

SUMMARY
Transcription factor activity and turnover are functionally linked, but the global patterns by which DNA-bound regulators are eliminated remain poorly understood. We established an assay to define the chromosomal location of DNA-associated proteins that are slated for degradation by the ubiquitin-proteasome system. The genome-wide map described here ties proteolysis in mammalian cells to active enhancers and to promoters of specific gene families. Nuclear-encoded mitochondrial genes in particular correlate with protein elimination, which positively affects their transcription. We show that the nuclear receptor corepressor NCoR1 is a key target of proteolysis and physically interacts with the transcription factor CREB. Proteasome inhibition stabilizes NCoR1 in a site-specific manner and restrains mitochondrial activity by repressing CREB-sensitive genes. In conclusion, this functional map of nuclear proteolysis links chromatin architecture with local protein stability and identifies proteolytic derepression as highly dynamic in regulating the transcription of genes involved in energy metabolism.
INTRODUCTION
Ever-finer maps are being drawn of DNA and its occupying transcriptional regulators and chromatin. This map is static by default and only describes the constellation of proteins and nucleic acids at a given time. However, many transcription factors are short-lived and selectively destroyed by the ubiquitin-proteasome system (UPS) upon assembly into functional DNA-bound complexes (Salghetti et al., 2000). Such proteolysis can have several consequences for gene expression. Simplified, it can restrict transcription by eliminating necessary factors, or it can increase expression by removing repressors (Lipford and Deshaies, 2003).
The quantitative contribution of local protein degradation on individual gene expression has not been evaluated on a genome-wide scale. We therefore sought to draw a dynamic map of protein turnover to assess how DNA-associated proteolysis correlates with specific genes and with chromatin composition. Our study had three goals. The first goal was to assess degradation of DNA-bound factors on a genome-wide scale. The second goal was to define sites of proteolysis in the context of gene expression and chromatin architecture. The third goal was to identify transcriptional regulators with high turnover dynamics and determine the impact of their degradation on relevant gene transcription.
The UPS eliminates proteins in a specific, step-wise manner (Ciechanover, 2012). Studies in S. cerevisiae demonstrated that the UPS regulates transcription and showed by chromatin immunoprecipitation (ChIP) that the proteasome physically interacts with DNA (Auld et al., 2006). A caveat of this approach is that some components of the proteasome regulate gene expression without involving protein turnover. Furthermore, the residence of the proteasome does not necessarily correspond with the location at which the “kiss of death,” the conjugation of ubiquitin chains, occurs. Other approaches to investigate effects of the UPS on gene expression involve the identification of target proteins by mass spectroscopy or the selective study of enzymes involved in ubiquitin transfer, in particular E3 ubiquitin ligases (Rubenstein and Hochstrasser, 2010). Importantly, these studies do not provide spatial information such as the DNA binding pattern of target proteins at the time of degradation. We therefore chose to directly examine the genomic sites of protein elimination. The distribution of proteasome-sensitive ubiquitin on DNA was used as an indicator of degradation initiation. By charting the nuclear locations of proteolysis and functionally linking proteasome activity to gene expression, we generated a genome-wide map of DNA-associated proteolysis.
This project revealed a correlation of DNA-bound protein degradation with active gene promoters and enhancers in mouse and human cells. In addition, proteolysis was associated with distinct gene ontologies and either promoted or suppressed transcription. Nuclear-encoded mitochondrial genes in particular showed signs of rapid protein turnover, which stimulated their expression. Utilizing integrative genomics, we identified the nuclear receptor corepressor NCoR1 as a major target of the UPS at these genes. Further, we defined biochemical interaction between NCoR1 and the transcription factor cyclic AMP response element-binding protein (CREB) at degradation sites. We therefore conclude that continuous elimination of NCoR1 is required to maintain transcript levels, and restraining its turnover by proteasome inhibition or depletion of the relevant ubiquitin ligase Siah2 diminishes mitochondrial function.
RESULTS
A Method to Detect DNA-Associated Protein Degradation
Ubiquitin not only marks proteins for degradation but is also involved in nonproteolytic functions–for instance, ubiquitin modifies histones H2A and H2B. Recent work suggests that the composition of ubiquitin chains on proteolytic substrates is variable (Xu et al., 2009), which makes it difficult to predict what type of chain induces degradation. We therefore defined degradative ubiquitination functionally by virtue of being sensitive to proteasome inhibition. Exposing cells to a brief pulse of the irreversible and specific inhibitor lactacystin leads to accumulation of degradation-prone substrates in their polyubiquitinated state. Such treatment results in a rapid redistribution of ubiquitin from its nonproteolytic to a proteasome-targeting function (Kim et al., 2011b). In particular, levels of monomeric ubiquitin on histones H2A and H2B decrease to be channeled toward the formation of degradative chains on proteins that are slated for elimination (Figure 1A and data not shown). For this study, we crosslinked and immunoprecipitated ubiquitin with DNA in the presence or absence of proteasome inhibition and mapped its location by sequencing. We defined genomic sites at which degradation was initiated by comparing the distribution of ubiquitin under proteolytic stress with that at steady state.
Figure 1. Ubiquitin Redistributes following Proteasome Inhibition.
