

Abstract
Baseline and posttreatment samples from hepatitis C virus (HCV) genotype (GT) 1-infected patients who received a combination of danoprevir and mericitabine from a phase II clinical study (INFORM-SVR) were analyzed. In addition to resistance monitoring, sequencing and phenotypic assays were combined with statistical analysis to identify potential novel amino acid substitutions associated with treatment outcome. The NS5B S282T substitution associated with mericitabine resistance was identified in 2/30 viral breakthrough patients and was replaced by wild-type viruses after cessation of drug treatment (during follow-up). The NS3 R155K substitution associated with danoprevir resistance was also observed in these 2 patients. All 69 GT 1a-infected patients who experienced viral breakthrough on treatment or relapsed during follow-up (relapsers) developed NS3 R155K. Among GT 1b-infected patients, substitutions at the danoprevir resistance locus NS3 D168 were observed in 15/20 subjects, whereas substitutions at the danoprevir resistance locus NS3 R155 were observed in 5/20 subjects. Interestingly, the baseline polymorphism NS5B Q47H was more prevalent in GT 1a-infected patients who achieved a sustained virologic response at follow-up week 24 (SVR24) than in non-SVR24 patients (2/13 versus 0/72), and a postbaseline NS3 S122G substitution was more prevalent in GT 1a-infected patients with viral breakthrough than in relapsers (4/22 versus 0/47). Neither substitution conferred resistance to danoprevir or mericitabine, but the substitutions reduced (NS5B Q47H) or improved (NS3 S122G) replication capacity by 2- to 4-fold. The NS5B S282T mericitabine-resistant variant was rare and did not persist once drug was discontinued. NS5B Q47H and NS3 S122G are two newly identified substitutions that affected replication capacity and were enriched in distinct treatment response groups. (This study has been registered at ClinicalTrials.gov under registration no. NCT01278134.)
INTRODUCTION
Hepatitis C virus (HCV) infection is one of the major causes of chronic liver disease, affecting over 170 million people worldwide. The standard of care (SOC) for patients infected with HCV genotype (GT) 1 is pegylated alfa interferon (IFN) and ribavirin (RBV) plus a protease inhibitor (PI), telaprevir or boceprevir (1), which provide sustained virological response rates (SVRs) ranging from 63 to 75% in treatment-naive patients (2–4). More recently, simeprevir, a new PI, was approved for the treatment of HCV in combination with PEG-IFN–RBV and has improved the SVR to ∼80% (5). However, the SOC is limited by reduced tolerability from IFN- and PI-related toxicities and the development of viruses resistant to the PI during treatment (1). Because of the high replication rate of HCV and the inherent error-prone nature of the HCV RNA-dependent RNA polymerase, drug-resistant HCV variants often emerge and are selected during treatment (6). The use of a combination of multiple direct-acting antiviral agents (DAAs) and/or the use of agents with a high barrier to resistance (i.e., agents to which viruses are shown to have a low incidence of resistance development in vitro and in vivo) can potentially reduce the emergence of resistance and improve the treatment response. Therefore, the development of all-oral, interferon-free direct-acting antiviral combination treatments has the potential to further improve the SVR and safety profiles of the current SOC and to address a significant unmet medical need.
Two oral DAAs currently in phase II clinical development targeting the HCV polymerase or protease are mericitabine (MCB) and danoprevir (DNV), respectively. In the phase I INFORM-1 study, treatment with a combination of MCB and DNV for 13 days produced a 3.7- to 5.2-log10 IU/ml decrease in HCV RNA levels in patients and prevented the emergence of resistance to either agent (7). Mericitabine is a triester prodrug of PSI-6130 (β-d-2′-deoxy-2′-fluoro-2′-C-methylcytidine; both the triester prodrug and the parent compound, PSI-6130, are referred to as “mericitabine” or “MCB” in this study). It is active across multiple HCV genotypes and has a high barrier to resistance (8–12). The in vitro-selected MCB-resistant mutant carrying the NS5B S282T substitution has not been detected to date in patients receiving MCB monotherapy or MCB–PEG-IFN–RBV triple therapy (9, 11, 12). Recently, a double NS5B mutant (the L159F L320F mutant) which confers low-level resistance to MCB was identified in a single patient who received MCB–PEG-IFN–RBV triple therapy for 24 weeks (12).
Danoprevir is a macrocyclic HCV protease inhibitor that has shown potent antiviral activity against HCV GTs 1, 4, 5, and 6 in vitro (13–15) and GTs 1 and 4 in vivo (16, 17). Resistance to DNV has been characterized in vitro (14) and in clinical trials (18–21). A subtype-specific (GT 1a versus GT 1b) response and resistance to DNV-containing regimens have been observed; the most frequently observed substitutions associated with DNV resistance are at NS3 protease position R155 for GT 1a, whereas substitutions at both R155 and D168 have been reported for the GT 1b subtype in DNV monotherapy (20) and triple therapy with ritonavir-boosted DNV (DNV/r), PEG-IFN, and RBV (21).
Identification of resistance-associated mutations by comparing sequence changes before and after in vitro selection or in vivo treatment has been the focus of many studies (for reviews, see references 22 and 23. In the present study, we report on the variants known to be resistant to MCB and DNV in the phase IIb INFORM-SVR study (ClinicalTrials.gov registration no. NCT01278134), which investigated the efficacy, safety, and tolerability of interferon-free treatment with MCB and DNV (ritonavir-boosted) in combination with RBV and without RBV in treatment-naive GT 1-infected patients. Furthermore, to broaden the scope of sequence analysis, we hypothesized that, in addition to known substitutions associated with resistance, there may be specific changes in the NS3 and/or NS5B regions associated with other aspects of the virus life cycle (such as viral fitness) which could have a role in treatment outcome (e.g., in patients who achieved sustained a virologic response at follow-up week 24 [SVR24] versus non-SVR24 patients and in patients who experience viral breakthrough versus patients who relapse during follow-up [relapsers]) and that these changes may preexist at the baseline or may be selected during treatment. We tested our hypothesis by statistical analysis of the clinical sequencing data from the INFORM-SVR study and subsequent mutagenesis and phenotypic characterization using the subgenomic replicon system.
MATERIALS AND METHODS
Study design and patient population.
HCV GT 1-infected treatment-naive patients received MCB (1,000 mg twice a day [BID]), 100 mg DNV BID plus 100 mg ritonavir BID (DNV/r) with RBV (1,000-mg [body weight, <75 kg] or 1,200-mg [body weight, ≥75 kg] daily split doses) in arm A or with RBV placebo in arm B for 12 to 24 weeks. Open-label PEG-IFN–RBV for 24 weeks was offered to some patients in arm B due to a higher-than-expected treatment failure rate. Only patients who received IFN-free DAA treatment were included in the resistance analysis for this report; patients who received PEG-IFN–RBV extension treatment were excluded. The study was conducted in full conformance with the principles of the Declaration of Helsinki and good clinical practice. The protocol and all amendments were reviewed and approved by local ethics committees and regulatory authorities. The study was conducted in full conformance with the principles of the “Declaration of Helsinki” or with the laws and regulations of the country in which the research was conducted. In the European Union (EU) and European Economic Area (EEA) countries, the study was in compliance with the EU Clinical Trial Directive. Written informed consent was obtained from all patients before any study-related activities occurred.
Samples for HCV resistance monitoring.
Genotypic and phenotypic characterization of the NS3/4A and NS5B coding regions was performed on samples from patients who experienced virologic failure, defined as follows. (i) Viral breakthrough patients showed either a sustained increase in the HCV RNA load of ≥1 log10 IU/ml (≥2 consecutive measurements) while on DAA treatment compared with the on-treatment nadir or had confirmed quantifiable HCV RNA (≥43 IU/ml) if the patient had previously been confirmed to have undetectable HCV RNA (<15 IU/ml) before the end of DAA treatment. Subjects who became HCV RNA positive (>15 IU/ml) at the last visit of DAA treatment were considered viral breakthrough patients. (ii) Patients with a partial response showed an initial HCV RNA decline (a ≥1-log10 IU/ml decrease from the baseline after 2 weeks of treatment or a ≥2-log10 IU/ml decrease from the baseline after 4 weeks of treatment), followed by stabilization (≥2 consecutive measurements within 0.5 log10 IU/ml of the nadir) while on DAA treatment, and/or experienced an HCV RNA load of ≥1,000 IU/ml by the end of DAA treatment of at least 4 weeks in duration. (iii) Patients with a nonresponse showed a decrease in the HCV RNA load of <1 log10 IU/ml from the baseline after 2 weeks of treatment or <2 log10 IU/ml after 4 weeks of treatment. (iv) Patients with relapses were those who responded to DAA treatment and then had a subsequent quantifiable viral load (≥43 IU/ml by ≥2 consecutive measurements) once DAA treatment was withdrawn.
