

Abstract
Heteroresistance is the coexistence of populations with differing nucleotides at a drug resistance locus within a sample of organisms. Although Sanger sequencing is the gold standard for sequencing, it may be less sensitive than deep sequencing for detecting fluoroquinolone heteroresistance in Mycobacterium tuberculosis. Twenty-seven fluoroquinolone monoresistant and 11 fluoroquinolone-susceptible M. tuberculosis isolates were analyzed by Sanger and Illumina deep sequencing. Individual sequencing reads were analyzed to detect heteroresistance in the gyrA and gyrB genes. Heteroresistance to fluoroquinolones was identified in 10/26 (38%) phenotypically fluoroquinolone-resistant samples and 0/11 (P = 0.02) fluoroquinolone-susceptible controls. One resistant sample was excluded because of contamination with the laboratory strain M. tuberculosis H37Rv. Sanger sequencing revealed resistance-conferring mutations in 15 isolates, while deep sequencing revealed mutations in 20 isolates. Isolates with fluoroquinolone resistance-conferring mutations by Sanger sequencing all had at least those same mutations identified by deep sequencing. By deep sequencing, 10 isolates had a single fixed (defined as >95% frequency) mutation, while 10 were heteroresistant, 5 of which had a single unfixed (defined as <95% frequency) mutation and 5 had multiple unfixed mutations. Illumina deep sequencing identified a higher proportion of fluoroquinolone-resistant M. tuberculosis isolates with heteroresistance than did Sanger sequencing. The heteroresistant isolates frequently demonstrated multiple mutations, but resistant isolates with fixed mutations each had only a single mutation.
INTRODUCTION
Fluoroquinolones are bactericidal against Mycobacterium tuberculosis. The etiology of fluoroquinolone resistance is incompletely explained in published studies. Mutations in DNA gyrase that modify the fluoroquinolone target in M. tuberculosis were identified in 56% of the phenotypically resistant isolates in a systematic review (1), although the rates were much higher in some individual studies (2–4). In that review, isolates in 32 of 41 studies were assessed for DNA gyrase mutations via Sanger sequencing (1). Sanger sequencing of the gyrase genes, gyrA and gyrB, is, to date, the gold standard in the identification of genotypic resistance mutations (5). However, Sanger sequencing is less sensitive than deep sequencing for low-frequency variants in M. tuberculosis; the former can detect minor allele frequencies of >10 to 15% (6, 7). Sanger sequencing is dependent on the presence of a variant above this threshold for effective amplification. Deep sequencing by the Illumina method, in contrast, amplifies individual DNA clusters and is more sensitive for the amplification of variants with as little as 1% frequency in the population (http://res.illumina.com/documents/products/technotes/technote_genotyping_rare_variants.pdf).
Heteroresistance refers to a population of organisms that are not homogeneous for a given nucleotide at a position implicated in drug resistance (8). Under antibiotic selection, the proportion of resistant organisms increases until a mutation becomes fixed, defined as a mutant allele frequency of >95% (9). Although a single sputum culture may not reflect the entire diversity of tuberculosis bacteria infecting a patient, heteroresistance at a resistance locus is sensitive for the detection of discordance in drug susceptibility at the site of disease.
Because M. tuberculosis is a haploid organism, its DNA should be represented by a single nucleotide at each position in the genome. Several etiologies are felt to underlie heteroresistance, including an infection with multiple strains of M. tuberculosis, a superinfection, or an ongoing evolution of mutations within an originally homogeneous strain (10). The presence of mixed infections or superinfections with a second strain is best assessed by molecular typing, as these infections will typically demonstrate mixed mycobacterial interspersed repetitive-unit (MIRU) typing, spoligotyping, or restriction fragment length polymorphism patterns (11). In the presence of a single molecular typing pattern, sequencing is necessary to identify heteroresistance at the genetic loci of interest. In this study, we compared Sanger sequencing of gyrA and gyrB with deep Illumina sequencing to determine the proportion of fluoroquinolone (FQ) monoresistant isolates with heteroresistance.
MATERIALS AND METHODS
Study population.
