack JA. 2004. The CCR5 and CXCR4 coreceptors–central to understanding the transmission and pathogenesis of human immunodeficiency virus type 1 infection. AIDS Res. Hum. Retroviruses 20:111–126. 10.1089/088922204322749567 [DOI] [PubMed] [Google Scholar]
- 36.Avdeef A. 2001. Physicochemical profiling (solubility, permeability and charge state). Curr. Top. Med. Chem. 1:277–351. 10.2174/1568026013395100 [DOI] [PubMed] [Google Scholar]
- 37.Koljonen M, Rousu K, Cierny J, Kaukonen AM, Hirvonen J. 2008. Transport evaluation of salicylic acid and structurally related compounds across Caco-2 cell monolayers and artificial PAMPA membranes. Eur. J. Pharm. Biopharm. 70:531–538. 10.1016/j.ejpb.2008.05.017 [DOI] [PubMed] [Google Scholar]
- 38.Sharma AK, George V, Valiathan R, Pilakka-Kanthikeel S, Pallikkuth S. 2013. Inhibitors of HIV-1 entry and integration: recent developments and impact on treatment. Recent Pat. Inflamm. Allergy Drug Discov. 7:151–161. 10.2174/1872213X11307020006 [DOI] [PubMed] [Google Scholar]
- 39.Klasse PJ. 2013. Structural biology. A new bundle of prospects for blocking HIV-1 entry. Science 341:1347–1348. 10.1126/science.1245384 [DOI] [PubMed] [Google Scholar]
- 40.Nozza S, Canducci F, Galli L, Cozzi-Lepri A, Capobianchi MR, Ceresola ER, Narciso P, Libertone R, Castelli P, Moioli M, D'Arminio Monforte A, Castagna A, ICONA Foundation. 2012. Viral tropism by geno2pheno as a tool for predicting CD4 decrease in HIV-1-infected naive patients with high CD4 counts. J. Antimicrob. Chemother. 67:1224–1227. 10.1093/jac/dkr600 [DOI] [PubMed] [Google Scholar]
- 41.Joly V, Fagard C, Grondin C, Descamps D, Yazdanpanah Y, Charpentier C, Colin de Verdiere N, Tabuteau S, Raffi F, Cabie A, Chene G, Yeni P, S 130 Apollo Trial Group 2013. Intensification of antiretroviral therapy through addition of enfuvirtide in naive HIV-1-infected patients with severe immunosuppression does not improve immunological response: results of a randomized multicenter trial (ANRS 130 Apollo). Antimicrob. Agents Chemother. 57:758–765. 10.1128/AAC.01662-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huet T, Kerbarh O, Schols D, Clayette P, Gauchet C, Dubreucq G, Vincent L, Bompais H, Mazinghien R, Querolle O, Salvador A, Lemoine J, Lucidi B, Balzarini J, Petitou M. 2010. Long-lasting enfuvirtide carrier pentasaccharide conjugates with potent anti-human immunodeficiency virus type 1 activity. Antimicrob. Agents Chemother. 54:134–142. 10.1128/AAC.00827-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Z, Zhou N, Sun Y, Ray N, Lataillade M, Hanna GJ, Krystal M. 2013. Activity of the HIV-1 attachment inhibitor BMS-626529, the active component of the prodrug BMS-663068, against CD4-independent viruses and HIV-1 envelopes resistant to other entry inhibitors. Antimicrob. Agents Chemother. 57:4172–4180. 10.1128/AAC.00513-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Radi M, Tintori C, Musumeci F, Brullo C, Zamperini C, Dreassi E, Fallacara AL, Vignaroli G, Crespan E, Zanoli S, Laurenzana I, Filippi I, Maga G, Schenone S, Angelucci A, Botta M. 2013. Design, synthesis, and biological evaluation of pyrazolo[3,4-d]pyrimidines active in vivo on the Bcr-Abl T315I mutant. J. Med. Chem. 56:5382–5394. 10.1021/jm400233w [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.