Baseline samples from all patients were sequenced. For the purpose of sequence determination, an HCV RNA load of ≥1,000 IU/ml was required.
Nucleotide sequence analyses of NS3/4A and NS5B coding regions from clinical isolates.
Extraction of HCV RNA from serum samples, amplification of the NS3/4A and NS5B coding regions, and nucleotide sequence analysis of the PCR products were performed at the DDL Diagnostic Laboratory (Voorburg, The Netherlands). Briefly, viral RNA was isolated from patient serum using a QIAamp MinElute virus spin kit (Qiagen, Hilden, Germany), and the NS3/4A (encompassing amino acids 1 to 685) or NS5B (encompassing amino acids 1 to 592) region was amplified by reverse transcription-PCR (RT-PCR) using proprietary DDL primers flanking the region of interest. The PCR products were sequenced using an ABI 3130xl genetic analyzer according to the manufacturer's instructions. For sequence analysis, strain H77 (GenBank accession number AF009606) was used as the reference for GT 1a and strain Con1 (GenBank accession number AJ238799) was used as the reference for GT 1b. For the purpose of sequence determination, an HCV RNA load of ≥1,000 IU/ml was required. The detection limit for minor variants from mixtures was ∼20%.
Clonal sequencing of the NS3/4A gene from clinical isolates was carried out by cloning the PCR product amplified from patient serum into a pCR2.1-TOPO vector (Invitrogen, Carlsbad, CA), and ∼100 clones were sequenced for each sample. The NS3 protease coding region from selected clones carrying wild-type sequences or R155K and D168E substitutions was subcloned into the GT 1b replicon shuttle vector for phenotypic analysis, as described below.
Cell culture and HCV inhibitors.
Cured Huh7 cells, generated by removal of the replicon by alfa IFN treatment for 2 weeks, which made them highly permissive for HCV replicon replication (24), were obtained from Ralf Bartenschlager (University of Heidelberg, Heidelberg, Germany). PSI-6130 (25) was obtained from Pharmasset, Inc. (Princeton, NJ), and danoprevir (26) was obtained from InterMune, Inc. (Brisbane, CA). Compound stocks (10 mM) were prepared in 100% dimethyl sulfoxide (DMSO). Alfa-2a IFN (Roferon-A) was manufactured by Hoffmann-La Roche (27). Stock solutions of alfa-2a IFN were diluted to 106 IU/ml in Dulbecco modified Eagle medium containing 10% (vol/vol) fetal bovine serum.
Site-directed mutagenesis and phenotypic analysis.
Site-directed mutagenesis was used to introduce mutations identified in NS3 and NS5B coding regions into GT 1a (H77) replicons using a QuikChange site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA).
Phenotypic characterization of clinical isolates was performed by measuring the drug susceptibility of chimeric replicons carrying patient-derived NS3 protease or the NS5B coding region at Monogram Biosciences Inc. (South San Francisco, CA) (28) for the majority of the patient samples or in-house for samples from patient 1 (9), using similar experimental procedures. Briefly, viral RNA was isolated from patient serum, and the NS3 protease or NS5B coding region was amplified by RT-PCR using primers containing restriction sites flanking the region of interest and cloned into replicon shuttle vectors carrying the luciferase reporter gene. For samples from patient 1 (GT 1a infected), which were phenotyped in-house, the NS5B coding region was cloned into the GT 1a replicon backbone to match the patient subtype, whereas the NS3 protease coding region was cloned into the GT 1b replicon because it was the only system available in-house which allowed efficient replication of NS3 chimeric replicons. For all samples phenotyped at Monogram Sciences, the GT 1b replicon backbone was used per the standard protocol established at the facility.
In vitro-transcribed RNAs from replicons containing site-directed mutations or coding regions from clinical isolates were electroporated into cured Huh7 cells, and inhibitor susceptibility was determined by evaluating the ability of chimeric replicons to replicate in the absence and presence of inhibitor at 72 to 96 h postelectroporation. The percent inhibition at each serially diluted inhibitor concentration was calculated as follows: [1 − (luciferase activity in the presence of inhibitor/luciferase activity in the absence of inhibitor)] × 100.
The inhibitor concentrations required to reduce virus replication by 50% (EC50s) were extrapolated from fitted curves by using the XLfit program (ID Business Solutions Ltd., Surrey, United Kingdom).
The replication capacity was determined in-house as the luciferase signal at 96 h posttransfection divided by the luciferase signal at 4 h posttransfection and expressed as the normalized replication efficiency compared to that of the reference replicons, which was set equal to 1. To determine the relative fitness of mutant replicons under DNV treatment, the replication capacity was measured under treatment with DNV at 0.1 nM, 0.8 nM, and 4 nM (1× EC50, 1× EC90, and 5× EC90 of the wild-type GT 1a replicon, respectively). To determine the relative fitness of mutant replicons under DNV and MCB combination treatment, the replication capacity was measured under treatment with 0.8 nM DNV and 1.4 μM MCB (each at 1× EC90).
HCV RNA amplification for 454 sequencing.
Reverse transcription of viral RNA isolated from patient serum was performed using random hexamers as primers following the manufacturer's instructions (Transcriptor high-fidelity cDNA synthesis kit; Roche Diagnostics, Indianapolis, IN).
Amplification of the NS5B region (generating a 985-nucleotide fragment for GT 1a-infected samples and a 916-nucleotide fragment for GT 1b-infected samples) and the NS3 region (generating a 750-nucleotide fragment for GT 1a-infected samples and an 806-nucleotide fragment for GT 1b-infected samples) was carried out in duplicate using a FastStart high-fidelity PCR system (Roche Diagnostics, Indianapolis, IN) under the following PCR conditions: 3 min at 94°C and then 30 cycles of 30 s at 94°C, 30 s at 52°C, and 1 min at 72°C, followed by a final 7-min extension step at 72°C. The duplicate products of this first-round PCR (PCR1) were pooled and then amplified in a nested PCR (PCR2) using 454 fusion primers containing custom 7-mer bar codes (designed to be able to pool amplified DNA from different patient samples while reducing the chance for cross-sample contamination) and the same cycling conditions used for PCR1. PCR2 produced 3 smaller overlapping amplicons (308- to 368-nucleotide size range) encompassing NS5B amino acids 244 to 496 and 2 smaller overlapping amplicons (244- to 356-nucleotide size range) encompassing NS3 amino acids 31 to 175 and 30 to 190 for GT 1a and GT 1b isolates, respectively.
Ultradeep pyrosequencing (UDPS) analysis using 454/Roche FLX.
Standard flowgram format (SFF) files were processed to generate paired files containing FASTA sequence reads and Phred-equivalent quality scores for each sequence library. To reduce sequence artifacts, we excluded reads shorter than 200 nucleotides and removed reads containing one or more bases with a quality score of <10 or a mean quality score of <25. Sequenced reads were demultiplexed using the 5′ primer and bar code sequences, and each UDPS read was aligned to the population-based sequence of each sample using the MosaikAligner program (http://bioinformatics.bc.edu). The technical error rate was estimated by sequencing two plasmids carrying HCV reference strains, GT 1a strain H77 and GT 1b strain Con1. Each UDPS read was aligned to the corresponding plasmid sequence, and the number of mismatches was counted. Assuming that errors occurred in a Poisson distribution and that samples contained an input viral copy number of ∼1,000, the limit of detection (LOD) in variant calling was established at 0.5% using published methods (29).
Statistical analysis.
To identify amino acid substitutions in NS3 or NS5B regions which might be associated with treatment response, the Fisher exact test was carried out to compare the prevalence of (i) a given baseline polymorphism in SVR24 patients versus non-SVR24 patients and (ii) a given postbaseline substitution in viral breakthrough versus relapser patients. Baseline polymorphisms were defined by comparing the NS3 or NS5B Sanger population sequence of a sample at the baseline to the GT 1a H77 reference sequence, whereas postbaseline substitutions were defined by comparing postbaseline Sanger population sequences to the corresponding sequence at the baseline. The prevalence of a given substitution was determined as the proportion of patients carrying the substitution. Due to the small number of GT 1b-infected patients in the study, the analysis was carried out with GT 1a-infected patients only. The P values from the Fisher exact test were also adjusted by the Benjamini-Hochberg procedure (30).
Nucleotide sequence accession numbers.
The sequences from clinical isolates have been deposited in GenBank, and the corresponding accession numbers are KJ529536 to KJ530550.
RESULTS
Detection of MCB and DNV dual resistance in patients who experienced viral breakthrough.
