

Abstract
Rifapentine is under active investigation as a potent drug that may help shorten the tuberculosis (TB) treatment duration. A previous rifapentine dose escalation study with daily dosing indicated a possible decrease in bioavailability as the dose increased and an increase in clearance over time for rifapentine and its active metabolite, desacetyl rifapentine. This study aimed to assess the effects of increasing doses on rifapentine absorption and bioavailability and to evaluate the clearance changes over 14 days. A population analysis was performed with nonlinear mixed-effects modeling. Absorption, time-varying clearance, bioavailability, and empirical and semimechanistic autoinduction models were investigated. A one-compartment model linked to a transit compartment absorption model best described the data. The bioavailability of rifapentine decreased linearly by 2.5% for each 100-mg increase in dose. The autoinduction model suggested a dose-independent linear increase in clearance of the parent drug and metabolite over time from 1.2 and 3.1 liters · h−1, respectively, after a single dose to 2.2 and 5.0 liters · h−1, respectively, after 14 once-daily doses, with no plateau being reached by day 14. In clinical trial simulations using the final model, rifapentine demonstrated less-than-dose-proportional pharmacokinetics, but there was no plateau in exposures over the dose range tested (450 to 1,800 mg), and divided dosing increased exposures significantly. Thus, the proposed compartmental model incorporating daily dosing of rifapentine over a wide range of doses and time-related changes in bioavailability and clearance provides a useful tool for estimation of drug exposure that can be used to optimize rifapentine dosing for TB treatment. (This study has been registered at ClinicalTrials.gov under registration no. NCT01162486.)
INTRODUCTION
Tuberculosis (TB) is a major global health problem and remains a leading cause of death from an infectious disease (1). The current first-line regimen for TB was developed decades ago, and 6 months of treatment is still required for cure (2). The long duration is challenging for patients and costly to TB programs. Rifapentine (RFP) is a cyclopentyl analogue of rifampin, the key sterilizing agent in the standard TB treatment regimen that kills bacteria by inhibiting DNA-dependent RNA polymerase. RFP has higher antimicrobial potency and a longer half-life than rifampin and was approved by the Food and Drug Administration (FDA) for treatment of TB at a dose of 600 mg twice weekly (in the intensive phase) and once weekly (in the continuation phase) (3). However, the relapse rate among patients in some patient populations treated with such an intermittent RFP regimen is unacceptably high, indicating that the optimal dosing regimen of RFP for TB treatment has yet to be fully characterized. Recent studies in a well-validated mouse model of TB disease have shown that the replacement of rifampin with RFP can shorten the treatment duration to 3 months or less when RFP is given daily and that RFP's treatment-shortening activity is dose dependent (4, 5). Daily dosing of RFP was well tolerated at doses ranging from 5 to 20 mg/kg of body weight in healthy volunteers (6). The replacement of rifampin with daily doses of RFP of up to 20 mg/kg is currently being investigated in several TB treatment trials. Though the bactericidal activity of rifamycins against Mycobacterium tuberculosis is assumed to correlate best with the area under the concentration-time curve (AUC)-to-MIC ratio (AUC/MIC) (7, 8), the target concentration associated with maximal sterilizing activity has not been definitively defined.
Following oral administration, RFP is converted by esterases to a major circulating but less active metabolite, desacetyl rifapentine (desRFP) (9, 10). Both the parent drug and the metabolite are primarily eliminated by biliary excretion (11, 12). The oral bioavailability of RFP increases when administered with food, and the magnitude of the increase varies by meal type (13, 14). Previous studies showed a less than proportional increase in exposure to RFP with increasing dose, including a noncompartmental analysis of multiple-dose data suggesting that a plateau in exposure was reached at a dose of 15 mg/kg (6, 15). This finding was particularly concerning, given that RFP's treatment-shortening properties are exposure dependent in the murine model and that a dose of 10 mg/kg daily did not significantly increase the proportion of patients with sputum culture conversion at 2 months compared to the proportion for patients receiving a standard dose of rifampin in a phase II clinical trial (16). In addition, evidence exists that RFP induces its own clearance (CL) when administered intermittently at doses higher than 600 mg (13, 17), but the relationship between dose, time, and autoinduction has not been fully characterized for RFP and desRFP when RFP is administered daily.
The main objective of this study was to develop an integrated population pharmacokinetic (PK) model for RFP and desRFP after daily dosing incorporating data from a broad range of doses. In the model, we quantified the dose- and time-dependent changes in clearance and bioavailability of RFP and desRFP with the goal of using this model together with data from previously published studies evaluating the effect of food on RFP concentrations to perform simulations to determine the most appropriate dosing strategy to increase RFP exposures for TB treatment.
MATERIALS AND METHODS
Study design.
A phase I, open-label, dose escalation trial of RFP given once daily was conducted among healthy volunteers (ClinicalTrials.gov registration no. NCT01162486). RFP was sequentially administered daily at doses of 5, 10, 15, and 20 mg/kg to independent cohorts with 6 to 7 subjects per dosing cohort to evaluate the maximal tolerated daily dose of RFP. A detailed description of the enrollment criteria has been reported elsewhere (6). Briefly, subjects received their assigned oral dose of RFP following a low-fat breakfast (865 kcal, 20% [20 g] fat, 5.1 g fiber) once daily for 14 days. All subjects provided written informed consent. The study was approved by the institutional review boards of the Johns Hopkins University School of Medicine and the Centers for Disease Control and Prevention.