M. tuberculosis isolates were obtained from a population-based study of patients with newly diagnosed, culture-confirmed tuberculosis reported to the Tennessee Department of Health from January 2002 to December 2010 (12, 13). The sample included 27 phenotypically fluoroquinolone-resistant isolates and 11 fluoroquinolone-susceptible controls. Susceptible controls were matched by lineage and, where possible, sublineage, using MIRU analysis and spoligotyping. If no exact match was available, an isolate with the most loci in agreement was chosen. Each isolate was grown in culture, and MIC testing for ofloxacin was performed. Large swipes of several colonies were taken from the same plates for DNA extraction. Sanger sequencing and deep sequencing were performed from the same DNA sample.
Phenotypic resistance testing.
A 1.0 McFarland suspension was prepared from 3-week-old M. tuberculosis colonies grown on Lowenstein-Jensen medium in the absence of ofloxacin. After confirmation of turbidity by nephelometry, the suspension served as the standard inoculum for all dilutions used in susceptibility testing. One hundred microliters each of 10−2 and 10−4 dilutions of the standard inoculum was plated on 7H10 agar containing ofloxacin. Control plates were also inoculated with bacilli in the absence of the drug. Phenotypic resistance was defined as >1% colony growth in the presence of 2 μg/ml ofloxacin. The MIC was determined by the first serial dilution showing <1% colony growth on the 7H10 agar (14).
Lineage determination.
Spoligotyping and 12-loci MIRU typing were performed by the Michigan Department of Community Health on all M. tuberculosis isolates in culture-confirmed tuberculosis cases in Tennessee from 2004 to 2010 and a select number of isolates from 2002 and 2003 (15). Lineage and sublineage were obtained from the CDC through the Tuberculosis Genotyping Information Management System (16).
Sanger sequencing.
For full methods, see Devasia et al. (13). DNA was extracted from a single M. tuberculosis colony grown on Lowenstein-Jensen medium. The entire gyrA and gyrB genes were amplified by PCR in separate reactions. The amplified DNA fragments were purified (MinElute gel extraction kit; Qiagen, Valencia, CA) and cloned using a TA Cloning kit (Invitrogen, Carlsbad, CA). Plasmids were sequenced via an ABI Prism 3730xl DNA analyzer (Applied Biosystems, Foster City, CA) using BigDye chemistry per the manufacturer's instructions. Applied Biosystems DNA sequencing software (version 5.0) was used to collect and analyze the raw data. Sequences were analyzed using DNA Baser (Heracle BioSoft, Pitesti, Romania). The gyrA and gyrB sequences for all isolates were assembled and compared to the gyrA and gyrB sequences in M. tuberculosis reference strain H37Rv (GenBank accession number AL123456.2).
Deep Illumina sequencing.
Genomic DNA was purified using a ZR bacterial/fungal DNA miniprep kit (Zymo Research, Irvine, CA). Samples were then heated to 94°C for 10 min. Whole-genome sequencing was performed at the Genome Sciences Resource at Vanderbilt University on an Illumina HiSeq 2500 system for 29 isolates; 9 isolates were sequenced at the Illumina Sequencing Core at Boston University on an Illumina GaIIx system. In both cases, identical library preparation was performed for fragmentation of DNA, end repair, and ligation of Illumina adapters. Ligated DNA was then selected for cluster generation and sequencing, which were performed per the Illumina manual on the HiSeq and GaIIx systems, respectively, for the isolates described above. Base calls were generated from the internal Casava pipeline software (Illumina, San Diego, CA).
The HiSeq output was in the FASTQ format. Output from the GaIIx was in the QSEQ format, which was transformed into a FASTQ file using a Perl script. Low-quality base tails and reads having less than two-thirds of the bases with quality scores of ≥30 in the first half of the read were removed prior to sequence analysis using Sickle (University of California-Davis) (17).
Sequences were then aligned, mapped, and compared to the reference H37Rv genome (GenBank accession number AL123456.2). The Burrows-Wheeler aligner (BWA) was used to align and merge the paired end reads (18). The alignments were then sorted and indexed using SAMtools (19). SnpEff was used to call single nucleotide polymorphisms (SNPs) and annotate output base changes (20).
Whole-genome sequencing was performed on a total of 38 specimens. In one sample, 827 heteroplasmic SNPs were identified with a common mutant to a reference ratio of approximately 1:4. When analyzed, the 827 SNPs differed from the H37Rv reference by only 5 SNPs, suggesting contamination of the sample with H37Rv. This sample was therefore excluded. The other 37 isolates showed no evidence of contamination or multiple strains.