A total of 169 patients (113 GT 1a-infected patients, 56 GT 1b-infected patients) received at least one dose of treatment of DNV/r and MCB with RBV (arm A) or without RBV (arm B). Thirty patients (24 GT 1a-infected patients, 6 GT 1b-infected patients) experienced viral breakthrough by the end of DAA treatment. No patients experienced a partial response or nonresponse while on DAA treatment. Sixty-three patients (48 GT 1a-infected patients, 15 GT 1b-infected patients) experienced a relapse after the end of DAA treatment.
Out of the 30 patients who experienced viral breakthrough, 3 had known NS5B substitutions associated with MCB resistance detected by population sequencing. The NS5B S282T substitution was detected at the time of premature discontinuation in two GT 1a-infected patients (patients 1 and 2) from arm B. The NS5B substitutions L159F (L159F and L159L as a mixture) and L320F were detected in a single GT 1a-infected patient (patient 3) from arm A at treatment week 24. Subsequent resequencing of the week 24 sample of this patient (as part of the procedure for phenotypic analysis) revealed only the L320F single substitution, suggesting that the L159F substitution was present at a low abundance in this sample. No additional serum from week 24 or other posttreatment samples from this patient were available for further analysis, such as by clonal sequencing; we therefore could not confirm the presence of the L159F and L320F double mutation in patient 3.
The three viral breakthrough patients with NS5B substitutions also had the NS3 R155K substitution that confers resistance to DNV. In patients 1 and 2, UDPS analysis (Fig. 1) concurrently showed a high prevalence (≥70%) of NS5B S282T and NS3 R155K at one or more time points (at follow-up week 4 for patient 1 and at the time of premature discontinuation and follow-up week 2 for patient 2), suggesting that a proportion of the viruses (at least ∼40%, according to Dirichlet's drawer principle) likely contained the NS5B S282T and NS3 R155K substitutions on the same viral genome.
FIG 1.
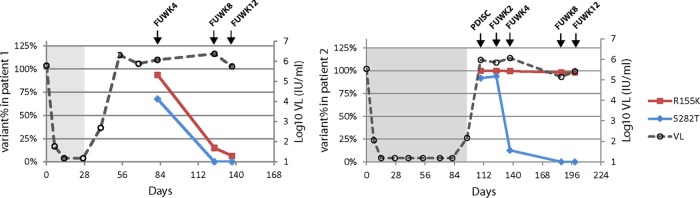
UDPS analysis of persistence of NS5B S282T and NS3 R155K substitutions after cessation of drug treatment (during follow-up). The percentage of NS5B S282T and NS3 R155K determined by UDPS is plotted over study days for patients 1 (A) and 2 (B). FUWK, follow-up week; PDISC, premature discontinuation; VL, viral load. The on-treatment period is shaded. The reversion of S282T to the wild type occurred in both patients after the cessation of drug treatment (during follow-up). The LOD by UDPS was 0.5% (see Materials and Methods for details).
The persistence of the NS5B S282T substitution after cessation of drug treatment (during follow-up) was investigated by UDPS. The NS5B S282T substitution was detected up to follow-up week 4 (Fig. 1) and replaced by wild-type virus at follow-up week 8 in both patients 1 and 2 (LOD of the assay = 0.5%). UDPS was also applied to the baseline samples and revealed a single 338-bp read with S282T out of 3,972 reads in patient 2 and none of 9,979 reads in patient 1. UDPS of the same NS5B fragment from the baseline samples of 11 additional GT 1a-infected breakthrough patients who did not develop the S282T substitution during treatment showed no read with the change (average number of reads per sample, 5,599; range, 2,315 to 11,698). Since patients 1 and 2 also had the NS3 R155K substitution, UDPS was used to investigate its persistence during follow-up. Patients 1 and 2 showed different kinetics of persistence of the NS3 R155K substitution (Fig. 1). In patient 1, the R155K abundance decreased from 94% at follow-up week 4 to 6.4% at follow-up week 12; in patient 2, R155K persisted at a high abundance (>95%) up to follow-up week 12.
Among the 63 relapser patients, no known substitutions associated with MCB resistance were detected by population sequencing.
Subtype-specific (GT 1a versus GT 1b) DNV resistance among patients who experienced viral breakthrough or relapse.
Among GT 1a-infected patients who experienced viral breakthrough or relapse in this study, the NS3 R155K substitution was found to be the major resistance pathway, detected in all patients with sequencing data at the point of breakthrough (n = 22) or relapse (n = 47). In contrast, among GT 1b-infected breakthrough (n = 5) and relapser (n = 15) patients with sequencing data, substitutions were more frequently observed at the NS3 D168 position (n = 15) than at the R155 position (n = 5); one GT 1b-infected relapser had no known changes associated with DNV resistance by population sequencing (Table 1). Substitutions at position V36 were found only in conjunction with R155K (V36A/M with R155K in 8 GT 1a-infected patients) or D168E (V36A/G with D168E in 2 GT 1b-infected patients). Substitutions at both positions R155 and D168 were observed in 4 GT 1a-infected patients and 1 GT 1b-infected patient (Table 1).
TABLE 1.
NS3 substitutions conferring DNV resistance observed in patients at the time of viral breakthrough or relapse
| Patient population | No. of patients with the following NS3 substitution(s): |
No. of patients for which: |
Total no. of patients evaluated | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| R155K | D168A/E/T | V36A/M R155K | V36A/G D168E | R155K D168A/E | R155Q D168A | None | Total | Assay was NDa | Assay failed | ||
| Breakthrough | |||||||||||
| GT 1a infected | 16 | 0 | 6 | 0 | 0 | 0 | 0 | 22 | 2 | 0 | 24 |
| GT 1b infected | 3 | 1 | 0 | 1 | 0 | 0 | 0 | 5 | 1 | 0 | 6 |
| Relapser | |||||||||||
| GT 1a infected | 41 | 0 | 2 | 0 | 4 | 0 | 0 | 47 | 0 | 1 | 48 |
| GT 1b infected | 1 | 11 | 0 | 1 | 0 | 1 | 1 | 15 | 0 | 0 | 15 |
ND, not done due to low viral load.
Phenotypic characterization of replicons containing NS3 and NS5B coding regions from clinical isolates.
NS3 and NS5B phenotypic analysis was performed on clinical isolates from the three GT 1a-infected patients who developed variants resistant to MCB and DNV. In the two patients (patients 1 and 2) where NS5B S282T was observed, samples carrying NS5B S282T (at the time of premature discontinuation of patient 1 and at the time of premature discontinuation and follow-up week 2 of patient 2) showed low-level phenotypic resistance to MCB (a 3- to 4-fold increase in the EC50 over the day 1 baseline EC50), as well as a 75- to 490-fold increase in the DNV EC50 compared to the day 1 baseline EC50, demonstrating the dual resistance phenotype of the isolates from these two patients (see Tables S1 and S2 in the supplemental material). In patient 3, who developed the substitutions NS5B L159F/L (mixture) and L320F at week 24, NS5B phenotyping showed a 1.6-fold change in the MCB EC50 over the day 1 baseline EC50 (1.3-fold over the EC50 for the GT 1b Con1 reference control). We have previously reported a 2.7-fold increase in the MCB EC50 with the L320F single mutant and a 4.3-fold increase with the L159F L320F double mutant using the GT 1a H77 replicon systems (12). The change in MCB susceptibility in the week 24 sample from patient 3 is consistent with the predominance of the L320F single mutant on the basis of sequencing analysis. The small differences (∼2-fold) in the MCB EC50 fold change between this study and the previous study (12) could be due to assay variation at the two facilities (Monogram Sciences versus in-house) or the different genetic backgrounds of the NS5B coding region (patient isolate versus H77 sequence). NS3 phenotyping showed an 844-fold increase in the DNV EC50, consistent with the presence of DNV-resistant variants in this sample (see Tables S1 and S2 in the supplemental material).