PK data collection.
Blood samples for PK analysis were collected after the 1st and 14th daily doses. PK sampling time points were predose and 0.5, 1, 2, 4, 5, 8, 12, and 24 h postdose (samples were also collected 34, 48, and 72 h after the 14th dose). Sampling time points were chosen to support primary noncompartmental analysis. For each individual, additional trough blood samples were collected after 5 and 9 doses of RFP. RFP and desRFP plasma concentrations were quantified using a validated liquid chromatography-mass spectrometry assay (6).
Population PK analysis.
A basic model structure was established using the full PK profile data after the first dose of RFP. Since RFP exhibits highly variable absorption, several absorption models were investigated, using previously reported methods (18, 19), including a zero-order absorption model, a first-order absorption model with a lag time, a sequential zero- and first-order model, and a transit absorption model. The transit compartment chain for the transit absorption model is depicted in Fig. 1 and in equations 1 to 3:
| (1) |
| (2) |
| (3) |
where A1 represents the amount of RFP in the absorption compartment, t is time, dose is the amount of RFP administered (in milligrams), F is bioavailability, ktr is a transit rate constant describing the flow of RFP between neighboring transit compartments, n is the number of transit compartments, ka is the first-order absorption rate constant, mean transit time (MTT) is the average amount of time (in hours) spent by a drug molecule traveling from the first transit compartment to the absorption compartment (equation 3), A2 is the amount of drug in the central compartment, CL is clearance, and V is the apparent volume of distribution.
FIG 1.
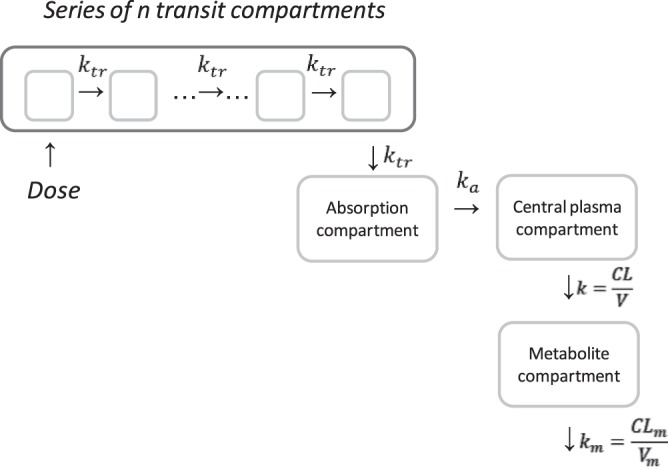
Rifapentine model structure. Abbreviations: ktr, transit rate constant; ka, absorption rate constant; n, number of transit compartments; k, rifapentine elimination rate constant; CL, rifapentine clearance; V, rifapentine volume of distribution; km, metabolite elimination rate constant; CLm, metabolite clearance; Vm, metabolite volume of distribution.
The relationship between bioavailability and dose was quantified at this stage to investigate the potential decrease in bioavailability with increased dose indicated by the observed trends in the data. Several models were tested, including exponential, sigmoidal maximum-effect (Emax), and linear models. Exponential and sigmoidal Emax models have the ability to characterize the plateau in bioavailability saturation eventually reached with increasing dose, which is not the case with the linear model described in equation 4.
| (4) |
where θpop is the relative bioavailability (Frel) in individuals who received RFP at a dose of 300 mg once daily, which was arbitrarily set at a value of 1. θdose,F is a slope term determining the relative change in RFP bioavailability for each 100-mg increase in the RFP dose. The available data did not support estimation of models more complex than the linear one.
Autoinduction models, including empirical and semimechanistic models, were examined to describe potential changes in CL and/or bioavailability over time. All tested models indicated that there was no clear concentration-dependent relationship between the magnitude of autoinduction and RFP plasma concentrations, therefore resulting in the same magnitude of CL increase with time across all dose levels. This was evident by only marginal improvements in likelihood and an inability to estimate the RFP 50% effective concentration parameter in all tested semimechanistic models. The observed autoinduction was sufficiently described with a linear model where CL was either a continuous function of time or a step function.
Finally, metabolite data were added to the model, and an assumption was made that the rate of metabolite formation was equal to the rate of RFP elimination. This is a commonly made assumption in the absence of intravenous data. Metabolite PKs were sufficiently described by a one-compartment model, as represented in equation 5 and Fig. 1,
| (5) |
where A3 is the amount of metabolite in the plasma, and CLm and Vm are the clearance and volume of distribution of the metabolite, respectively.
Model building and evaluation criteria.
All data were analyzed using the nonlinear mixed effects approach available in the NONMEM program (version 7.1). The first-order conditional estimation with interaction (FOCEI) method was employed throughout the analysis of the PK data. The individual parameters were assumed to be log-normally distributed, and proportional error was employed for description of residual variability. The model-building procedure was guided by the likelihood ratio test, diagnostic plots, and internal model validation techniques, including visual and numerical predictive checks.
Clinical trial simulations.