Read level analysis.
The SAMtools mpileup command was used to generate a pileup report that detailed all nucleotide calls at each position for each sample. A custom Perl script was used to generate counts based on strand and base calls at each position on both the forward and reverse strands. Given that false-positive reads are rare in the setting of marking duplicates and performing local realignment, at least 2 reads on both the forward and reverse strands were required to consider a position for analysis (21). Heteroresistance was defined as a resistant allele proportion between 1% and 95% (9). The means of values on the forward and reverse strands were obtained. In this analysis, we evaluated only the gyrA and gyrB genes.
RESULTS
Isolates varied based upon phenotypic resistance to fluoroquinolones, phylogeny, and molecular typing as shown in Table 1. The population included 38 isolates, 34 of which were from the Euro-American lineage; the remainder were of East Asian lineage. Within the Euro-American isolates, the most common sublineages were X (6), Haarlem (4), Latin American Mediterranean (LAM) (3), and S (1).
TABLE 1.
Characteristics for the 37 M. tuberculosis isolates included in the primary analysisa
| OFXb MIC | Study no. | Lineage | MIRU |
|---|---|---|---|
| 1 | 15 | Euro-American | 224315153224 |
| 1 | 16 | Euro-American | 223326153323 |
| 1 | 17 | Euro-American | 223125163324 |
| 1 | 18 | Euro-American | 223226153321 |
| 1 | 19 | Euro-American/X | 224225153324 |
| 1 | 20 | Euro-American | 233326153321 |
| 1 | 21 | Euro-American | 222325143321 |
| 1 | 22 | East Asian | 222325173543 |
| 1 | 23 | Euro-American/X | 225325153224 |
| 1 | 24 | Euro-American/LAMc | 224225163321 |
| 1 | 25 | Euro-American/Haarlem | 225225153323 |
| 4 | 29 | Euro-American | 223426153322 |
| 8 | 2 | Euro-American/Haarlem | 225313153321 |
| 8 | 28 | Euro-American/X | 123325153323 |
| 8 | 45 | Euro-American | 223325153322 |
| 16 | 38 | Euro-American | 223326153221 |
| 64 | 1 | Euro-American | 224225153323 |
| 64 | 6 | Euro-American/X | 224324155323 |
| 64 | 7 | Euro-American/X | 225325153224 |
| 64 | 8 | Euro-American | 223225153325 |
| 64 | 11 | East Asian | 223325183533 |
| 64 | 12 | Euro-American/S | 2w3325153225 |
| 64 | 13 | Euro-American/LAM | 223225163321 |
| 64 | 14 | Euro-American/LAM | 224225163321 |
| 64 | 40 | Euro-American | 223226153321 |
| 64 | 41 | Euro-American | 221125143323 |
| 64 | 44 | Euro-American | 233315163321 |
| 128 | 9 | Euro-American/Haarlem | 224325153323 |
| 128 | 27 | East Asian | 222325173543 |
| 128 | 36 | East Asian | 222325173543 |
| 256 | 4 | Euro-American | 223125153222 |
| 256 | 5 | Euro-American/X | 224425153324 |
| 256 | 10 | Euro-American | 124325153224 |
| 256 | 26 | Euro-American/Haarlem | 225325153323 |
| 256 | 35 | Euro-American | 223125163324 |
| 256 | 37 | Euro-American/X | 124325133324 |
| 256 | 42 | Euro-American | 224315153323 |
Isolates are sorted by ofloxacin MIC and then by sample number.
OFX, ofloxacin.
LAM, Latin American Mediterranean.
Study isolates were assessed as shown in Fig. 1. Ofloxacin drug susceptibility testing performed prior to sequencing revealed 26 isolates with and 11 isolates without phenotypic resistance. Fifteen of 26 resistant isolates had known resistance-conferring mutations in gyrA or gyrB identified by Sanger sequencing. Illumina deep sequencing on the 37 isolates confirmed the presence of resistance-conferring mutations in the 15 phenotypically resistant samples with mutations on Sanger sequencing and also identified known resistance-conferring mutations in gyrA or gyrB in an additional 5 phenotypically resistant isolates. The remaining 6 isolates had no known resistance-conferring mutations on deep sequencing.
FIG 1.