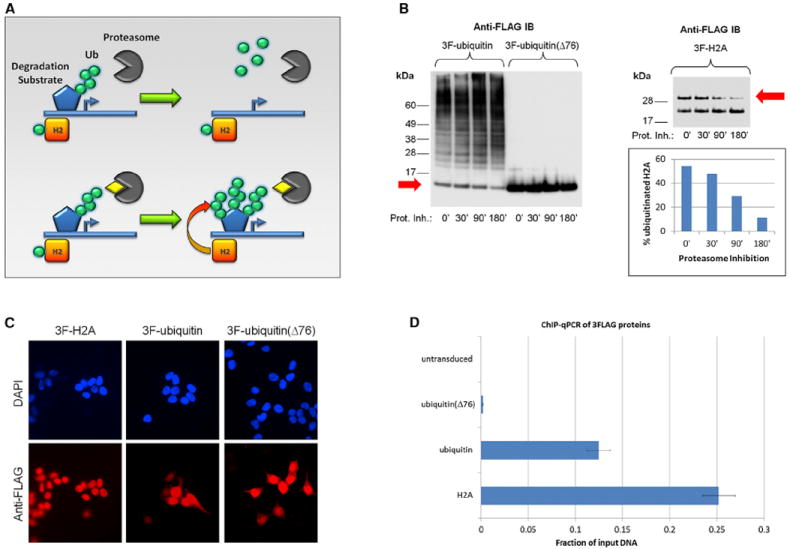
(A) Top: polyubiquitination (Ub, green circles) initiates degradation. Bottom: proteasome inhibition (yellow rhombus) increases ubiquitination on elimination-prone substrates, whereas nondegradative ubiquitination, e.g., on histones (H2), remains unaffected or even decreases.
(B) Left: immunoblotting (IB) of 3FLAG-ubiquitin after proteasome inhibition for up to 3 hr (25 μM lactacystin) in HEK293T cells. Ubiquitin conjugation increases up to 80%, whereas free ubiquitin decreases (red arrow). Nonfunctional ubiquitin (Δ76) fails to modify proteins (right lanes). Right: monoubiquitination of 3FLAG-H2A decreases upon treatment (red arrow), quantified by densitometry (bottom).
(C) The 3FLAG epitope does not interfere with the distribution of tagged proteins in HEK293T cells by immunohistochemistry (red for FLAG epitope; nuclei in blue).
(D) ChIP-qPCR of HEK293T cells after transfection with 3FLAG-H2A (positive control), 3FLAG-ubiquitin, or 3FLAG-ubiquitin(Δ76). Recovery of crosslinked DNA is 60-fold reduced in mutant compared to wild-type ubiquitin (probed for GAPHD promoter). Bars represent mean ± SD.
See also Figure S1.
Ubiquitin was tagged with an N-terminal 3FLAG epitope and displayed no signs of toxicity. Furthermore, tagged ubiquitin was a competent covalent modifier and distributed normally (Figure 1B, left, and Figure 1C). Upon proteasome inhibition, ubiquitin accumulated at degradation-prone substrates, whereas its unconjugated form decreased (Figure 1B, left, red arrow) (Kim et al., 2011b; Wagner et al., 2011). Treatment also reduced the levels of monoubiquitination at histone H2A, which is consistent with the expected redistribution (Figure 1B, right, red arrow). Ubiquitin is an 8.6 kDa molecule that can easily enter the nucleus. Given its abundance, we were concerned that crosslinking of free ubiquitin with DNA could lead to high background. To address this, we performed ChIP-qPCR analyses with a dysfunctional ubiquitin mutant lacking the C-terminal G76. Nonspecific crosslinking was responsible for less than 2% of the specific DNA recovery achieved with functional ubiquitin (Figure 1D and data not shown). Association of DNA with ubiquitin is therefore mostly dependent upon DNA binding of modified proteins and not of ubiquitin itself. We treated cells with proteasome inhibitor or with DMSO as solvent control (referred to as “untreated”) for short periods to minimize secondary effects. Treatment for 3 to 6 hr showed comparable results in ChIP experiments (data not shown). This duration did not significantly affect the intracellular location of ubiquitin, the viability and phenotype of the cells in our study, or their cell-cycle distribution (Figures S1A–S1D available online).
Degradative Ubiquitination Correlates with Active Genomic Regions
Our next goal was to identify ubiquitination sites on a global level in the presence of active or inactivated proteasome. We performed ChIP-on-chip experiments with 3FLAG-ubiquitin transduced human HEK293T cells and with human mesenchymal progenitor cells derived from the H9 embryonic stem cell line (Figure S2A). Inspecting 24,633 promoters, there was substantial overlap in ubiquitin-associated promoters between lactacystin-treated and untreated cells (58.3% for HEK293T cells and 34.1% and 40.8% for two independent experiments with H9-derived mesenchymal cells; Figure S2B). However, genes of promoters that were exclusively associated with degradative ubiquitination in the treated sample were more highly expressed than those that were uniquely linked to steady-state ubiquitination (p = 0.000878 for HEK293T cells and p = 3.2 × 10−8 for H9-derived cells; two-sided Wilcoxon rank-sum test; Figure S2C). These findings connect degradation with high transcription levels and are consistent with data in S. cerevisiae, in which DNA binding of the proteasome correlates with active genomic regions (Auld et al., 2006).
In order to create an unbiased genome-wide map of DNA-linked degradation, we combined ChIP with next-generation sequencing (ChIP-seq). We analyzed the well-established mouse preadipocyte cell line 3T3-L1 in its undifferentiated form, a cell type that may be described as analogous to the human H9-derived mesenchymal cells. 3T3-L1 cells are widely used to study adipogenesis and metabolism in vitro. Several groups have mapped the genome of 3T3-L1 cells, and global chromatin profiles are available (Mikkelsen et al., 2010). We performed two independent experiments and analyzed a total of 33,164 3FLAG-ubiquitin peaks with and 46,044 peaks without addition of proteasome inhibitor. 36.6% of the ubiquitin peaks overlapped between the two experiments with lactacystin treatment, and 63.2% of peaks overlapped between the two untreated experiments. This manuscript focuses on peaks that were reproducible within their respective treatment condition between both experiments. An analysis of all peaks produced similar results (data not shown).