Clinical trial simulations were performed to evaluate different dosing strategies for increasing RFP exposure. Two main scenarios were evaluated: a dosing frequency change from once daily to twice daily and the effect of high-fat food. More specifically, we evaluated AUC levels after administration of 15 and 20 mg/kg for 14 days. The choice of the doses and clinical trial simulation design were chosen to support AIDS Clinical Trials Group (ACTG) clinical trial A5311, which was planned to evaluate and confirm strategies for increasing exposure without an additional increase in the actual daily dose. All simulations were performed using the final model structure and final parameter estimates, including all levels of variability (between-subject and within-subject variability). The quantified relationship between dose and bioavailability was utilized from the established model, while the magnitude of the effect of food on bioavailability was assessed from the literature (13). Additional clinical trial simulations were performed to predict PK results from the ongoing phase II study of dose-escalating daily RFP in patients with drug-sensitive TB (Tuberculosis Trials Consortium [TBTC] study 29X). We evaluated steady-state exposures (after at least 3 weeks of daily treatment) following daily dosing at 10, 15, and 20 mg/kg with food. In these simulations, we utilized the dosing algorithms used in study 29X for weight-based dosing assignment. To do so, we assumed that the patient weight in the virtual population used in clinical trial simulations is uniformly distributed from 45 to 85 kg. For the purposes of these simulations, we assumed that autoinduction is complete by the end of week 2 of daily RFP administration; therefore, the CL at steady state is equal to the estimated CL at day 14 from the model. If this assumption does not hold, the study 29X exposures would be overpredicted.
The main aim with clinical trial simulations was to project exposures from the above-mentioned clinical studies; therefore, no particular PK target was utilized at this stage.
RESULTS
The final database included PK data from 26 healthy subjects (6, 7, 7, and 6, respectively, from the 5-, 10-, 15-, and 20-mg/kg dosing cohorts). Two subjects from the 10-mg/kg cohort and one subject from the 15-mg/kg cohort contributed only partial PK data because of early study discontinuation. The median age, weight, and height of the subjects were 47 years (range, 23 to 59 years), 79.9 kg (range, 59.1 to 99.3 kg), and 174 cm (range, 157 to 198 cm), respectively; 5 of the 26 subjects were female. The median RFP doses by cohort were 450, 750, 1,200, and 1,725 mg once daily, with doses ranging from 450 mg to 1,800 mg. The clinical and safety results of the trial as well as PK summary data by dosing cohort were described previously (6).
In total, 503 RFP and 405 desRFP plasma concentration measurements were available for the analysis. The absorption phase of RFP exhibited high variability as the dose increased from 5 mg/kg to 20 mg/kg. RFP PKs were described by a one-compartment model with first-order absorption and elimination linked to the transit compartment absorption model (Fig. 1). Parent and metabolite data were fitted simultaneously. The model PK parameters and variability estimates for RFP and desRFP from the final model are presented in Table 1. The transit compartment model substantially improved the model fit compared to the fit for the other absorption models. For example, compared to the first-order absorption model with a lag time, the improvement in likelihood was evident by a decrease in the objective function value of 69 (P < 10−14) accompanied by visual improvement in goodness-of-fit plots. The absorption rate constant was 2 h−1, and the mean transit time and number of transit compartments were 1.1 h and 10, respectively. After a single dose, RFP apparent clearance (CL/F) and volume of distribution (V/F) were estimated to be 1.2 liters · h−1 and 41 liters, respectively. Metabolite apparent clearance (CLm/Fm) and volume of distribution (Vm/Fm) were estimated to be 3.1 liters · h−1and 15.5 liters, respectively. After evaluating exponential, sigmoidal Emax, and linear models, we found that the linear model best described both the RFP and desRFP data, with an estimated decrease in RFP bioavailability of 2.5% occurring with each 100-mg increase in RFP dose beginning at the lowest dose tested (Fig. 2). The relative fraction metabolized (Fm) increased by 4.9% with each 100-mg increase in RFP dose. Between-subject variabilities in CL, V, MTT, and CLm were 36.9%, 23.8%, 55.4%, and 56.6%, respectively.
TABLE 1.
Population PK parameter estimates of RFP and desRFP
| Parameter | Estimate | % SE |
|---|---|---|
| CL (liters/h) | 1.18 | |
| V (liters) | 41 | |
| ka (h−1) | 2 | |
| MTT (h) | 1.1 | |
| No. of transit compartments | 10 | |
| θdose,F | 0.025 | |
| CLm/Fm (liters · h−1) | 3.1 | 22.3 |
| Vm/Fm (liters) | 15.5 | 18.3 |
| θdose,Fm | 0.049 | 57.6 |
| IIVa (variances plus % CV) | ||
| ƞCL | 0.136 (36.9) | |
| ƞV | 0.0566 (23.8) | |
| ƞMTT | 0.307 (55.4) | |
| ƞCLm | 0.32 (56.6) | 32.2 |
| Covariance for ƞCL∼ ƞV (covariance and correlation) | 0.0684 (0.780) | |
| Residual variability | ||
| Additive error for RFP (mg · liter−1) | 0.48 | 14.4 |
| Proportional error for RFP | 0.26 | 3.0 |
| Additive error for desRFP (mg · liter−1) | 0.072 | 17.8 |
| Proportional error for desRFP | 0.40 | 2.5 |
IIV, interindividual variability. Values of IIV are shown in parentheses as % CV; etas represent the random variable with zero mean that distinguishes individual pharmacokinetic parameters from the population mean (the variances of etas are data before the parentheses).