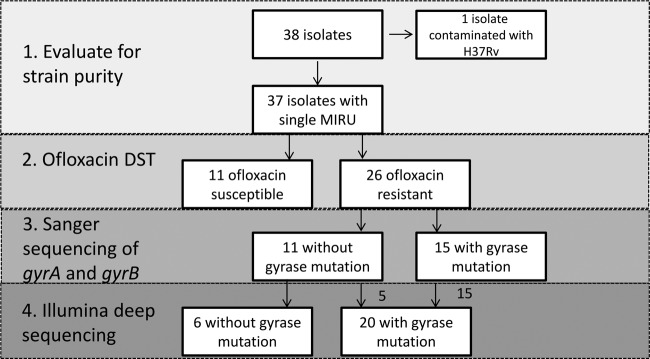
Genomic evaluation of M. tuberculosis study isolates. MIRU, mycobacterial interspersed repeat units; DST, drug susceptibility testing.
The exact proportions and sites of heteroresistance in DNA gyrase are shown in Table 2. On deep sequencing, heteroresistance was observed in 10 of 26 (38%) isolates phenotypically resistant to ofloxacin, compared to 0 of 11 isolates susceptible to ofloxacin (Fisher's exact test, P = 0.02). Of heteroresistant isolates, 5 had a single unfixed mutation, and 5 had multiple unfixed mutations. No isolates contained a heteroresistant position and a fixed resistance-conferring mutation. No isolates with fixed mutations had multiple mutations in gyrA or gyrB. Heteroresistance was identified at 2 loci in gyrB and 5 loci in gyrA. Heteroresistance frequencies shown are from read-level evaluation of deep sequencing data and ranged between 0.04 and 0.84. The proportion of heteroresistance plotted against the detection of heteroresistance by Sanger sequencing is shown in Fig. 2.
TABLE 2.
Proportion of heteroresistance at resistance-conferring loci in gyrA (Rv0006) and gyrB (Rv0005)a
| Ofloxacin MIC | Study no. | Mutation by Sanger sequencing | Proportion of heteroresistance at: |
Total | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Rv0005 site (mutation) |
Rv0006 site (mutation) |
|||||||||
| 1613 T (N538I) | 1616 A (T539N) | 269 T (A90V) | 271 C (S91P) | 280 A (D94N) | 280T (D94Y) | 281 G (D95G) | ||||
| 1 | 15 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 1 | 16 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 1 | 17 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 1 | 18 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 1 | 19 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 1 | 20 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 1 | 21 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 1 | 22 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 1 | 23 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 1 | 24 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 1 | 25 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 4 | 29 | 0 | 0.23b | 0.09 | 0.22 | 0 | 0 | 0.45 | 0.99 | |
| 8 | 2 | A90V | 0 | 0 | 0.84 | 0 | 0 | 0 | 0 | 0.84 |
| 8 | 28 | D94N | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 |
| 8 | 45 | 0 | 0 | 0 | 0 | 0 | 0 | 0.09 | 0.09 | |
| 16 | 38 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 64 | 1 | D94G | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 |
| 64 | 6 | D94G/D94N | 0 | 0 | 0 | 0 | 0.49 | 0 | 0.5 | 0.99 |
| 64 | 7 | D94Y | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 |
| 64 | 8 | D94G | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 |
| 64 | 11 | D94G | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 |
| 64 | 12 | D94G | 0 | 0 | 0 | 0 | 0 | 0 | 0.48 | 0.48 |
| 64 | 13 | D94Y | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 |
| 64 | 14 | S91P | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 |
| 64 | 40 | 0 | 0 | 0.01 | 0 | 0 | 0 | 0 | 0.01 | |
| 64 | 41 | D94G | 0 | 0 | 0 | 0 | 0 | 0 | 0.37 | 0.37 |
| 64 | 44 | N538I | 0.98 | 0 | 0 | 0 | 0 | 0 | 0 | 0.98 |
| 128 | 9 | D94N | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 |
| 128 | 27 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 128 | 36 | 0 | 0 | 0 | 0 | 0.01 | 0 | 0 | 0.01 | |
| 256 | 4 | D94G/D94N | 0 | 0 | 0 | 0 | 0.82 | 0 | 0.17 | 0.99 |
| 256 | 5 | D94G/D94N | 0 | 0 | 0 | 0 | 0.28 | 0 | 0.19 | 0.47 |
| 256 | 10 | D94G | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 |
| 256 | 26 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 256 | 35 | 0 | 0 | 0.19 | 0 | 0 | 0 | 0.01 | 0.2 | |
| 256 | 37 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 256 | 42 | 0 | 0 | 0 | 0 | 0 | 0 | 0.04 | 0.04 | |
Isolates are sorted by MIC and then by sample number. The proportion of heteroresistance was <1% in samples 36 and 40.