To assess the regulatory relationship between degradation and transcript levels, we evaluated the expression of genes whose transcription start sites (TSS) are located within 3 kilobases of ubiquitin peaks. We observed a tight connection between proteasome-sensitive ubiquitination and active gene expression (Figures 2A, 2B, and S2D; p = 1.95 × 10−142; two-sided Wilcoxon rank-sum test with Bonferroni correction).
Figure 2. Degradation Is Enriched at Highly Expressed Genes.
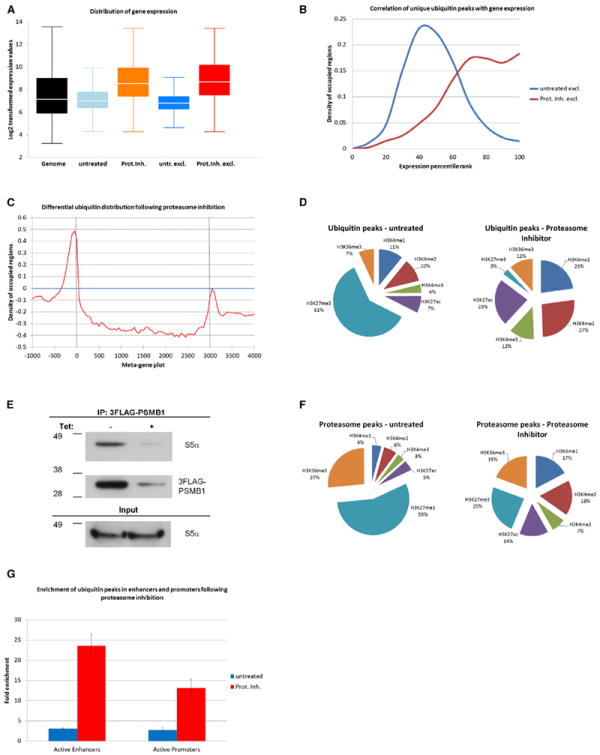
(A) Log2-normalized expression values of genes with TSS within 3 kbp of ubiquitin peaks. ChIP-seq was performed with 3FLAG-ubiquitin in 3T3-L1 cells that were mock treated or treated with lactacystin. The first box plot depicts the expression range of the entire genome. Next shown are the plots for ubiquitin-associated genes in untreated or lactacystin-treated cells (1,218 and 1,755 genes, respectively). The two right-most plots depict genes that are exclusively associated with ubiquitin in untreated or treated cells (892 and 1,429 genes, respectively).
(B) Expression ranks of genes associated with ubiquitination. Considered were 892 genes unique to untreated cells and 1,429 genes unique to treated cells. Proteasome-sensitive ubiquitination is associated with higher transcription (p = 1.95 × 10−142).
(C) Metagenomic change in ubiquitin distribution following lactacystin treatment (genes scaled to 3 kbp size). TSS and termination site are marked.
(D) Correlation of ubiquitin enrichment with chromatin marks in untreated and treated 3T3-L1 cells. Regional overlaps between ubiquitin peaks and histone H3 modifications are shown.
(E) IB after IP of 3FLAG-PSMB1 in 3T3-L1 cells. 3F-PSMB1 expression was regulated by a repressor (Tet).
(F) Regional overlaps of H3 chromatin marks with 3FLAG-PSMB1 ChIP-seq peaks.
(G) Enrichment of ubiquitin at active promoters (H3K4me3/H3K27ac) and enhancers (H3K4me1/H3K27ac) (bars represent mean ± SD). Proteasome-sensitive ubiquitination is associated with active enhancers and promoters (p = 8.17 × 10−318 and p = 8.36 × 10−319).
See also Figures S2 and S3.
We next investigated whether ubiquitin peaks cluster in regions relevant for transcription. To this purpose, we depicted the metagenomic changes in ubiquitination following proteasome inhibition. Treatment produced signal enrichments immediately upstream of the TSS (Figures 2C and S3A), implying that the promoters of active genes are sites of vigorous protein turnover. Differential analysis also suggests that steady-state ubiquitination is more prevalent over the gene body. To more precisely correlate the location of ubiquitinated substrates with chromatin architecture, we compared our binding sites with a map of histone H3 modifications (Mikkelsen et al., 2010). Steady-state ubiquitination frequently occurred at regions that are trimethylated at H3K27 (Figure 2D, left), which is considered a repressive modification (Kouzarides, 2007). Following treatment with lactacystin, we observed a redistribution of ubiquitin toward active histone marks, including acetylated H3K27 and methylated H3K4 (Figures 2D, right, and Figures S3B and S3C). The majority of ubiquitination sites overlapped with at least one of the six H3 chromatin states studied (83.4% in untreated and 96.9% in treated cells).