FIG 2.

Observed relationship between interindividual variability in bioavailability and oral dose (left) and study arm (right). The line represents the Lowess smooth through the data.
While the single-dose model described the RFP PK data after the first RFP dose very well using a visual predictive check, the observed RFP plasma concentrations were overpredicted when the established single-dose model was used to predict concentrations after multiple daily doses of RFP (not shown), suggesting changes in either CL or bioavailability over time. Through examination of the difference in RFP bioavailability and CL between the single-dose and daily dosing data, we found that there was a decrease in bioavailability and an increase in CL ranging from −13.0% to −27.4% and from 21.2% to 44.4%, respectively, without evident trends with increasing dose for multiple-dose RFP compared to single-dose RFP (Fig. 3). Since the change in bioavailability with time was not as significant as that in CL, the induction model for CL with time was included in the model. All the autoinduction models investigated suggested a similar autoinduction pattern of a linear increase of CL/F and CLm/Fm over time independently of the dose administered, increasing from the baseline values of 1.2 liters · h−1 and 3.0 liters · h−1 for RFP and desRFP, respectively, after a single dose to 2.1 liters · h−1 and 5.0 liters · h−1 for RFP and desRFP, respectively, after 14 daily doses. After the inclusion of the time-varying effect on clearance for daily dosing of RFP, the visual predictive check for the multiple-dose PK profile was substantially improved, with the observed 95th, 50th, and 5th percentiles of both the RFP and desRFP plasma concentrations falling well within the 95% confidence interval (CI) of each percentile from the model prediction on day 14 (Fig. 4). For the single-dose PK profile, the 5th and 95th percentiles had minor under- and overprediction at 4 to 6 h and after 6 h, respectively, but the overall predictability of the model was acceptable.
FIG 3.
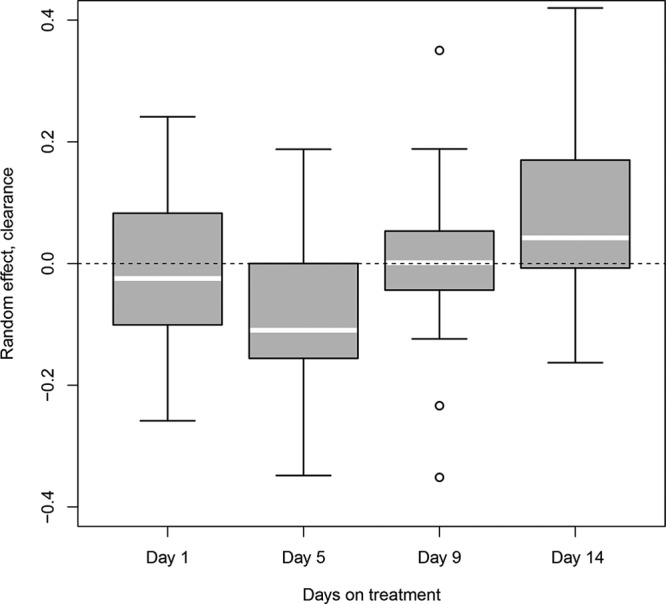
Observed relationship between subject-specific Bayesian estimates of clearance (ETA) estimated at days 1, 5, 9, and 14 on treatment. If within-subject variability in clearance is truly random, there would not be a visible trend with time. White line within each box, median population value.
FIG 4.

Visual predictive check for blood levels of rifapentine (left) and metabolite (right) for different days on treatment. Solid black lines, median of the observed data; dotted black lines, 5th and 95th percentiles of the observed data; middle gray shaded area, simulated median with uncertainty (for 500 repetitions of the visual predictive check); lower and upper gray shaded areas, simulated 5th and 95th percentiles with uncertainty, respectively.
Since we found that RFP bioavailability decreased and the relative fraction of RFP metabolized increased as the dose increased, we hypothesized that for the same total daily dose, splitting the dose so that it is given twice daily rather than once daily would result in increases in RFP exposures. We performed clinical trial simulations using the established model structure and the final parameter estimates to find more efficient dosing regimens. The medians, along with the 2.5th and 97.5th percentiles of the RFP AUC from time zero to 24 h (AUC0–24), from the simulated dosing regimens are shown in Table 2. Splitting a once-daily dose into twice-daily administration increased the RFP exposure, with median AUCs increasing from 433 μg · h/ml (95% CI, 213, 850 μg · h/ml) with once-daily dosing at 15 mg/kg to 522 μg · h/ml (95% CI, 258, 1,090 μg · h/ml) with twice-daily dosing at 7.5 mg/kg and from 496 μg · h/ml (95% CI, 246, 1,005 μg · h/ml) with once-daily dosing at 20 mg/kg to 678 μg · h/ml (95% CI, 343, 1,381 μg · h/ml) with twice-daily dosing at 10 mg/kg. The twice-daily administration of RFP at 7.5 mg/kg even resulted in a slightly higher level of RFP exposure than the once-daily administration of RFP at 20 mg/kg. The simulated plasma concentration-time profiles of RFP following 14 days of daily administration at 20 mg/kg and twice-daily administration at 10 mg/kg are shown in Fig. 5. Previous studies have shown that food, especially high-fat meals, enhances RFP oral bioavailability (13, 14). Here we also simulated the scenario of twice-daily administration of RFP at 10 mg/kg following high-fat meals, where we assumed an additional increase in exposure of 30% with a high-fat meal. The RFP AUC was further increased, with a median of 881 μg · h/ml (95% CI, 446 to 1,795 μg · h/ml), which is almost doubled compared with that of once-daily administration at 20 mg/kg in the absence of a high-fat meal (Table 2).