NS3 clinical isolates from 18 patients who experienced viral breakthrough (n = 6) or relapse (n = 12) were subjected to phenotypic analysis (see Tables S2 and S3 in the supplemental material). A comparison of the EC50s while the patients were on treatment or at follow-up to those at the baseline showed a significant increase (11- to >1,000-fold compared to the day 1 baseline EC50s), confirming that substitutions at positions V36, R155, and D168 conferred resistance to DNV in GT 1a- and GT 1b-infected patients. It was noted that follow-up samples from patient 12 (infected with GT 1b), which did not have any known DNV-resistant variants, showed 6- to 11-fold increases in the EC50 over the day 1 baseline EC50, but the EC50s were only 2- to 3-fold greater than those for the Con1 reference control due to the low EC50 (0.27-fold that for Con1) of the day 1 baseline sample (see Table S3 in the supplemental material). The posttreatment changes in the NS3/4A region in this sample over the day 1 baseline sequence were N77S, M175L, R/S470S, and I/V668V, with prevalences of 2%, 0%, 3.5%, and 39%, respectively, among GT 1b-infected baseline samples from the INFORM-SVR study and of 0.6%, 0.6%, 4%, and 49%, respectively, among GenBank GT 1b sequences. The substitution NS3 M175L has been reported to confer a low level of resistance to the PI boceprevir (31). To evaluate its role in DNV resistance, NS3 M175L was introduced into the GT 1b Con1 replicon by site-directed mutagenesis and showed no effect on DNV susceptibility (a 1.1-fold change in the EC50 over that for Con1). Taken together, the evidence suggests that the follow-up samples from patient 12 were susceptible to DNV. The day 1 baseline EC50s for isolates from patients 2, 13, and 17 were 3- to 6-fold higher than the EC50 for the Con1 reference control. No known substitutions associated with DNV resistance or polymorphisms common to these three baseline samples were identified. Variations in baseline susceptibility to PIs among clinical isolates have been reported (32) and could be due to nonspecific differences in the virus genetic background.
Clonal sequencing and phenotypic analysis of clinical isolates carrying NS3 R155K and D168E substitutions in a patient sample showing a biphasic DNV dose-response.
NS3 phenotyping of the follow-up week 12 sample from patient 1 showed a biphasic dose-response curve for DNV (Fig. 2). The first-phase EC50 (EC50-1) value was calculated (GraphPad Prism, San Diego, CA) to be 0.09 ± 0.06 nM (mean ± standard deviation [SD], n = 4), which was similar to the EC50 of the Con1 reference control (0.11 ± 0.05 nM, n = 10) and the EC50 of the same patient's day 1 baseline isolate (0.24 ± 0.04 nM, n = 8). The second-phase EC50 (EC50-2) was 17.7 ± 0.9 nM (n = 4), an increase of 73-fold over the day 1 baseline EC50; however, no known substitutions associated with DNV resistance were detected in this sample by population sequencing (see Table S2 in the supplemental material).
FIG 2.
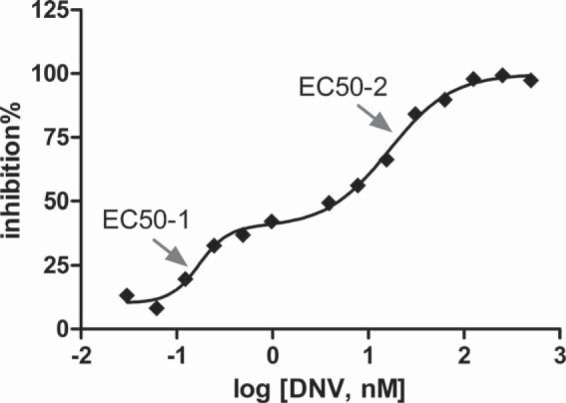
Biphasic DNV dose-response curve obtained by NS3 phenotyping of the patient 1 follow-up week 12 sample. The percentage of replicon inhibition is plotted against the log10 DNV concentration, and the data were fitted using the biphasic program in GraphPad Prism (version 5) for Windows (GraphPad Software, San Diego, CA). The arrows indicate EC50s calculated for the first and second phases. One representative of 4 independent experiments is shown.
We hypothesized that this biphasic behavior was due to the presence of mixtures of wild-type and minor species of resistant viruses not detectable by population sequencing. To test the hypothesis, clonal sequencing of the NS3 protease region from the follow-up week 12 serum sample was carried out prior to transfection. Clonal sequencing was also carried out by amplifying and cloning of replicon RNAs harvested at 96 h after RNA transfection in the phenotypic assay. To address the concern of contamination by input transfected RNA, a replication-deficient control replicon in which the GND motif replaced the GDD motif in NS5B (24) was included. Results showed that PCR products were amplified only from RNAs isolated from replication-competent replicons at 96 h posttransfection (data not shown), suggesting that active replication of replicon RNA is required for detection by RT-PCR under these conditions. Prior to transfection, clonal sequencing of the follow-up week 12 sample showed that both the D168E (13.7%) and R155K (5.6%) single mutants were present as minor variants in this sample (n = 124 clones). At 96 h posttransfection, clonal sequencing of replicon RNA isolated from transfected cells detected only the D168E sequence at a frequency of 18.2% (n = 77 clones), similar to the frequency seen in the serum sample prior to transfection. The absence of the R155K substitution posttransfection was consistent with the hypothesis that replicons carrying the R155K substitution were outgrown by the wild-type and D168E replicons during transfection and that the D168E mutant was largely responsible for the reduced susceptibility to DNV observed in the phenotypic assay at the population level (i.e., EC50-2).
To evaluate the replication capacity of the R155K and D168E mutants, 6 individual clones carrying a single NS3 sequence were isolated from the follow-up week 12 sample (Table 2): two clones had the wild-type sequence over the entire NS3 protease region, two clones had the R155K substitution, and two clones had the D168E substitution. No other changes were present in the NS3 protease region in the four clones with substitutions. The two clones carrying R155K indeed showed a lower replication capacity than the clones with D168E (5.0 to 7.2% versus 38%). In a drug susceptibility assay, reduced susceptibility to DNV was observed with clones carrying the R155K or D168E substitution (∼300-fold and ∼100-fold over that at the baseline, respectively).
TABLE 2.
Replication capacity and susceptibility to DNV of replicons containing individual NS3 sequences cloned from the follow-up week 12 sample of patient 1
| Samplea | NS3 substitution | Replication capacity (%) over that at day 1 | Mean ± SD DNV EC50 (nM)b | Fold increase in EC50 compared with that: |
|
|---|---|---|---|---|---|
| At day 1 | For Con1 | ||||
| Con1 (GT 1b) | WTc | 0.14 ± 0.06 (17) | |||
| Day 1 pool | WT | 100 | 0.19 ± 0.09 (9) | 1.3 | |
| Clone C6 | WT | 69 | 0.35 ± 0.15 (4) | 1.9 | 2.5 |
| Clone E4 | WT | 66 | 0.21 ± 0.07 (3) | 1.1 | 1.5 |
| Clone A4-1 | R155K | 7.2 | 58 ± 26 (7) | 313 | 414 |
| Clone E8 | R155K | 5.0 | 56 ± 15 (9) | 298 | 400 |
| Clone E5-1 | D168E | 38 | 18 ± 6 (4) | 94 | 128 |
| Clone F3-1 | D168E | 38 | 20 ± 6 (4) | 108 | 143 |
The NS3 protease coding region was cloned into the Con1 (GT 1b) replicon genetic background.
Values in parentheses are numbers of experiments.
WT, wild type.
Analysis of baseline polymorphisms that may be associated with SVR24.
To systematically explore the relationship between NS3/4A and NS5B baseline polymorphisms and treatment outcome, the Fisher exact test was performed to identify baseline variants that were differentially observed in GT 1a-infected SVR24 patients (n = 13) versus non-SVR24 patients (n = 72). A total of 256 and 248 distinct baseline polymorphisms were tested in NS3/4A and NS5B, respectively. After adjustment for multiple hypotheses by the Benjamini-Hochberg procedure, all P values were >0.05. However, we proceeded to evaluate several NS5B substitutions using P equal to 0.05 (by the Fisher exact test) as the cutoff. The rationale for using the P values from the Fisher exact test to prioritize potential response-associated substitutions for experimental testing is described in the Discussion.
No baseline polymorphism in NS3/4A was found to be different between SVR24 and non-SVR24 patients at the level of P equal to 0.05. One NS5B baseline polymorphism (Q47H) was found in 15% of GT 1a-infected SVR24 patients (2/13) but in 0% of non-SVR24 patients (P = 0.02) (Table 3). NS5B Q47H was introduced into the replicon of the same subtype as the patient samples (GT 1a) by site-directed mutagenesis, and drug susceptibility testing was performed. As controls, two NS5B amino acid changes (S90D and S180G) which did not show enrichment in SVR24 patients by the Fisher exact test (P > 0.05) were tested in parallel. The amino acid changes Q47H, S90D, and S180G in NS5B had no effect on MCB or DNV EC50 or EC90 values (Table 4 and data not shown). However, among the isolates with the 3 amino acid changes (NS5B Q47H, S90D, and S180G), those with the Q47H change had the lowest replication capacity in the GT 1a replicon system (replication capacities for isolates with the 3 amino acid changes, 0.25 ± 0.11, 0.37 ± 0.09, and 1.1 ± 0.4 relative to that for the reference replicon, respectively; n = 3 to 5). Statistical analysis showed no significant correlation between the presence of Q47H and the baseline viral load among the 13 SVR24 patients (P > 0.05, Mann-Whitney test).