Heteroresistant positions are shown in bold type.
FIG 2.
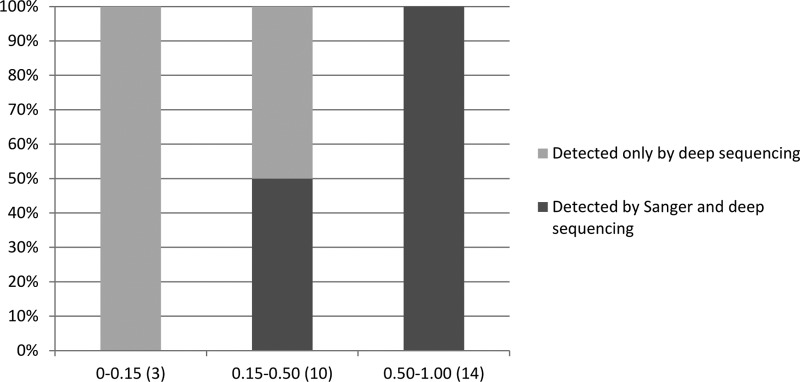
Comparison of gyrA and gyrB resistance-conferring mutations plotted by the proportion of reads at each locus as detected by Sanger sequencing and Illumina deep sequencing. The proportion of heteroresistance by deep sequencing (number of isolates per group) is shown on the x axis, and the percentage of isolates with heteroresistance detected by Sanger sequencing is shown on the y axis.
A comparison of the exact mutations detected by Sanger sequencing and deep sequencing is shown in Table 2. All 10 isolates with fixed mutations in DNA gyrase were identified by Sanger sequencing. Sanger sequencing detected multiple mutations in 3 samples. Deep sequencing revealed resistance-conferring mutations in 20/26 isolates. In 4 of these 20, multiple mutations reported to confer resistance were identified. Average read depth across gyrA and gyrB varied from 129 to 530, with a median of 307 and an interquartile range of 139.
DISCUSSION
In this study, we identified a high proportion (38%) of heteroresistance among fluoroquinolone-resistant M. tuberculosis isolates by deep sequencing. Previous studies have employed different methods to assess heteroresistance to fluoroquinolones, including Sanger sequencing of gyrA, Sanger sequencing of gyrA and gyrB plus spoligotyping, and a line probe assay. Proportions of heteroresistance were 23%, 21%, and 20%, respectively. Two of these studies contained only isolates resistant to multiple drugs, and in the third, 23/35 samples were resistant to at least one drug (6, 22, 23). In contrast, our M. tuberculosis isolates were resistant only to fluoroquinolones.
Both deep sequencing and Sanger sequencing detected isolates with heteroresistance; however, deep sequencing identified heteroresistance in 6 more isolates than Sanger sequencing. A direct comparison between the two methods is possible since the same aliquot of DNA was used to perform both tests. The detection of heteroresistance in 15% of samples by Sanger sequencing is consistent with that in previous studies. However, the detection of heteroresistance in 38% of samples by deep sequencing suggests that heteroresistance is more common than previously reported and that deep sequencing offers improved sensitivity over Sanger sequencing for heteroresistance detection. There are no studies using deep sequencing to detect heteroresistance to other antimycobacterial drugs, so it is not possible to compare proportions of heteroresistance between antimycobacterial drugs.
The use of cultured samples for deep sequencing may, in fact, select for more fit mutants, which may result in an underestimate of heteroresistance in this study. As sequencing technologies improve, rapid diagnostic testing for drug resistance directly from patient samples may allow better estimates of the frequency and proportion of heteroresistance. The proportion of heteroresistance needed to result in treatment failure is not currently known. In our study, one isolate with phenotypic fluoroquinolone resistance demonstrated only 4% heteroresistance and another had 9%. The Xpert MTB/RIF assay requires a frequency of at least 65% to detect a heteroresistant mutation (24). Despite our focus on fluoroquinolones rather than on rifampin, the Xpert platform rather than the mutation under study limits the detection of resistance mutations in samples with low proportions of resistant organisms. Thus, a test requiring a similar proportion of resistant mutants would have identified only 60% of the isolates we found to be genotypically resistant. This finding suggests a need for highly sensitive genotypic resistance testing for fluoroquinolones, especially since these same samples exhibiting very low levels of heteroresistance for a known resistance mutation were phenotypically resistant to fluoroquinolone.