TABLE 2.
Predicted multiple-dose AUC0–24 from clinical trial simulations using different simulated RFP dosing strategiesa
| Dose | AUC0–24 (μg · h/ml) |
||
|---|---|---|---|
| Median | 2.5th percentile | 97.5th percentile | |
| Once-daily dosing | |||
| 15 mg/kg | 433 | 213 | 850 |
| 20 mg/kg | 496 | 246 | 1,005 |
| 20 mg/kg with a high-fat meal | 645 | 320 | 1,306 |
| Twice-daily dosing | |||
| 7.5 mg/kg | 522 | 258 | 1,090 |
| 10 mg/kg | 678 | 343 | 1,381 |
| 10 mg/kg with a high-fat meal | 881 | 446 | 1,795 |
The effect of a high-fat meal was assumed to exhibit an additional increase in exposure of 30%, on the basis of the findings of Zvada et al. (13).
FIG 5.

Clinical trial simulations of once-daily (QD) versus twice-daily (BID) dosing to be tested in ACTG study A5311 as a strategy of increasing RPT exposure.
We also performed clinical trial simulations to predict exposures that might be expected in TBTC study 29X, an ongoing dose-ranging study of RFP among patients with drug-sensitive pulmonary TB. The predicted range of exposures is shown in Fig. 6. Our results indicate that by doubling the once-daily dose, the exposure will increase less than proportionally (1.8 times) but will not reach a plateau. The estimated median exposures for the 10-mg/kg, 15-mg/kg, and 20-mg/kg study arms were, respectively, 252 μg · h/ml (5th and 95th percentiles, 132 and 516 μg · h/ml, respectively), 330 μg · h/ml (5th and 95th percentiles, 173 and 662 μg · h/ml, respectively), and 400 μg · h/ml (5th and 95th percentiles, 218 to 779 μg · h/ml, respectively). If we account for the potential additional increase in exposure due to the effect of high-fat food, which was administered in study 29X, the estimated median exposures for the 10-mg/kg, 15-mg/kg, and 20-mg/kg study arms were, respectively, 336 μg · h/ml (5th and 95th percentiles, 163 and 666 μg · h/ml, respectively), 439 μg · h/ml (5th and 95th percentiles, 216 and 852 μg · h/ml, respectively), and 525 μg · h/ml (5th and 95th percentiles, 276 and 979 μg · h/ml, respectively).
FIG 6.

Predicted AUC distribution in patient study 29X. Predictions were based on the final model without (left panel) and with (right panel) the potential effect of high-fat food. Each box represents the simulated distribution of AUCs following the dosing criteria used in study 29X. White line within each box, population median; circles, outliers.
DISCUSSION
Rifapentine is a rifamycin with potent activity against M. tuberculosis that is being evaluated as an agent to shorten the duration of TB treatment. Using a nonlinear mixed-effects modeling approach applied to rich PK data collected from individuals receiving daily doses of RFP ranging from 450 mg to 1,800 mg, we demonstrated that the bioavailability of RFP decreases with increasing dose beginning at the lowest dose tested. In addition, RFP pharmacokinetics are time dependent, but the magnitude of autoinduction does not vary with concentration. Increases in clearance over time did not reach a clear plateau after the first 14 days of dosing, suggesting that steady state may not yet be achieved at 2 weeks. Clinical trial simulations demonstrate that splitting the dose or taking the dose with food could substantially increase daily RFP exposures, which is a significant finding, given that RFP's activity appears to correlate best with AUC/MIC and that current dosing achieves concentrations that are still on the steep part of the dose-response curve. The dose-response relationship was reported in mouse studies (20), guinea pig studies (21), and patient early bactericidal activity studies (22). Most importantly, preliminary results from the recently completed phase IIb TBTC trial 29X in patients suggests a pronounced exposure-response relationship, where the time to culture conversion on solid and liquid media was best predicted by rifapentine exposure (23). Our clinical trial simulations suggest that dose splitting and administration of drug with high-fat food can be utilized as effective strategies to optimally achieve PK-pharmacodynamic targets, once they are firmly established.
The population pharmacokinetics of single-dose RFP among patients with pulmonary TB disease have been described previously (24), and the effects of different food types on single-dose RFP exposures have also been evaluated using mathematical models (13). This analysis of single-dose and multiple-dose data from subjects receiving a broader range of RFP doses allowed exploration of the relationship between dose and bioavailability and the time and concentration dependency of clearance. A model incorporating parent and metabolite data showed that the relative fraction of RFP metabolized increased by 4.9% with each 100-mg increase in dose, suggesting possible saturation of other elimination routes (e.g., renal).