Proteasomes have been observed to cluster with polyubiquitinated proteins in the cytoplasm. This attraction is promoted by ubiquitin-binding proteins such as S5α, which are located in the lid subunits (Sakata et al., 2012). The lids are noncovalently associated with the core of the proteasome that contains the catalytic subunits. To test whether proteasome and substrate also colocalize in the nucleus, we performed ChIP-seq with a 3FLAG-tagged version of the catalytic proteasome subunit β1 (PSMB1). The N-terminal tag did not interfere with integration into the holoproteasome, as assessed by coimmunoprecipitation and immunoblots (Figure 2E). Also, previous studies showed that inhibitors do not dissociate the holoenzyme and that the majority of proteasomes are fully assembled when bound to DNA (Geng and Tansey, 2012; Kriegenburg et al., 2008). Based on ChIP-seq, location of the proteasome mirrored ubiquitination sites, including a remarkable redistribution following lactacystin treatment. At steady state, the proteasome remained in genomic regions rich in trimethylated H3K27 (Figure 2F, left). After inhibition, we observed increased clustering with active markers, including methylated H3K4 and acetylated H3K27 (Figure 2F, right). This further validated our assay by showing that lactacystin treatment and the accumulation of ubiquitin on degradation-prone substrates is followed by an equivalent mobilization of the proteasome to such affected genomic regions.
The complexity of gene expression in metazoans is thought to be largely driven by enhancer elements. Given the variable genomic distance between enhancers and their target genes, functional annotations have been notoriously difficult. However, it was recently proposed that these DNA elements can be defined by a distinct combination of chromatin signatures. Enhancers and promoters generally display H3K4 mono- or trimethylation, respectively, and by considering the status of H3K27 acetylation versus methylation, active and inactive regions can be distinguished (Heintzman et al., 2009). We determined the relative enrichment of degradative ubiquitination and found strong correlations with both active enhancers and active promoters (Figure 2G; p = 8.17 × 10−318 and p = 8.36 × 10−319, respectively; two-sided Fisher’s exact test).
Taken together, our data suggest that protein turnover mirrors gene activity. Degradation is linked to specific chromatin states and is enriched at regulatory sites.
Site-Specific Degradation Promotes Expression of CREB Target Genes
Our next goal was to investigate the functional impact of local degradation on target gene expression. To minimize secondary effects on genes that are downstream of proteasome-sensitive signaling pathways, the shortest effective treatment duration for lactacystin was used. Although a 3 hr regimen was sufficient to observe redistribution of ubiquitin by ChIP, we had to extend this treatment to 6 hr for significant changes to transpire in overall gene expression. Extension of the lactacystin pulse increased the log2-normalized gene expression variance by 5.1-fold (Figure 3A). We previously noted that degradation peaks correlate with enhancer sequences. However, rapid changes in gene expression upon proteasome inhibition were more common when protein turnover was located close to TSS and were likely promoter associated (Figure 3B).
Figure 3. DNA-Linked Proteolysis Impacts Specific Genes.
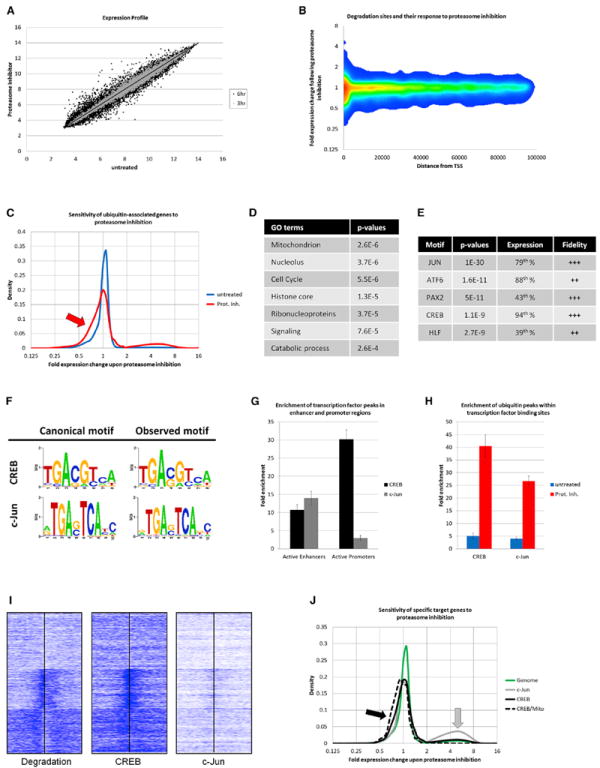
(A) Log2-normalized genome-wide expression in untreated or lactacystin-treated 3T3-L1 cells. Gray dots indicate 3 hr treatment, and black dots indicate 6 hr treatment.
(B) Sensitivity of genes to lactacystin (y axis) in relationship to distance of closest degradation site (x axis).
(C) Response of genes to lactacystin treatment. Plotted are genes with TSS within 3 kbp of the closest steady state (blue) or degradative ubiquitin peak (red). Proteasome inhibition represses some degradation-associated genes (red arrow).
(D) Gene ontologies associated with degradative ubiquitination. Considered were 1,755 genes with ubiquitin peaks within 3 kbp of TSS.
(E) Motif enrichments in regions of degradative ubiquitination, expression ranks of transcription factors, and quality of the observed motifs.
(F) Motifs for CREB and c-Jun in regions of DNA-associated protein turnover.
(G) Enrichment of 3FLAG-c-Jun and endogenous CREB at active enhancers and promoters.
(H) Correlation of ubiquitin peaks and CREB and c-Jun binding. Bars represent mean ± SD.
(I) Heatmap of ubiquitination peaks (left), CREB (middle), and c-Jun (right) at 1,755 promoters associated with degradation. Midlines represent TSS; shown are 3 kbp up- and downstream.