Deep sequencing quantified multiple coexisting mutations, including several not identified by Sanger sequencing. In 4 of 5 isolates with multiple resistance-conferring mutations, the frequencies of mutant alleles summed to 1 despite mutant alleles located in different loci or even different genes (gyrA versus gyrB). The relative fitness costs of gyrase mutations are not known in M. tuberculosis; however, in studies of Streptococcus, Campylobacter, and Neisseria gonorrhoeae these mutations are associated with a fitness cost (25–27). Notably, only single resistance-conferring mutations were fixed in each isolate. In our population, D94G was the most prevalent fixed gyrase mutation, followed by D94N, D94Y, and S91P. However, the D94G mutation was also the most prevalent unfixed mutation, suggesting that no resistance mutation was associated with disproportionate adaptation to growth in laboratory conditions. Interestingly, 6 isolates with phenotypic ofloxacin resistance had no gyrase mutations identified. This may suggest loss of less fit, unfixed gyrase mutations after sputum collection or an alternative mechanism of fluoroquinolone resistance.
Heteroresistance must be distinguished from mixed-strain infections. In our population, we performed both spoligotyping and 12-loci MIRU-variable-number tandem repeat (VNTR) typing. Typing was done with a loop of bacteria from multiple colonies, increasing the sensitivity for detecting mixed infection. All isolates included in the analysis resulted in a single pattern by MIRU and spoligotyping, making the presence of a single lineage more likely. Additionally, the analysis of heteroresistant positions revealed a mixture in only one isolate, which had been contaminated with H37Rv.
This study was limited by the need for a large amount of DNA, requiring use of cultured organisms. Sequencing directly from sputum might have possibly captured a broader array of organisms but generated less DNA and presumably had a decreased ability to detect mutations. The use of cultured organisms may favor those better adapted to grow under laboratory conditions. Furthermore, our study was performed in a low disease burden area with monoresistant isolates. However, Sun et al. showed that heteroresistance was present in 3 patients demonstrating multidrug resistance (MDR) and treatment failure after heavy treatment exposure (9). This suggests that a substantial proportion of resistance is not transmitted in MDR TB cases. An additional study is needed to identify the rate of heteroresistance in MDR TB cases. Our study was also limited by the low number of fluoroquinolone-resistant isolates in our study area, the state of Tennessee. However, all of the isolates were fluoroquinolone monoresistant, which allows evaluation of mutations associated only with fluoroquinolone resistance. In addition, gyrase-binding assays have not been performed for known gyrase resistance-conferring mutations; such information would be helpful for understanding the relative fitness of mutant strains.
In summary, through deep sequencing, we have identified a higher proportion of heteroresistance in fluoroquinolone-resistant M. tuberculosis isolates than in previously published studies. The fact that almost half of the heteroresistant mutations detected in our sample would not have been detected by a test requiring 65% frequency of a resistance mutation in a sequenced population, as in the case of the Xpert MTB/RIF, suggests that alternate strategies are needed to capture a complete picture of genotypic resistance in future point-of-care tests. Furthermore, nearly 20% of isolates studied harbored multiple resistance-conferring mutations in gyrase. In all such cases, multiple resistance mutations coexisted only in the absence of a fixed mutation. Single mutations were identified only in samples demonstrating fixed mutations, suggesting that the fitness cost is high when there is more than one fixed, fluoroquinolone resistance-conferring mutation.
ACKNOWLEDGMENTS
We acknowledge James Galagan, Chris Mawhinney, and Matt Petersen from Boston University for whole-genome sequencing of a portion of the samples.
This work was supported by the National Institutes of Health (grants NIAID R01 AI063200, K24 065298, and T32 AI 07474-17).
T.R.S. has received research grants through Vanderbilt for HIV cohort studies from Pfizer, Bristol Myers Squibb, and Virco, serves on a data safety monitoring board for Otsuka, and has received consultancy fees from Sanofi-Aventis. The other authors report no conflicts of interest.