Rifamycins are unique in their sterilizing activity against M. tuberculosis. Patients with multidrug-resistant (MDR) TB, that is, TB that is resistant to isoniazid and rifampin, must be treated for 18 to 24 months rather than 6 months to achieve cure without relapse. However, when rifampin was first developed, its manufacture was prohibitively expensive, and dosing was not optimized for maximal effectiveness (25). Rather, the exposures achieved with standard dosing of rifampin appear to be at the low end of the dose-response curve (7, 26). In addition, the marked variability in the bioavailability of rifampin among patients, together with higher-than-dose-proportional exposures at the current minimally effective dose, likely serves to amplify the interindividual differences in the antimicrobial effect of rifampin. Recent studies indicate that increases in rifampin dose result in concentrations that are more than dose proportional; specifically, a 3-fold increase in dose from 10 to 30 mg/kg daily results in a 7-fold increase in the AUC0–24 and enhanced early bactericidal activity for each increase in dose (27). Studies of high-dose rifampin for shortening of the treatment for TB are in the planning stages.
RFP, a newer rifamycin, has a lower MIC against M. tuberculosis and can shorten TB treatment to 2 to 3 months when substituted for rifampin and used at high daily doses in combination with standard first-line drugs in the mouse model (4). However, in a clinical trial comparing rifampin to RFP, each dosed at 10 mg/kg daily, the proportion of patients with pulmonary TB who converted their sputum cultures to negative after 2 months of treatment was similar between the treatment arms (16). The reasons for the lack of a clear superiority of RFP over rifampin in that study (TBTC study 29) are unclear, but lower-than-expected RFP concentrations may have played a role. To evaluate the safety and PKs of higher daily RFP doses, a dose escalation trial (TBTC study 29B) was conducted in healthy adults. In TBTC study 29B, it appeared that there was no increase in RFP exposure with an increase in dose from 15 to 20 mg/kg daily (6), corroborating previous work showing a less than proportional increase in exposure with increases in single-dose RFP (15). In the initial study 29B analyses, patients were grouped by dosing cohort and noncompartmental analysis techniques were used. However, the more detailed analysis of those study 29B data using nonlinear mixed-effects modeling presented here reveals that, even beginning at the lowest administered dose (in this case, 450 mg daily), the bioavailability of RFP decreases by about 2.5% with every 100-mg increase in dose in a linear fashion, suggesting that while increases in dose are likely to result in less-than-dose-proportional increases in exposures, there will not be a plateau in exposure with increasing dose over the dose range tested. To achieve concentrations at least as high as those associated with significant treatment shortening in the mouse model, then, either higher-dose RFP is needed (and is currently being tested in TBTC study 29X; see Fig. 6 for clinical trial simulations of expected exposures) or dosing strategies to increase exposures can be employed. Clinical trial simulations suggest that dividing the dose or taking RFP with food will result in higher concentrations, and these strategies are currently being tested in a phase I study (ClinicalTrials.gov registration no. NCT01574638). Divided dosing (i.e., dosing more than once daily) would have operational implications for administration of directly observed TB therapy, but knowledge of how to optimize RFP exposures is nevertheless important.
Autoinduction has been described previously with daily dosing of rifamycins, including rifampin and rifapentine (17, 28). With autoinduction of metabolizing enzymes or transporters, both bioavailability and clearance can theoretically be affected. In our population PK models, RFP clearance appeared to be time dependent, whereas time had a less significant effect on bioavailability. In addition, there was no clear relationship between dose and clearance or dose and the effects of time on clearance. Further, the time to maximal autoinduction with daily dosing could not be estimated with this study, as increases in clearance were seen up until the final PK sampling day after 14 days of dosing. Longer dosing will be required to determine when RFP concentrations are at steady state. The additional limitation of the current model is that it cannot be used to predict the magnitude of autoinduction with an intermittent dosing schedule (e.g., twice and thrice weekly). To do so, our current data would need to be enriched with the data from such studies; this would support implementation of a more mechanistic model where the relationship between plasma rifapentine concentration and the magnitude of autoinduction would be established in a way similar to that in a model for rifampin recently reported by Smythe et al. (29).
Conclusions.
In this study, nonlinear mixed-effects modeling was used to analyze rich multiple-dose parent and metabolite PK data from healthy adults receiving a broad range of daily RFP doses. Bioavailability decreased linearly with increasing dose, clearance was time but not concentration dependent, and steady state may not yet have been achieved after 2 weeks of daily dosing because autoinduction of clearance was increasing up to that point. Clinical trial simulations suggest alternative dosing strategies that may increase exposures and suggest that despite less-than-dose-proportional PKs, a plateau in exposures over the dose range being tested is not expected. These data can be used to inform dose optimization for upcoming treatment-shortening trials employing daily RFP for TB.
ACKNOWLEDGMENTS
We acknowledge the Tuberculosis Trials Consortium (TBTC) of the Centers for Disease Control and Prevention (CDC) for its support of this secondary analysis of TBTC study 29B data. Support was also provided by K23AI080842 (to K.E.D.) and UCSF-CTSI grant number KL2 TR000143-07 (to R.M.S.).
We thank the participants in TBTC study 29B who provided blood samples and data for this analysis.
The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention.