(J) Response of the entire genome (green curve) or specific target genes to lactacystin (targets defined by TSS within 3 kbp of binding sites). c-Jun targets are partially upregulated following proteasome inhibition (gray curve and arrow). CREB-associated genes show a trend for downregulation (black curve) that is significant for nuclear mitochondrial CREB targets (dotted curve and black arrow; p = 6.12 × 10−6).
See also Figure S3.
To define target genes of local proteolysis, degradation peaks that were within 3 kilobases of TSS were selected, and the sensitivity of these genes to 6 hr of lactacystin treatment was assessed. Compared to the entire genome (data not shown) or to steady-state ubiquitination sites (Figure 3C, blue curve), degradation-associated genes were overall repressed by proteasome inhibition (Figure 3C, red curve and arrow, and Figure S3D; p = 4.68 × 10−26). In other words, protein turnover close to TSS has a positive effect on the expression of a significant fraction of genes. A possible bias could be introduced if lactacystin had a generally toxic effect on gene expression, which would disproportionally affect short-lived messenger RNAs (mRNAs). However, this was not the case, as degradation-associated transcripts were of average half-life (Figure S3E) (Schwanhäusser et al., 2011). Instead, the products of these genes were significantly enriched for certain ontologies (Figures 3D and S3F), including nuclear-encoded mitochondrial components, ribosomes, and other RNA-interacting proteins, cell-cycle regulators, and nucleosome subunits (Figure S3G).
The ultimate goal of this topographical analysis is to identify the DNA-bound proteins that are targeted for degradation. By scanning sites of proteasome-dependent ubiquitination for known transcription factor binding motifs, we found enrichment of several DNA motifs, the top five of which are shown (Figure 3E). Utilizing a metric based on the significance of motif occurrence, the expression level of the transcription factor in question, and the quality of the observed DNA sequence motif, we picked c-Jun and CREB as the most promising candidates for being degraded at high levels. Within the regions scanned, we found more than 2,000 occurrences of the c-Jun binding sequence and more than 4,000 hits for CREB with excellent conservation of the observed motifs (Figure 3F). To directly assess any connection between these two transcription factors and degradation sites, ChIP-seq for endogenous CREB and for 3FLAG-c-Jun was performed. Whereas c-Jun preferentially bound to enhancer sequences, CREB was a more promoter-correlated transcription factor (Figure 3G). Both factors were associated with degradation sites (both p < 10−317, Two-sided Fisher’s exact test), with the relative enrichment of CREB outperforming that of c-Jun by 51.7% (Figure 3H). The DNA occupancy heatmap underscored the superior overlap of CREB binding with degradation sites compared to c-Jun (Figure 3I).
To investigate a functional link between CREB, c-Jun binding, and protein turnover, we compared the lactacystin sensitivity of genes within three kilobases of these transcription factors. Several c-Jun target genes were upregulated following proteasome inhibition (Figure 3J, gray curve and arrow). On the other hand, a proportion of CREB target genes was downregulated following lactacystin treatment (Figure 3J, black curve). Nuclear-encoded mitochondrial genes were significantly enriched for CREB binding (p = 3.7 × 10−40, Fisher’s exact test), and CREB was associated with 30.6% of all annotated nuclear mitochondrial genes (390 out of 1,274) (Huang da et al., 2009). Overall, CREB-linked genes were not significantly up- or down-regulated by proteasome inhibition, but the mitochondrial fraction was clearly repressed (Figure 3J, dotted curve and black arrow; p = 6.12 × 10−6 comparing mitochondrial to nonmitochondrial CREB target genes).
Taken together, we showed that promoter-linked proteolysis stimulates the transcription of a substantial number of genes and, importantly, that these genes are enriched for certain functional ontologies, with mitochondrial components being the most prevalent. We could also demonstrate that subsets of CREB target genes significantly correlate with protein turnover.
CREB and NCoR1 Interact at Particularly Proteasome-Sensitive Genes
These results suggest that CREB elimination may be responsible for several of the defined DNA-linked degradation peaks. However, the following lines of evidence argue against that inference. First, genome-wide binding of CREB showed no dramatic change upon proteasome inhibition (Figures S4A–S4E and data not shown). Second, CREB is considered an activating transcription factor, especially at mitochondrial target genes (Altarejos and Montminy, 2011). Thus, its stabilization should increase the expression of relevant genes. We therefore examined whether a CREB corepressor, rather than CREB itself, is stabilized by proteasome inhibition. To identify a potential corepressor, we searched for DNA-binding motifs in regions where CREB and degradation peaks overlapped. Among other motif signatures, we found enrichment for ZBTB33, a protein that lends its DNA-binding domain to the nuclear receptor core-pressor NCoR1 (Figure 4A; p = 1.59 × 10−12, Z test) (Yoon et al., 2003). NCoR1 is a large adaptor module (271 kDa) that is involved in silencing of nuclear receptor target genes and is a known substrate of the UPS (Perissi et al., 2010). Conditional NCoR1 knockout increases insulin sensitivity and mitochondrial function by about 30% (Li et al., 2011; Yamamoto et al., 2011). Given that proteasome inhibition repressed nuclear-encoded mitochondrial genes and that these genes significantly overlapped with degradation sites (Figures 3C, 3D, and 3J), we investigated NCoR1 for mediating lactacystin-induced gene repression. Re-examining the genome-wide map of NCoR1 binding sites in 3T3-L1 cells, published by others (Raghav et al., 2012), revealed that NCoR1 preferentially bound to promoters compared to enhancers, similar to CREB (Figure 4B, left). Furthermore, NCoR1 binding sites correlated with degradation peaks, especially when overlapping with CREB sites (Figure 4B, right). 47.6% (1,749) of genes that were within 3 kilo-bases of CREB were also within the same range of NCoR1 binding sites (Figure 4C). Of these, 23.2% (405) overlapped with genes in range of degradation sites. Such triple-associated genes were significantly repressed by proteasome inhibition (Figure 4D; p = 4.54 × 10−5) and enriched for nuclear-encoded mitochondrial genes (p = 0.00016, Fisher’s exact test).