TABLE 3.
Prioritization of NS3 and NS5B substitutions based on association with treatment responsea
| Coding region | Baseline polymorphism in SVR24 vs non-SVR24 patients |
Postbaseline substitutions in breakthrough vs relapser patients |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Substitution | No. of patients with polymorphism/total no. of patients tested (%) |
P value | Prevalence in GenBankb | Substitution | No. of patients with substitution/total no. of patients tested (%) |
P value | Prevalence in GenBank | |||
| SVR24 patients | Non-SVR24 patients | Breakthrough | Relapser | |||||||
| NS5B | Q47H | 2/13 (15) | 0/72 (0) | 0.02 | 3/169 (1.7) | L159F | 7/22 (32) | 0/48 (0) | 0.0001 | 0/169 (0) |
| S90D | 3/13 (23) | 5/72 (6.9) | 0.1 | 5/169 (3.0) | S189N | 3/22 (14) | 0/48 (0) | 0.03 | 0/169 (0) | |
| S180G | 1/13 (8) | 1/72 (1.4) | 0.28 | 3/169 (1.7) | ||||||
| NS3/4A | V36M | 5/22 (23) | 2/47 (4.3) | 0.03 | 1/308 (0.3) | |||||
| S122G | 4/22 (18) | 0/47 (0) | 0.008 | 12/308 (3.9) | ||||||
| A383G | 2/22 (9.1) | 0/47 (0) | 0.1 | 12/308 (3.9) | ||||||
Data for substitutions with P values (by the Fisher exact test) of <0.05 are in bold.
Curated HCV sequence database (37). Data represent the number of matches with sequences in GenBank/total number of sequences tested (percent).
TABLE 4.
Replication capacity and susceptibility to DNV and MCB of replicons containing NS3 and NS5B substitutions
| Mutant and substitution(s)a | Replication capacity (%) | DNV |
MCB |
||
|---|---|---|---|---|---|
| Mean ± SD EC50 (nM)b | EC50/WTc | Mean ± SD EC50 (μM)b | EC50/WT | ||
| NS3 mutants | |||||
| GT1a WT (H77) | 100 | 0.095 ± 0.03 (20) | 1 | 0.20 ± 0.09 (25) | 1 |
| NS3 R155K | 10 | 37.7 ± 9.7 (4) | 399 | 0.26 ± 0.05 (5) | 1.3 |
| NS3 V36M | 94 | 0.13 ± 0.04 (5) | 1.4 | 0.28 ± 0.11 (6) | 1.4 |
| NS3V36M R155K | 12 | 96.0 ± 34.4 (7) | 1,015 | 0.22 ± 0.07 (8) | 1.1 |
| NS3 S122G | 188 | 0.11 ± 0.03 (5) | 1.2 | 0.33 ± 0.10 (6) | 1.6 |
| NS3 S122G R155K | 45 | 32.3 ± 13.5 (5) | 341 | 0.28 ± 0.03 (5) | 1.4 |
| NS3 A383G | 105 | 0.11 ± 0.02 (3) | 1.2 | 0.32 ± 0.04 (4) | 1.5 |
| NS3 A383G R155K | 15 | 41.9 ± 4.2 (3) | 443 | 0.33 ± 0.11 (4) | 1.6 |
| NS3 and NS5B double mutants | |||||
| NS5B L159F | 3.0 | 0.05 ± 0.02 (4) | 0.5 | 0.49 ± 0.16 (7) | 2.4 |
| NS3 R155K NS5B L159F | 0.4 | 43.9 ± 4.6 (3) | 464 | 0.64 ± 0.25 (4) | 3.1 |
| NS5B S189N | 93 | 0.10 ± 0.03 (4) | 1.1 | 0.24 ± 0.05 (4) | 1.2 |
| NS3 R155K NS5B S189N | 13 | 27.9 ± 6.6 (4) | 295 | 0.23 ± 0.10 (4) | 1.2 |
| NS5B mutants | |||||
| NS5B Q47H | 25 | 0.06 ± 0.02 (3) | 0.7 | 0.20 ± 0.06 (3) | 1 |
| NS5B S90D | 37 | 0.05 ± 0.005 (3) | 0.5 | 0.18 ± 0.07 (5) | 0.9 |
| NS5B S180G | 109 | 0.11 ± 0.04 (5) | 1.1 | 0.19 ± 0.04 (5) | 0.9 |
Introduced as site-directed mutations into the H77 (GT 1a) replicon genetic background.
Values in parentheses are numbers of experiments.
EC50/WT, fold change in EC50 relative that for the wild type.
Analysis of postbaseline substitutions that may be associated with viral breakthrough.
Similar to the analysis of baseline polymorphisms, the Fisher exact test was carried out to identify postbaseline substitutions that were differentially observed in GT 1a-infected breakthrough versus relapser patients, regardless of whether they conferred resistance to MCB or DNV. A total of 314 and 308 distinct postbaseline substitutions were tested in NS3/4A and NS5B, respectively. Two substitutions in NS3 (V36M and S122G) and 2 in NS5B (L159F and S189N) were found to be more prevalent in breakthrough patients than relapsers, with P being <0.05 by the Fisher exact test (Table 3). After adjustment by the Benjamini-Hochberg procedure, all P values were >0.05. Experimental testing was performed for the 4 substitutions identified from the Fisher exact test, as well as one control amino acid change (NS3 A383G) which did not meet the condition of P being <0.05. GT 1a mutant replicons carrying single substitutions as well as substitutions in combination with NS3 R155K were constructed, since all GT 1a-infected breakthrough and relapser patients had the NS3 R155K substitution.
Of the three NS3 substitutions identified by this analysis (Table 4), V36M as a single substitution had little effect on the DNV EC50, but the isolate with the V36M R155K double substitution showed a 2.5-fold increase in the DNV EC50 over that for the isolate with the R155K single substitution. The substitutions S122G and A383G showed no effect on DNV susceptibility either as single substitutions or as substitutions in combination with R155K. All mutants remained fully susceptible to MCB. To determine the relative fitness of NS3 mutant replicons under DNV treatment, the replication capacity was measured under treatment with various doses of DNV. As expected, replicons carrying substitutions associated with DNV resistance (NS3 V36M, R155K, and V36M R155K) replicated with a higher efficiency under increasing concentrations of DNV than the respective DMSO-treated controls (Fig. 3). NS3 S122G increased the replication capacity by ∼2-fold relative to that for the GT 1a wild-type control in the presence and absence of DNV, and the S122G R155K double mutant had an ∼4-fold increased replication capacity over that for the R155K mutant in the presence and absence of DNV. No change in replication capacity was observed with the control NS3 A383G when it was present either as a single substitution or as a substitution in combination with R155K.
FIG 3.
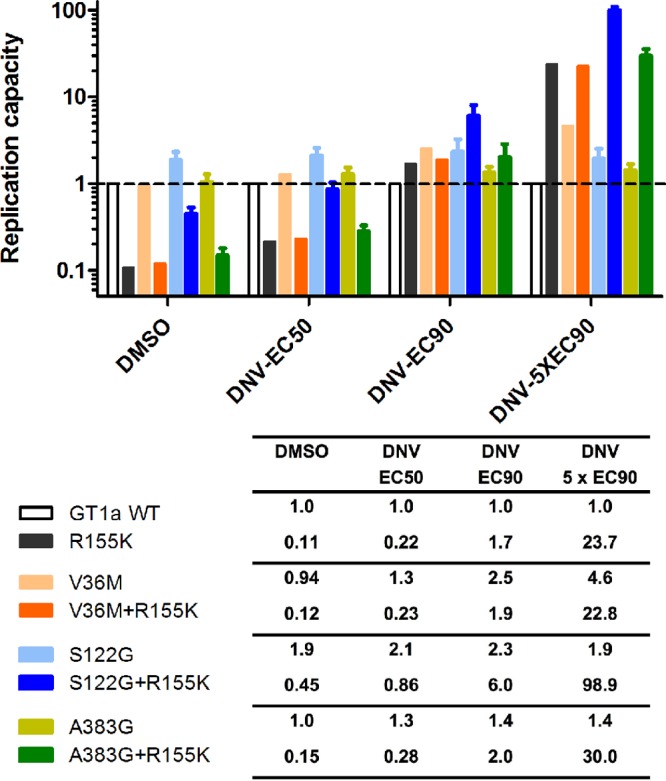
Replication capacity of NS3 mutant replicons under treatment with various concentrations of DNV. Replicon cells transfected with NS3 mutant constructs were dosed with 0.5% DMSO as a control or DNV, as indicated. Replication capacity was determined as the luciferase signal at 96 h posttransfection divided by that at 4 h posttransfection. The replication capacity of mutant replicons is expressed as a fraction of that of the GT 1a H77 reference replicon, which is set to a value of 1. Error bars represent standard deviations from 3 to 8 experiments. WT, wild type.