Footnotes
Published ahead of print 10 March 2014
REFERENCES
- 1.Zaman K. 2010. Tuberculosis: a global health problem. J. Health Popul. Nutr. 28:111–113. 10.3329/jhpn.v28i2.4879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chan ED, Iseman MD. 2002. Current medical treatment for tuberculosis. BMJ 325:1282–1286. 10.1136/bmj.325.7375.1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blumberg HM, Burman WJ, Chaisson RE, Daley CL, Etkind SC, Friedman LN, Fujiwara P, Grzemska M, Hopewell PC, Iseman MD, Jasmer RM, Koppaka V, Menzies RI, O'Brien RJ, Reves RR, Reichman LB, Simone PM, Starke JR, Vernon AA. 2003. American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America: treatment of tuberculosis. Am. J. Respir. Crit. Care Med. 167:603–662. 10.1164/rccm.167.4.603 [DOI] [PubMed] [Google Scholar]
- 4.Rosenthal IM, Zhang M, Williams KN, Peloquin CA, Tyagi S, Vernon AA, Bishai WR, Chaisson RE, Grosset JH, Nuermberger EL. 2007. Daily dosing of rifapentine cures tuberculosis in three months or less in the murine model. PLoS Med. 4:e344. 10.1371/journal.pmed.0040344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang T, Zhang M, Rosenthal IM, Grosset JH, Nuermberger EL. 2009. Short-course therapy with daily rifapentine in a murine model of latent tuberculosis infection time. Am. J. Respir. Crit. Care Med. 180:1151–1157. 10.1164/rccm.200905-0795OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dooley KE, Bliven-Sizemore EE, Weiner M, Lu Y, Nuermberger EL, Hubbard WC, Fuchs EJ, Melia MT, Burman WJ, Dorman SE. 2012. Safety and pharmacokinetics of escalating daily doses of the antituberculosis drug rifapentine in healthy volunteers. Clin. Pharmacol. Ther. 91:881–888. 10.1038/clpt.2011.323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jayaram R, Gaonkar S, Kaur P, Suresh BL, Mahesh BN, Jayashree R, Nandi V, Bharat S, Shandil RK, Kantharaj E, Balasubramanian V. 2003. Pharmacokinetics-pharmacodynamics of rifampin in an aerosol infection model of tuberculosis. Antimicrob. Agents Chemother. 47:2118–2124. 10.1128/AAC.47.7.2118-2124.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gumbo T, Louie A, Deziel MR, Liu W, Parsons LM, Salfinger M, Drusano GL. 2007. Concentration-dependent Mycobacterium tuberculosis killing and prevention of resistance by rifampin. Antimicrob. Agents Chemother. 51:3781–3788. 10.1128/AAC.01533-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakajima A, Fukami T, Kobayashi Y, Watanabe A, Nakajima M, Yokoi T. 2011. Human arylacetamide deacetylase is responsible for deacetylation of rifamycins: rifampicin, rifabutin, and rifapentine. Biochem. Pharmacol. 82:1747–1756. 10.1016/j.bcp.2011.08.003 [DOI] [PubMed] [Google Scholar]
- 10.Heifets LB, Lindholm-Levy PJ, Flory MA. 1990. Bactericidal activity in vitro of various rifamycins against Mycobacterium avium and Mycobacterium tuberculosis. Am. Rev. Respir. Dis. 141:626–630. 10.1164/ajrccm/141.3.626 [DOI] [PubMed] [Google Scholar]
- 11.Emary WB, Toren PC, Mathews B, Huh K. 1998. Disposition and metabolism of rifapentine, a rifamycin antibiotic, in mice, rats, and monkeys. Drug Metab. Dispos. 26:725–731 [PubMed] [Google Scholar]
- 12.Reith K, Keung A, Toren PC, Cheng L, Eller MG, Weir SJ. 1998. Disposition and metabolism of 14C-rifapentine in healthy volunteers. Drug Metab. Dispos. 26:732–738 [PubMed] [Google Scholar]
- 13.Zvada SP, Van Der Walt JS, Smith PJ, Fourie PB, Roscigno G, Mitchison D, Simonsson US, McIlleron HM. 2010. Effects of four different meal types on the population pharmacokinetics of single-dose rifapentine in healthy male volunteers. Antimicrob. Agents Chemother. 54:3390–3394. 10.1128/AAC.00345-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chan SL, Yew WW, Porter JH, McAdam KP, Allen BW, Dickinson JM, Ellard GA, Mitchison DA. 1994. Comparison of Chinese and Western rifapentines and improvement of bioavailability by prior taking of various meals. Int. J. Antimicrob. Agents 3:267–274. 10.1016/0924-8579(94)90054-X [DOI] [PubMed] [Google Scholar]
- 15.Weiner M, Bock N, Peloquin CA, Burman WJ, Khan A, Vernon A, Zhao Z, Weis S, Sterling TR, Hayden K, Goldberg S. 2004. Pharmacokinetics of rifapentine at 600, 900, and 1,200 mg during once-weekly tuberculosis therapy. Am. J. Respir. Crit. Care Med. 169:1191–1197. 10.1164/rccm.200311-1612OC [DOI] [PubMed] [Google Scholar]