Figure 4. CREB and Corepressor NCoR1 Interact at Proteasome-Sensitive Promoters.
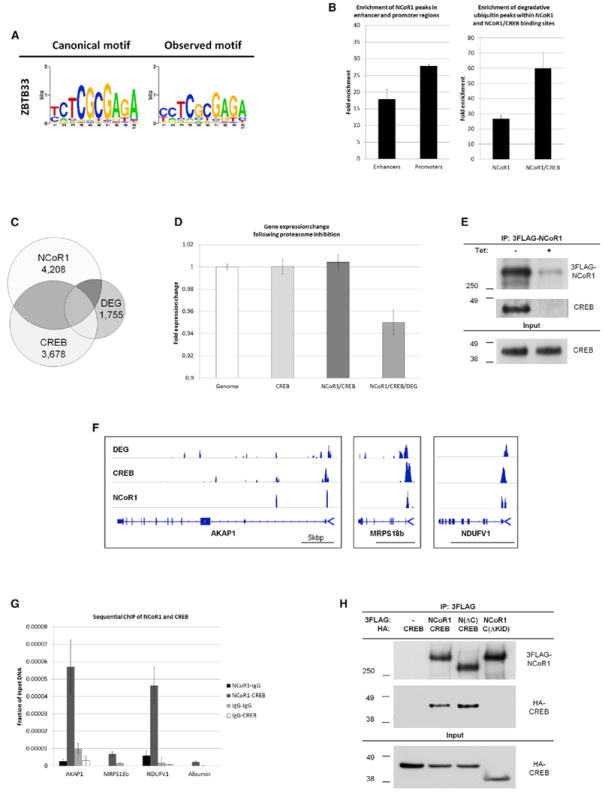
(A) Sites of CREB binding and overlapping degradation revealed enrichment of the ZBTB33 motif, a DNA-binding factor that associates with NCoR1.
(B) Global distribution of NCoR1 favors binding at active promoters (left: sites defined as in Figure 2G). Right: NCoR1 is enriched at degradation sites. Enrichment for degradation at NCoR1- and CREB-associated genes is 48.3% stronger than enrichment at genes associated with CREB only (Figure 3H). Bars represent mean ± SD.
(C) Venn diagram of genes with TSS within 3 kbp of NCoR1, CREB, and degradative ubiquitin peaks (DEG).
(D) Gene sensitivity to lactacystin treatment (bars represent mean ± SEM). Genes that are enriched for the combination of NCoR1, CREB, and degradation peaks are repressed by proteasome inhibition (p = 4.54 × 10−5).
(E) IB after IP of 3FLAG-NCoR1. 3F-NCoR1 expression in 3T3-L1 cells was controlled by a repressor (Tet).
(F) ChIP-seq traces of degradative ubiquitination, CREB, and NCoR1 at three nuclear mitochondrial genes. Black bars represent 5 kbp.
(G) Sequential ChIP-qPCR was performed with isotype control, anti-NCoR1, and anti-CREB antibodies in the sequence depicted. NCoR1 and CREB associated in all three mitochondrial promoters (p < 0.008). Bars represent mean ± SD.
(H) IB after IP of 3FLAG-NCoR1 and HA-CREB transfected human HEK293T cells. N(ΔC), a mutant of NCoR1 missing the three RIDs at the C terminus was used in lane three and C(ΔKID), a mutant lacking the KID domain of CREB, was used in lane four.
See also Figure S4.
Association does not prove interaction, and the promoters of distinct cell fractions could be occupied by either NCoR1 or by CREB in a mutually exclusive fashion. To investigate whether these two factors biochemically interact, we performed nuclear coimmunoprecipitation without crosslinking. The results demonstrate that overexpressed NCoR1 and CREB can engage in complex formation with each other (Figure 4E).
We next examined in more detail three randomly picked mitochondrial genes that were repressed by proteasome inhibition and associated with degradation: AKAP1, MRPS18b, and NDUFV1. ChIP-seq tracks at these three genes showed overlapping peaks between degradative ubiquitination, CREB, and NCoR1 close to the TSS (Figure 4F). We confirmed co-occupancy of endogenous NCoR1 and CREB at these promoters by sequential ChIP (Figure 4G; p < 0.008, two-sided Student’s t test).
NCoR1 is best known for its role in repressing unliganded nuclear hormone receptors (Perissi et al., 2010). The interaction with nuclear hormone receptors is mediated by NCoR1’s C-terminal receptor interaction domains (RID). To address which regions are involved in binding to CREB, we deleted all three RIDs. This mutant form of NCoR1 still robustly interacted with CREB (Figure 4H, third lane). On the other hand, deletion of the kinase-inducible domain (KID) in CREB markedly reduced binding to NCoR1 (Figure 4H, fourth lane). The KID is required for full activation of CREB via phosphorylation of S133, and its involvement in NCoR1 binding suggests a link between transcriptional activity and repression.