The three NS5B replicon mutants from this analysis remained fully susceptible to DNV (Table 4). The NS5B substitution L159F had a small effect (∼2-fold) on MCB susceptibility, as previously shown (12). No change in MCB susceptibility was seen in an isolate with the NS5B S189N double substitution. In the replicon fitness study under combined treatment with DNV and MCB (each at 1× EC90), the NS3 R155K and NS5B L159F single mutants showed 7- to 8-fold increases in replication capacity relative to that for their respective DMSO-treated controls. The NS5B L159F NS3 R155K double mutant showed a larger increase (45-fold) in replication capacity over that for the DMSO-treated control (Fig. 4). The isolate with the NS5B S189N substitution had a replication capacity similar to that of the GT 1a wild type, and the replication capacity did not change upon DNV and MCB treatment. No effect on replication capacity compared to that seen with the R155K mutation alone was observed when S189N was combined with R155K. A plausible explanation for the lack of an effect from S189N is that this amino acid substitution (P = 0.03) may represent a false-positive result from the statistical analysis; it is also possible that the relatively small effect of S189N on HCV replication cannot be detected using the replicon system.
FIG 4.
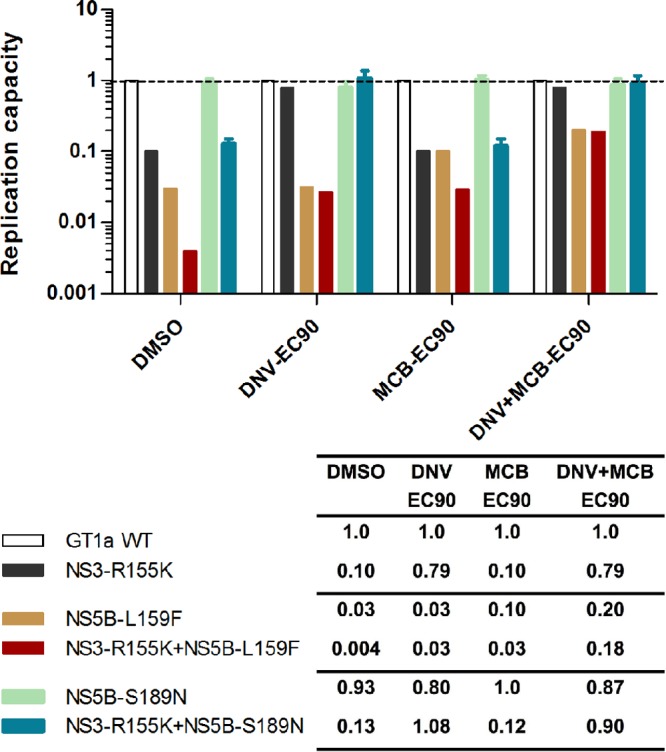
Replication capacity of NS5B mutant replicons under treatment with various concentrations of DNV and MCB. Replicon cells transfected with NS5B mutant constructs (with or without NS3 R155K) were dosed with 0.5% DMSO as a control or DNV and MCB, as indicated. Replication capacity was determined as the luciferase signal at 96 h posttransfection divided by that at 4 h posttransfection. The replication capacity of mutant replicons is expressed as a fraction of that of the GT 1a H77 reference replicon, which is set to a value of 1. Error bars represent standard deviations from 3 to 8 experiments.
DISCUSSION
In this study, genotyping and phenotyping analysis was carried out not only to monitor the emergence of HCV resistance to DNV and MCB but also, in combination with statistical analysis, to identify and characterize novel amino acid substitutions that may be associated with viral fitness and play a role in treatment outcome.
The NS5B S282T substitution is the only one that conferred resistance to MCB during selection in the in vitro replicon system and has not been detected to date in patients receiving MCB monotherapy or MCB–PEG-IFN–RBV triple therapy (8, 9, 11, 12). In the INFORM-1 study, where patients received MCB plus DNV for up to 13 days, clonal analysis of 10 patients showed that S282T was not present at the baseline, nor was it selected during treatment (33). In this larger study with a longer duration of treatment with MCB plus DNV, we identified two GT 1a-infected breakthrough patients who developed the NS5B S282T variant during treatment. The S282T mutant sequence was replaced by the wild-type sequence, as determined by UDPS, by follow-up week 8 in both patients, a finding which is in agreement with the reported low fitness of NS5B S282T mutants in replicon studies (8). The DNV-resistant NS3 R155K variant was also detected in these two patients. However, the persistence of NS3 R155K during follow-up was different in the two patients: in patient 1, the NS3 sequences had mostly reverted to the wild-type sequence by follow-up week 12, but the R155K variant persisted as the dominant variant in patient 2 throughout the follow-up period. It is of interest to note that the prevalence of resistant variants with changes in NS5B (S282T) and NS3 (R155K) can decrease at different rates in patients after the cessation of treatment, suggesting that independent reversion at each site rather than outgrowth of an archived wild-type sequence (at both sites) had occurred.
To address the prevalence of NS5B S282T prior to treatment, UDPS analysis of baseline sequences was carried out and found 1/∼3,000 reads with S282T in patient 1. A very low level of preexisting S282T (0.05%) was reported in the baseline sample of one patient who relapsed after receiving sofosbuvir monotherapy (34). In our UDPS study, because only a single read with S282T was detected in one baseline sample (but not in patient 2 or any of the other 11 patients analyzed by UDPS), we cannot rule out factors such as contamination and PCR polymerase error as potential causes of the false-positive results. Further studies of additional patients which generate more reads with longer lengths will be essential to establish a definitive conclusion, and the reproducibility and clinical relevance of such extremely low levels of preexisting variants remain to be determined.
Recently, an NS5B mutant with the double mutations L159F and L320F, which confer low-level resistance to MCB and sofosbuvir (a structurally related nucleoside inhibitor), has been reported (12). In this study, NS5B L159F/L (mixture) and L320F were detected in one GT 1a-infected breakthrough patient, and phenotypic analysis showed a minimal change (1.6-fold) in MCB susceptibility, similar to what was observed with the L320F single mutant (12). This result could be explained by the absence or low abundance of the L159F L320F double mutant in the sample (see Results). We and others have reported that low-fitness mutants such as the L159F L320F double mutant must be present at high frequencies to cause an observable decrease in susceptibility in the phenotypic assay (12, 33, 35). Taken together, our findings show that resistance to MCB is rare in patients receiving an IFN-free DAA treatment regimen and reaffirmed the high barrier to MCB resistance.
Amino acid substitutions conferring resistance to DNV were observed in all patients of this study who experienced viral breakthrough or relapse, except for one GT 1b-infected patient. Our study confirmed the subtype-specific pattern (GT 1a versus GT 1b) observed with DNV resistance (20, 21). The NS3 R155K substitution was the major resistance pathway in the GT 1a subtype, detected in all GT 1a-infected breakthrough and relapser patients. In GT 1b-infected patients, substitutions at the NS3 D168 position were more frequently observed than the R155K change. The R155K replacement of arginine (R) with lysine (K) requires 1 nucleotide change in the GT 1a genetic background, whereas 2 nucleotide changes are needed in GT 1b. Therefore, GT 1b viruses present a higher genetic barrier than GT 1a viruses to protease inhibitors that select R155K as a major resistance pathway. However, the NS3 R155K and D168E mutant viruses can coexist in the same patient, as demonstrated by clonal sequencing and phenotyping of the follow-up week 12 sample from patient 1. Our data also suggest that the competition between coexisting mutant and wild-type viruses is a complex process determined by the viral replication fitness of different variants. In the study of the biphasic dose-response to DNV of the follow-up week 12 sample (Fig. 2), the proportion of resistant viruses carrying D168E was <20%; one would thus expect a large proportion (∼80%) of the inhibition (corresponding to the prevalence of wild-type viruses in the mixture) to occur at low drug concentrations. However, the plateau for the first phase of inhibition was reached at ∼40% inhibition. As shown by Verbinnen et al. (35), in mixtures containing wild-type and resistant viruses with different replication capacities, the contributions of the wild-type viruses (as reflected by the plateau for the first phase of inhibition) can be different from what was expected on the basis of the replicon replication level measured when the replicons were transfected alone. It has been postulated that the discrepancy could arise from the dynamic competition between wild-type and mutant replicons for cellular resources when cotransfected into the same cells in the absence and/or presence of serially diluted inhibitors (35).