- 16.Dorman SE, Goldberg S, Stout JE, Muzanyi G, Johnson JL, Weiner M, Bozeman L, Heilig CM, Feng PJ, Moro R, Narita M, Nahid P, Ray S, Bates E, Haile B, Nuermberger EL, Vernon A, Schluger NW. 2012. Substitution of rifapentine for rifampin during intensive phase treatment of pulmonary tuberculosis: study 29 of the tuberculosis trials consortium. J. Infect. Dis. 206:1030–1040. 10.1093/infdis/jis461 [DOI] [PubMed] [Google Scholar]
- 17.Dooley K, Flexner C, Hackman J, Peloquin CA, Nuermberger E, Chaisson RE, Dorman SE. 2008. Repeated administration of high-dose intermittent rifapentine reduces rifapentine and moxifloxacin plasma concentrations. Antimicrob. Agents Chemother. 52:4037–4042. 10.1128/AAC.00554-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Savic RM, Jonker DM, Kerbusch T, Karlsson MO. 2007. Implementation of a transit compartment model for describing drug absorption in pharmacokinetic studies. J. Pharmacokinet. Pharmacodyn. 34:711–726. 10.1007/s10928-007-9066-0 [DOI] [PubMed] [Google Scholar]
- 19.Wilkins JJ, Savic RM, Karlsson MO, Langdon G, McIlleron H, Pillai G, Smith PJ, Simonsson US. 2008. Population pharmacokinetics of rifampin in pulmonary tuberculosis patients, including a semimechanistic model to describe variable absorption. Antimicrob. Agents Chemother. 52:2138–2148. 10.1128/AAC.00461-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosenthal IM, Tasneen R, Peloquin CA, Zhang M, Almeida D, Mdluli KE, Karakousis PC, Grosset JH, Nuermberger EL. 2012. Dose-ranging comparison of rifampin and rifapentine in two pathologically distinct murine models of tuberculosis. Antimicrob. Agents Chemother. 56:4331–4340. 10.1128/AAC.00912-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dutta NK, Illei PB, Peloquin CA, Pinn ML, Mdluli KE, Nuermberger EL, Grosset JH, Karakousis PC. 2012. Rifapentine is not more active than rifampin against chronic tuberculosis in guinea pigs. Antimicrob. Agents Chemother. 56:3726–3731. 10.1128/AAC.00500-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sirgel FA, Fourie PB, Donald PR, Padayatchi N, Rustomjee R, Levin J, Roscigno G, Norman J, McIlleron H, Mitchison DA. 2005. The early bactericidal activities of rifampin and rifapentine in pulmonary tuberculosis. Am. J. Respir. Crit. Care Med. 172:128–135. 10.1164/rccm.200411-1557OC [DOI] [PubMed] [Google Scholar]
- 23.Savic RM, Weiner M, MacKenzie W, Helig C, Dooley K, Engle M, Nsubuga P, Phan H, Peloquin C, Dorman S, for the Tuberculosis Trials Consortium of the Centers for Disease Control and Prevention 2013. Pharmacokinetic-pharmacodynamic analysis of rifapentine in patients during intensive phase treatment for tuberculosis from Tuberculosis Trial Consortium studies 29 and 29X, abstr 11 Abstr. 6th Int. Workshop Pharmacol. Tuberc. Drugs, Denver, CO [Google Scholar]
- 24.Langdon G, Wilkins J, McFadyen L, McIlleron H, Smith P, Simonsson US. 2005. Population pharmacokinetics of rifapentine and its primary desacetyl metabolite in South African tuberculosis patients. Antimicrob. Agents Chemother. 49:4429–4436. 10.1128/AAC.49.11.4429-4436.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Ingen J, Aarnoutse RE, Donald PR, Diacon AH, Dawson R, Plemper van Balen G, Gillespie SH, Boeree MJ. 2011. Why do we use 600 mg of rifampicin in tuberculosis treatment? Clin. Infect. Dis. 52:e194–e199. 10.1093/cid/cir184 [DOI] [PubMed] [Google Scholar]
- 26.Diacon AH, Patientia RF, Venter A, van Helden PD, Smith PJ, McIlleron H, Maritz JS, Donald PR. 2007. Early bactericidal activity of high-dose rifampin in patients with pulmonary tuberculosis evidenced by positive sputum smears. Antimicrob. Agents Chemother. 51:2994–2996. 10.1128/AAC.01474-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boeree M, Diacon A, Dawson R, Venter A, du Bois J, Narunsky K, Hoelscher M, Gillespie S, Phillips P, Aarnoutse R, PanACEA Consortium 2013. What is the “right” dose of rifampiny? abstr 148LB Abstr. Conf. Retroviruses Opportunistic Infect., Atlanta, GA [Google Scholar]
- 28.Acocella G, Pagani V, Marchetti M, Baroni GC, Nicolis FB. 1971. Kinetic studies on rifampicin. I. Serum concentration analysis in subjects treated with different oral doses over a period of two weeks. Chemotherapy 16:356–370 [DOI] [PubMed] [Google Scholar]
- 29.Smythe W, Khandelwal A, Merle C, Rustomjee R, Gninafon M, Bocar Lo M, Sow OB, Olliaro PL, Lienhardt C, Horton J, Smith P, McIlleron H, Simonsson US. 2012. A semimechanistic pharmacokinetic-enzyme turnover model for rifampin autoinduction in adult tuberculosis patients. Antimicrob. Agents Chemother. 56:2091–2098. 10.1128/AAC.05792-11 [DOI] [PMC free article] [PubMed] [Google Scholar]