Taken together, our data argue for an interaction between CREB and the corepressor NCoR1 at specific promoters. These sites are concentrated at nuclear-encoded mitochondrial genes. Their susceptibility to proteasome inhibition may reflect a requirement for continuous corepressor degradation.
Proteolysis of NCoR1 by Siah2 Derepresses Mitochondrial Genes
To verify functional consequence of the interaction between CREB and NCoR1, we examined gene expression following NCoR1 knockdown by RNA interference (for knockdown efficiencies, see Figures S4F–S4H). Compared to the entire genome, CREB target genes were significantly derepressed by NCoR1 deletion (Figure 5A, black curve and arrow; p = 4.54 × 10−5). The subset of mitochondrial CREB targets showed a trend for even stronger activation relative to all CREB-associated genes (Figure 5A, dotted curve; p = 2.79 × 10−5; two-sided Wilcoxon rank-sum test). As a control, c-Jun target genes were not affected (Figures S4I and S4J). We also observed robust upregulation of degradation-associated genes in the absence of NCoR1 (data not shown; p = 6.99 × 10−14), arguing for a connection between local protein turnover and NCoR1.
Figure 5. NCoR1 and Siah2 Antagonistically Regulate Mitochondrial CREB Target Genes.
(A) 3T3-L1 cells were transduced with NCoR1-specific or scrambled shRNA. CREB target genes (black curve) and CREB-associated nuclear mitochondrial genes (dotted curve) are derepressed following NCoR1 depletion (black arrow).
(B) Sensitivity plot of 1,274 mitochondrial genes to NCoR1 knockdown (blue curve), Siah2 knockdown (orange curve), and Siah2/NCoR1 double knockdown (black curve) compared to scrambled shRNA.
(C) Derepression by NCoR1 knockdown compared to double knockdown does not apply to the entire genome (black curve) but to specific gene entities such as nuclear mitochondrial genes (gray curve).
(D) Expression of 1,274 mitochondrial genes following lactacystin treatment. NCoR1 single as well as Siah2/NCoR1 double knockdown cells show repression (<1 compared to untreated cells with the respective shRNA constructs). Sensitivity to inhibition is greater in cells with NCoR1 depletion only (x axis), and the regression curve (dotted line) is diverging from the diagonal (black line).
(E) ChIP of 3FLAG-NCoR1 and endogenous CREB shows binding to the promoters of AKAP1, MRPS18b, and NDUFV1. NCoR1 accumulates following proteasome inhibition (analysis by qPCR; p < 0.03).
(F) Expression of genes based on RT-qPCR in cells transduced with scrambled shRNA (“3T3L1”) or cells depleted for NCoR1, Siah2, and Siah2/NCoR1. Expression is significantly reduced by lactacystin and Siah2 knockdown and increased by NCoR1 knockdown (p < 0.032). Combined Siah2/NCoR1 knockdown had no effect.
(G) qPCR of three promoters after sequential ChIP of 3FLAG-NCoR1 in the first round.
(H) Followed by precipitation of polyubiquitin in the second round. Cells were treated with lactacystin and either transduced with scrambled shRNA or depleted for Siah2.
(I) Gene expression following CREB knockdown. CREB target genes (blue)–especially those co-overlapping with NCoR1 binding and degradation peaks (red)–are derepressed (black arrow; p = 9.42 × 10−24).
(J) ChIP of endogenous NCoR1 shows depletion following CREB knockdown (p < 0.04).
(K) CREB target genes are upregulated following CREB knockdown (p < 0.05).
(L) Levels of Ser133-phosphorylated CREB are not affected by NCoR1 or Siah2 knockdown.
(M) Acetylation of H3K27 at the promoters of AKAP1, MRPS18b, and NDUFV1 is increased in NCoR1 knockdown cells and decreased in Siah2 knockdown cells (p < 0.005). All bars represent mean ± SD.
See also Figures S4 and S5.
The nuclear proteolysis map presented here is unlikely to offer a complete record of all DNA-associated degradation events. Because our data suggest that continued transcription of some mitochondrial genes requires protein turnover, we next examined the entire set of 1,274 annotated nuclear mitochondrial genes and found strong derepression in NCoR1 knockdown cells (Figure 5B, blue curve; p = 2.92 × 10−11). This set of genes was also downregulated by proteasome inhibition (data not shown; p = 1.31 × 10−57). Therefore, although our examination of nuclear proteolysis only attributed degradation to 12.2% of mitochondrial genes, it accurately defined these genes in general as antagonistically regulated by NCoR1 and the proteasome.
We next sought to determine whether NCoR1 itself is targeted for degradation at these sites. Previous work has defined Siah2 as an E3 ligase that ubiquitinates NCoR1 for elimination (Zhang et al., 1998). Cells that were depleted for NCoR1 (Figure 5B, blue curve) showed higher expression of mitochondrial genes compared to NCoR1/Siah2 double-knockdown cells (black curve; p = 9.26 × 10−37). Knockdown of Siah2 alone resulted in repression of mitochondrial genes (Figure 5B, orange curve; p = 6.33 × 10−82). The response to depletion of NCoR1 versus NCoR1 in combination with Siah2 did not apply to the entire genome but significantly affected nuclear-encoded mitochondrial genes (Figure 5C; p = 7.24 × 10−84). Ratios greater than one on the × coordinate in Figure 5C indicate genes that are either more upregulated or less downregulated in the single compared to the double-knockdown cells.