To systematically analyze the full repertoire of HCV baseline polymorphisms and on-treatment substitutions in relation to the treatment response, we combined statistical analysis of sequencing data with site-directed mutagenesis and phenotypic testing. Potential baseline polymorphisms and on-treatment substitutions associated with the treatment response were prioritized by the Fisher exact test using P equal to 0.05 as the cutoff. In general, P values can be adjusted by the use of procedures such as the Benjamini-Hochberg correction to control for false discovery; on the other hand, these procedures can increase the probability of incorrectly accepting the null hypothesis as false negative (36). Because the aim of our statistical analysis was to prioritize a set of testable hypotheses for experimental verification, we believe that it would be important to avoid the risk of accidental elimination of potentially interesting substitutions (false-negative results). Following this line of reasoning, we adopted the P values from the Fisher exact test as a tool to identify candidate amino acid changes for subsequent experimental testing.
Using the method outlined above, we have identified and characterized two novel amino acid substitutions from GT 1a-infected patients that were enriched in distinct treatment responses and affected replication capacity. The Q47H baseline polymorphism in NS5B was more prevalent in SVR24 patients than non-SVR24 patients; the postbaseline substitution S122G in NS3 was more prevalent in breakthrough patients than relapsers. Both substitutions are relatively rare natural variants; among GenBank and INFORM-SVR GT 1a baseline sequences, Q47H was present at a frequency of 1.7% in both populations, and S122G was present in the two populations at frequencies of 3.9% and 12.5%, respectively. Neither substitution conferred resistance to DNV or MCB in replicon studies. However, replicons carrying these two substitutions showed a significant impact on replication capacity. Q47H in NS5B (which was more prevalent in SVR24 patients) reduced the replicon capacity compared to that for the wild-type GT 1a replicon, suggesting that a lower viral fitness at the baseline may contribute to a more favorable treatment outcome. It would be interesting to test the hypothesis by assessing the association of the replication capacity of baseline samples with the SVR in future clinical studies. The S122G substitution in NS3 improved the replication capacity of the R155K mutant (a mutant with low fitness) under DNV treatment, which may explain its prevalence in breakthrough patients. In summary, this study suggests that a systematic analysis of sequence data from sets of clinical data on DAA combinations can provide valuable insights into viral fitness in relation to the treatment response, complementary to what is currently known from studies on drug resistance.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by Hoffmann-La Roche.
All of us were employees of Hoffmann-La Roche at the time of the study.
We thank Sophie Le Pogam for coordinating the 454 sequencing collaboration and helpful discussions, Severine Margeridon-Thermet and Tommy Liu for 454 sequencing data and quality control, and Mariola Ilnicka, Katherine Piso, and Victoria Baronas for excellent technical assistance.
Footnotes
Published ahead of print 17 March 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02672-13.
REFERENCES
- 1.Ghany MG, Nelson DR, Strader DB, Thomas DL, Seeff LB. 2011. An update on treatment of genotype 1 chronic hepatitis C virus infection: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 54:1433–1444. 10.1002/hep.24641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jacobson IM, McHutchison JG, Dusheiko G, Di Bisceglie AM, Reddy KR, Bzowej NH, Marcellin P, Muir AJ, Ferenci P, Flisiak R, George J, Rizzetto M, Shouval D, Sola R, Terg RA, Yoshida EM, Adda N, Bengtsson L, Sankoh AJ, Kieffer TL, George S, Kauffman RS, Zeuzem S. 2011. Telaprevir for previously untreated chronic hepatitis C virus infection. N. Engl. J. Med. 364:2405–2416. 10.1056/NEJMoa1012912 [DOI] [PubMed] [Google Scholar]
- 3.Poordad F, McCone J, Jr, Bacon BR, Bruno S, Manns MP, Sulkowski MS, Jacobson IM, Reddy KR, Goodman ZD, Boparai N, DiNubile MJ, Sniukiene V, Brass CA, Albrecht JK, Bronowicki JP. 2011. Boceprevir for untreated chronic HCV genotype 1 infection. N. Engl. J. Med. 364:1195–1206. 10.1056/NEJMoa1010494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sherman KE, Flamm SL, Afdhal NH, Nelson DR, Sulkowski MS, Everson GT, Fried MW, Adler M, Reesink HW, Martin M, Sankoh AJ, Adda N, Kauffman RS, George S, Wright CI, Poordad F. 2011. Response-guided telaprevir combination treatment for hepatitis C virus infection. N. Engl. J. Med. 365:1014–1024. 10.1056/NEJMoa1014463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jacobson IM, Dore GJ, Foster GR, Fried MW, Manns MP, Marcellin P, Poordad F, Araujo ES, Peeters M, Lenz O, Ouwerkerk-Mahadevan S, De La Rosa G, Kalmeijer R, Sinha R, Beumont-Mauviel M. 2013. Simeprevir (TMC435) with peginterferon/ribavirin for treatment of chronic HCV genotype 1 infection in treatment-naïve patients: efficacy in difficult-to-treat patient sub-populations in the QUEST 1 and 2 phase III trials. Hepatology 58:756A–757A [Google Scholar]
- 6.Pawlotsky JM. 2006. Hepatitis C virus population dynamics during infection. Curr. Top. Microbiol. Immunol. 299:261–284. 10.1007/3-540-26397-7_9 [DOI] [PubMed] [Google Scholar]
- 7.Gane EJ, Roberts SK, Stedman CA, Angus PW, Ritchie B, Elston R, Ipe D, Morcos PN, Baher L, Najera I, Chu T, Lopatin U, Berrey MM, Bradford W, Laughlin M, Shulman NS, Smith PF. 2010. Oral combination therapy with a nucleoside polymerase inhibitor (RG7128) and danoprevir for chronic hepatitis C genotype 1 infection (INFORM-1): a randomised, double-blind, placebo-controlled, dose-escalation trial. Lancet 376:1467–1475. 10.1016/S0140-6736(10)61384-0 [DOI] [PubMed] [Google Scholar]
- 8.Ali S, Leveque V, Le Pogam S, Ma H, Philipp F, Inocencio N, Smith M, Alker A, Kang H, Najera I, Klumpp K, Symons J, Cammack N, Jiang WR. 2008. Selected replicon variants with low-level in vitro resistance to the hepatitis C virus NS5B polymerase inhibitor PSI-6130 lack cross-resistance with R1479. Antimicrob. Agents Chemother. 52:4356–4369. 10.1128/AAC.00444-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Le Pogam S, Seshaadri A, Ewing A, Kang H, Kosaka A, Yan JM, Berrey M, Symonds B, De La Rosa A, Cammack N, Najera I. 2010. RG7128 alone or in combination with pegylated interferon-alpha2a and ribavirin prevents hepatitis C virus (HCV) replication and selection of resistant variants in HCV-infected patients. J. Infect. Dis. 202:1510–1519. 10.1086/656774 [DOI] [PubMed] [Google Scholar]
- 10.Pawlotsky JM, Najera I, Jacobson I. 2012. Resistance to mericitabine, a nucleoside analogue inhibitor of HCV RNA-dependent RNA polymerase. Antivir. Ther. 17:411–423. 10.3851/IMP2088 [DOI] [PubMed] [Google Scholar]
- 11.Le Pogam S, Yan JM, Kosaka A, Ji Y, Gonzaludo N, Ewing A, Klumpp K, Najera I. 2010. No evidence of drug resistance or baseline S282T resistance mutation among GT 1 and GT 4 HCV infected patients on nucleoside polymerase inhibitor RG7128 and peg-IFN/RBV combination treatment for up to 12 weeks: interim analysis from the PROPEL study. Hepatology 52:701A–702A [Google Scholar]
- 12.Tong X, Le Pogam S, Li L, Haines K, Piso K, Baronas V, Yan JM, So SS, Klumpp K, Najera I. 2014. In vivo emergence of a novel mutant L159F/L320F in the NS5B polymerase confers low-level resistance to the HCV polymerase inhibitors mericitabine and sofosbuvir. J. Infect. Dis. 209:668–675. 10.1093/infdis/jit562 [DOI] [PubMed] [Google Scholar]