Footnotes
Published ahead of print 31 March 2014
REFERENCES
- 1.Maruri F, Sterling TR, Kaiga AW, Blackman A, van der Heijden YF, Mayer C, Cambau E, Aubry A. 2012. A systematic review of gyrase mutations associated with fluoroquinolone-resistant Mycobacterium tuberculosis and a proposed gyrase numbering system. J. Antimicrob. Chemother. 67:819–831. 10.1093/jac/dkr566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen J, Chen Z, Li Y, Xia W, Chen X, Chen T, Zhou L, Xu B, Xu S. 2012. Characterization of gyrA and gyrB mutations and fluoroquinolone resistance in Mycobacterium tuberculosis clinical isolates from Hubei Province, China. Braz. J. Infect. Dis. 16:136–141. 10.1016/S1413-8670(12)70294-5 [DOI] [PubMed] [Google Scholar]
- 3.Chernyaeva E, Fedorova E, Zhemkova G, Korneev Y, Kozlov A. 2013. Characterization of multiple and extensively drug resistant Mycobacterium tuberculosis isolates with different ofloxacin-resistance levels. Tuberculosis (Edinb.) 93:291–295. 10.1016/j.tube.2013.02.005 [DOI] [PubMed] [Google Scholar]
- 4.Jnawali HN, Hwang SC, Park YK, Kim H, Lee YS, Chung GT, Choe KH, Ryoo S. 2013. Characterization of mutations in multi- and extensive drug resistance among strains of Mycobacterium tuberculosis clinical isolates in Republic of Korea. Diagn. Microbiol. Infect. Dis. 76:187–196. 10.1016/j.diagmicrobio.2013.02.035 [DOI] [PubMed] [Google Scholar]
- 5.Köser CU, Feuerriegel S, Summers DK, Archer JA, Niemann S. 2012. Importance of the genetic diversity within the Mycobacterium tuberculosis complex for the development of novel antibiotics and diagnostic tests of drug resistance. Antimicrob. Agents Chemother. 56:6080–6087. 10.1128/AAC.01641-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hofmann-Thiel S, van Ingen J, Feldmann K, Turaev L, Uzakova GT, Murmusaeva G, van Soolingen D, Hoffmann H. 2009. Mechanisms of heteroresistance to isoniazid and rifampin of Mycobacterium tuberculosis in Tashkent, Uzbekistan. Eur. Respir. J. 33:368–374. 10.1183/09031936.00089808 [DOI] [PubMed] [Google Scholar]
- 7.Rohlin A, Wernersson J, Engwall Y, Wiklund L, Bjork J, Nordling M. 2009. Parallel sequencing used in detection of mosaic mutations: comparison with four diagnostic DNA screening techniques. Hum. Mutat. 30:1012–1020. 10.1002/humu.20980 [DOI] [PubMed] [Google Scholar]
- 8.Rinder H, Mieskes KT, Löscher T. 2001. Heteroresistance in Mycobacterium tuberculosis. Int. J. Tuberc. Lung Dis. 5:339–345 [PubMed] [Google Scholar]
- 9.Sun G, Luo T, Yang C, Dong X, Li J, Zhu Y, Zheng H, Tian W, Wang S, Barry CE, 3rd, Mei J, Gao Q. 2012. Dynamic population changes in Mycobacterium tuberculosis during acquisition and fixation of drug resistance in patients. J. Infect. Dis. 206:1724–1733. 10.1093/infdis/jis601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ford C, Yusim K, Ioerger T, Feng S, Chase M, Greene M, Korber B, Fortune S. 2012. Mycobacterium tuberculosis—heterogeneity revealed through whole genome sequencing. Tuberculosis (Edinb.) 92:194–201. 10.1016/j.tube.2011.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martín A, Herranz M, Navarro Y, Lasarte S, Ruiz Serrano MJ, Bouza E, Garcia de Viedma D. 2011. Evaluation of the inaccurate assignment of mixed infections by Mycobacterium tuberculosis as exogenous reinfection and analysis of the potential role of bacterial factors in reinfection. J. Clin. Microbiol. 49:1331–1338. 10.1128/JCM.02519-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Devasia RA, Blackman A, Gebretsadik T, Griffin M, Shintani A, May C, Smith T, Hooper N, Maruri F, Warkentin J, Mitchel E, Sterling TR. 2009. Fluoroquinolone resistance in Mycobacterium tuberculosis: the effect of duration and timing of fluoroquinolone exposure. Am. J. Respir. Crit. Care Med. 180:365–370. 10.1164/rccm.200901-0146OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Devasia R, Blackman A, Eden S, Li H, Maruri F, Shintani A, Alexander C, Kaiga A, Stratton CW, Warkentin J, Tang YW, Sterling TR. 2012. High proportion of fluoroquinolone-resistant Mycobacterium tuberculosis isolates with novel gyrase polymorphisms and a gyrA region associated with fluoroquinolone susceptibility. J. Clin. Microbiol. 50:1390–1396. 10.1128/JCM.05286-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.CLSI. 2011. Susceptibility testing of mycobacteria, nocardiae and other aerobic actinomycetes; approved standard—2nd ed. M24–A2. Clinical and Laboratory Standards Institute, Wayne, PA: [PubMed] [Google Scholar]
- 15.Mazars E, Lesjean S, Banuls AL, Gilbert M, Vincent V, Gicquel B, Tibayrenc M, Locht C, Supply P. 2001. High-resolution minisatellite-based typing as a portable approach to global analysis of Mycobacterium tuberculosis molecular epidemiology. Proc. Natl. Acad. Sci. U. S. A. 98:1901–1906. 10.1073/pnas.98.4.1901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghosh S, Moonan PK, Cowan L, Grant J, Kammerer S, Navin TR. 2012. Tuberculosis genotyping information management system: enhancing tuberculosis surveillance in the United States. Infect. Genet. Evol. 12:782–788. 10.1016/j.meegid.2011.10.013 [DOI] [PubMed] [Google Scholar]
- 17.Joshi NA, Fass JN. 2011. Sickle: a sliding-window, adaptive, quality-based trimming tool for FastQ files. https://github.com/najoshi/sickle
- 18.Li H, Durbin R. 2010. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26:589–595. 10.1093/bioinformatics/btp698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cingolani P, Platts A, Wang LE, Coon LM, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. 2012. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6:80–92. 10.4161/fly.19695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Q, Guo Y, Li J, Long J, Zhang B, Shyr Y. 2012. Steps to ensure accuracy in genotype and SNP calling from Illumina sequencing data. BMC Genomics 13(Suppl 8):S8. 10.1186/1471-2164-13-S8-S8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Streicher EM, Bergval I, Dheda K, Bottger EC, Gey van Pittius NC, Bosman M, Coetzee G, Anthony RM, van Helden PD, Victor TC, Warren RM. 2012. Mycobacterium tuberculosis population structure determines the outcome of genetics-based second-line drug resistance testing. Antimicrob. Agents Chemother. 56:2420–2427. 10.1128/AAC.05905-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang X, Zhao B, Liu L, Zhu Y, Zhao Y, Jin Q. 2012. Subpopulation analysis of heteroresistance to fluoroquinolone in Mycobacterium tuberculosis isolates from Beijing, China. J. Clin. Microbiol. 50:1471–1474. 10.1128/JCM.05793-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blakemore R, Story E, Helb D, Kop J, Banada P, Owens MR, Chakravorty S, Jones M, Alland D. 2010. Evaluation of the analytical performance of the Xpert MTB/RIF assay. J. Clin. Microbiol. 48:2495–2501. 10.1128/JCM.00128-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rozen DE, McGee L, Levin BR, Klugman KP. 2007. Fitness costs of fluoroquinolone resistance in Streptococcus pneumoniae. Antimicrob. Agents Chemother. 51:412–416. 10.1128/AAC.01161-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han J, Wang Y, Sahin O, Shen Z, Guo B, Shen J, Zhang Q. 2012. A fluoroquinolone resistance associated mutation in gyrA affects DNA supercoiling in Campylobacter jejuni. Front. Cell Infect. Microbiol. 2:21. 10.3389/fcimb.2012.00021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kunz AN, Begum AA, Wu H, D'Ambrozio JA, Robinson JM, Shafer WM, Bash MC, Jerse AE. 2012. Impact of fluoroquinolone resistance mutations on gonococcal fitness and in vivo selection for compensatory mutations. J. Infect. Dis. 205:1821–1829. 10.1093/infdis/jis277 [DOI] [PMC free article] [PubMed] [Google Scholar]