- 13.Gottwein JM, Scheel TK, Jensen TB, Ghanem L, Bukh J. 2011. Differential efficacy of protease inhibitors against HCV genotypes 2a, 3a, 5a, and 6a NS3/4A protease recombinant viruses. Gastroenterology 141:1067–1079. 10.1053/j.gastro.2011.06.004 [DOI] [PubMed] [Google Scholar]
- 14.Imhof I, Simmonds P. 2011. Genotype differences in susceptibility and resistance development of hepatitis C virus to protease inhibitors telaprevir (VX-950) and danoprevir (ITMN-191). Hepatology 53:1090–1099. 10.1002/hep.24172 [DOI] [PubMed] [Google Scholar]
- 15.Seiwert SD, Andrews SW, Jiang Y, Serebryany V, Tan H, Kossen K, Rajagopalan PT, Misialek S, Stevens SK, Stoycheva A, Hong J, Lim SR, Qin X, Rieger R, Condroski KR, Zhang H, Do MG, Lemieux C, Hingorani GP, Hartley DP, Josey JA, Pan L, Beigelman L, Blatt LM. 2008. Preclinical characteristics of the hepatitis C virus NS3/4A protease inhibitor ITMN-191 (R7227). Antimicrob. Agents Chemother. 52:4432–4441. 10.1128/AAC.00699-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Everson G, Cooper C, Hezode C, Shiffman ML, Yoshida E, Beltran-Jaramillo T, Ferenci P, Zeuzem S, Brunda M, Shulman N, Navarro M, Voulgari A, Najera I, Le Pogam S, Yetzer ES. 2012. Rapid and sustained achievement of undetectable HCV RNA during treatment with ritonavir-boosted danoprevir/PEG-IFNa-2a/RBV in HCV genotype 1 or 4 patients: Dauphine week 12 interim analysis. J. Hepatol. 56:S466. 10.1016/S0168-8278(12)61189-1 [DOI] [Google Scholar]
- 17.Gane EJ, Rouzier R, Stedman C, Wiercinska-Drapalo A, Horban A, Chang L, Zhang Y, Sampeur P, Najera I, Smith P, Shulman NS, Tran JQ. 2011. Antiviral activity, safety, and pharmacokinetics of danoprevir/ritonavir plus PEG-IFN alpha-2a/RBV in hepatitis C patients. J. Hepatol. 55:972–979. 10.1016/j.jhep.2011.01.046 [DOI] [PubMed] [Google Scholar]
- 18.Le Pogam S, Navarro M, Bu L, Voulgari A, Ilnicka M, Yan JM, Klumpp K. 2012. Low rate of on-treatment resistance to danoprevir boosted by ritonavir (DNVR) combined with PEG-IFNA-2a/ribavirin: 12 week interim analysis from Dauphine study. J. Hepatol. 56:S472. 10.1016/S0168-8278(12)61203-3 [DOI] [Google Scholar]
- 19.Le Pogam S, Yan JM, Chhabra M, Ilnicka M, Ji Y, Zhang Y, Shulman N, Klumpp K, Nájera I. 2011. Low prevalence of danoprevir resistance identified in GT1b HCV patients with prior null response treated with danoprevir plus low-dose ritonavir plus PEGIFNa-2a(40kd)/RBV for 12 weeks. J. Hepatol. 54:S485. 10.1016/S0168-8278(11)61229-4 [DOI] [Google Scholar]
- 20.Lim SR, Qin X, Susser S, Nicholas JB, Lange C, Herrmann E, Hong J, Arfsten A, Hooi L, Bradford W, Najera I, Smith P, Zeuzem S, Kossen K, Sarrazin C, Seiwert SD. 2012. Virologic escape during danoprevir (ITMN-191/RG7227) monotherapy is hepatitis C virus subtype dependent and associated with R155K substitution. Antimicrob. Agents Chemother. 56:271–279. 10.1128/AAC.05636-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Le Pogam S, Navarro M, Chi B, Voulgari A, Klumpp K, Najera I. 2012. Overall low rate of resistance to danoprevir (DNV) in HCV genotype (G) 1/4 patients treated with ritonavir-boosted danoprevir (DNVr) plus peginterferon alfa-2a (40KD)/ribavirin (P/R) in the DAUPHINE study. Hepatology 56:571A–572A [Google Scholar]
- 22.Thompson AJ, McHutchison JG. 2009. Antiviral resistance and specifically targeted therapy for HCV (STAT-C). J. Viral Hepat. 16:377–387. 10.1111/j.1365-2893.2009.01124.x [DOI] [PubMed] [Google Scholar]
- 23.Kieffer TL, Kwong AD, Picchio GR. 2010. Viral resistance to specifically targeted antiviral therapies for hepatitis C (STAT-Cs). J. Antimicrob. Chemother. 65:202–212. 10.1093/jac/dkp388 [DOI] [PubMed] [Google Scholar]
- 24.Lohmann V, Hoffmann S, Herian U, Penin F, Bartenschlager R. 2003. Viral and cellular determinants of hepatitis C virus RNA replication in cell culture. J. Virol. 77:3007–3019. 10.1128/JVI.77.5.3007-3019.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clark JL, Hollecker L, Mason JC, Stuyver LJ, Tharnish PM, Lostia S, McBrayer TR, Schinazi RF, Watanabe KA, Otto MJ, Furman PA, Stec WJ, Patterson SE, Pankiewicz KW. 2005. Design, synthesis, and antiviral activity of 2′-deoxy-2′-fluoro-2′-C-methylcytidine, a potent inhibitor of hepatitis C virus replication. J. Med. Chem. 48:5504–5508. 10.1021/jm0502788 [DOI] [PubMed] [Google Scholar]
- 26.Jiang Y, Andrews SW, Condroski KR, Buckman B, Serebryany V, Wenglowsky S, Kennedy AL, Madduru MR, Wang B, Lyon M, Doherty GA, Woodard BT, Lemieux C, Do MG, Zhang H, Ballard J, Vigers G, Brandhuber BJ, Stengel P, Josey JA, Beigelman L, Blatt L, Seiwert SD. 2014. Discovery of danoprevir (ITMN-191/R7227), a highly selective and potent inhibitor of hepatitis C virus (HCV) NS3/4A protease. J. Med. Chem. 57:1753–1769. 10.1021/jm400164c [DOI] [PubMed] [Google Scholar]
- 27.Hoffmann-La Roche Inc. 2008. Roferon-A prescribing information, revised. Hoffmann-La Roche Inc., Nutley, NJ [Google Scholar]
- 28.Reeves JD, Petropoulos CJ, Huang W. 6 June 2013. Methods for the phenotypic detection of HCV inhibitor resistant subpopulations. Patent application US20130302782
- 29.Wang C, Mitsuya Y, Gharizadeh B, Ronaghi M, Shafer RW. 2007. Characterization of mutation spectra with ultra-deep pyrosequencing: application to HIV-1 drug resistance. Genome Res. 17:1195–1201. 10.1101/gr.6468307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B Stat. Methodol. 57:289–300 [Google Scholar]
- 31.Chase R, Skelton A, Xia E, Curry S, Liu S, McMonagle P, Huang HC, Tong X. 2009. A novel HCV NS3 protease mutation selected by combination treatment of the protease inhibitor boceprevir and NS5B polymerase inhibitors. Antiviral Res. 84:178–184. 10.1016/j.antiviral.2009.09.003 [DOI] [PubMed] [Google Scholar]
- 32.Bae A, Sun SC, Qi X, Chen X, Ku K, Worth A, Wong KA, Harris J, Miller MD, Mo H. 2010. Susceptibility of treatment-naive hepatitis C virus (HCV) clinical isolates to HCV protease inhibitors. Antimicrob. Agents Chemother. 54:5288–5297. 10.1128/AAC.00777-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Le Pogam S, Yan JM, Chhabra M, Ilnicka M, Kang H, Kosaka A, Ali S, Chin DJ, Shulman NS, Smith P, Klumpp K, Najera I. 2012. Characterization of hepatitis C virus (HCV) quasispecies dynamics upon short-term dual therapy with the HCV NS5B nucleoside polymerase inhibitor mericitabine and the NS3/4 protease inhibitor danoprevir. Antimicrob. Agents Chemother. 56:5494–5502. 10.1128/AAC.01035-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hedskog C, Gontcharova V, Han B, Martin R, Miller MD, Mo H, Svarovskaia ES. 2013. Evolution of the HCV viral population from the one patient with S282T detected at relapse after sofosbuvir monotherapy, abstr A9 Abstr. Int. Workshop HIV Hepatitis Virus Drug Resist. Curative Strategies, Toronto, Ontario, Canada [Google Scholar]
- 35.Verbinnen T, Jacobs T, Vijgen L, Ceulemans H, Neyts J, Fanning G, Lenz O. 2012. Replication capacity of minority variants in viral populations can affect the assessment of resistance in HCV chimeric replicon phenotyping assays. J. Antimicrob. Chemother. 67:2327–2337. 10.1093/jac/dks234 [DOI] [PubMed] [Google Scholar]
- 36.Feise RJ. 2002. Do multiple outcome measures require p-value adjustment? BMC Med. Res. Methodol. 2:8. 10.1186/1471-2288-2-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuiken C, Yusim K, Boykin L, Richardson R. 2005. The Los Alamos hepatitis C sequence database. Bioinformatics 21:379–384. 10.1093/bioinformatics/bth485 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.