To determine whether the observed antagonism between NCoR1 and Siah2 was connected by the proteasome, we assessed the sensitivity of mitochondrial genes in single versus double-knockdown cells to proteasome inhibition. Although both cell types repressed mitochondrial genes upon lactacystin treatment, this response was attenuated in double-knockdown cells. Increased resistance of mitochondrial genes to lactacystin with Siah2 depletion resulted in an off-diagonal shift when plotted against NCoR1 single-knockdown cells (Figure 5D; p = 9.53 × 10−4). The reduced dynamic range indicates a role of Siah2 in proteasome-dependent derepression.
To substantiate our proposed model mechanistically, we analyzed the promoters of the three mitochondrial genes AKAP1, MRPS18b, and NDUFV1. ChIP-qPCR confirmed DNA binding (data not shown) and revealed that the levels of NCoR1, but not CREB, increased following lactacystin treatment (Figures 5E and S5A; p < 0.03, two-sided Student’s t test). In agreement with our previous microarray-based results, AKAP1, MRPS18b, and NDUFV1 were repressed by protea-some inhibition (p < 0.032, two-sided Student’s t test), activated upon NCoR1 depletion (p < 0.0015, two-sided Student’s t test), and repressed by Siah2 knockdown (p < 0.03, two-sided Student’s t test) but were not significantly affected by the combined depletion of NCoR1 and Siah2 (Figure 5F). Proteasome inhibition also reduced expression in NCoR1 single-knockdown cells, but the transcript levels after treatment were higher compared to treated native or treated double-knockdown cells (p < 0.01, two-sided Student’s t test). This residual effect may be caused by the incomplete removal of NCoR1 by RNA interference or by compensating factors. Consistent with a direct effect of NCoR1 turnover on the expression of AKAP1, MRPS18b, and NDUFV1, its promoter occupancy was significantly increased in Siah2-depleted cells (Figure 5G; p < 0.014, two-sided Student’s t test). To test whether this correlated with reduced levels of ubiquitinated NCoR1 in the absence of Siah2, we performed sequential ChIP-qPCR first for NCoR1 and then for polyubiquitin. We observed the expected decrease in ubiquitinated NCoR1 at these promoters, indicating that continuous ubiquitination and degradation of NCoR1 occurs in a Siah2-mediated fashion (Figure 5H; p < 0.013, two-sided Student’s t test). Concomitantly, knockdown of NCoR1 was accompanied by a reduced level of site-specific ubiquitination, suggesting that the corepressor is a major substrate of the UPS at these promoters (Figure S5B; p < 0.023, two-sided Student’s t test). We did not observe such a decline of ubiquitin at genes that were unresponsive to proteasome inhibition (data not shown).
We further validated the interaction between NCoR1 and CREB by depleting CREB through small hairpin RNA (shRNA). Many CREB target genes were upregulated in the absence of specific stimulation (Figure 5I, blue curve; p = 9.42 × 10−24). This derepression was especially high at genes that were also co-occupied by NCoR1 and degradation peaks (Figure 5I, red curve; p = 0.0368 compared to all CREB target genes). We also found strong positive correlation between the effects of NCoR1 and CREB knockdown on mitochondrial NCoR1 target genes (407 genes, Figures S5C–S5F; p = 8.83 × 10−24 by linear regression; Pearson’s coefficient ρ = 0.4673). CREB occupancy at the promoters of AKAP1, MRPS18b, and NDUFV1 was reduced by >77% in knockdown cells (data not shown). On the other hand, ChIP with an antibody against a region that is shared between CREB, ATF-1, and CREM showed no difference in CREB knockdown cells (data not shown). These observations suggest that CREB-like proteins rescue expression of CREB target genes in knockdown cells (Blendy et al., 1996) but fail to compensate for corepressor recruitment (Figures 5J and 5K; p < 0.04 and p < 0.05, two-sided Student’s t test).
Knockdown of NCoR1 or Siah2 had no effect on the phosphorylation level of CREB (Figure 5L), arguing that repression is achieved downstream of CREB activation. Phosphorylated CREB can recruit the histone acetyl transferase CBP/p330. To examine a potential antagonism on the chromatin level between the coactivator CBP/p300 and the HDAC-associated corepressor NCoR1 (You et al., 2013), we studied levels of H3K27 acetylation. This histone modification was significantly increased in the absence of NCoR1 and decreased in the absence of Siah2 (Figure 5M; p < 0.005, two-sided Student’s t test).
Combined, our results demonstrate that NCoR1 represses CREB target genes and specifically nuclear mitochondrial genes by deacetylating chromatin. This function is adjusted by the continuous degradation of NCoR1, mediated by the E3 ubiquitin ligase Siah2.
Dynamic Antagonism between NCoR1 and Siah2 Balances Mitochondrial Function
The conditional knockout of NCoR1 in muscle cells results in 20%–30% increased oxidative function (Yamamoto et al., 2011). To formally prove that the expression changes in NCoR1-depleted 3T3-L1 cells also translate into altered mitochondria function, we performed electrophysiological tests. The potentiometric dye DiOC6(3) is a marker for mitochondrial membrane potential (Δψm) (Perry et al., 2011). We analyzed cells with unaltered gene expression or following depletion of NCoR1 alone or in combination with Siah2. NCoR1 knockdown did not change the number of mitochondria based on fluorescent labeling or DNA quantification (Figures 6A, top, and S6A). However, the Δψm increased following NCoR1 single but not Siah2/NCoR1 double knockdown (Figure 6A, bottom, and Figures S6B and S6C; p < 0.04, two-sided Student’s t test). Conversely, overexpression of NCoR1 by transfection or extended proteasome inhibition reduced the Δψm (Figures S6D–S6F).
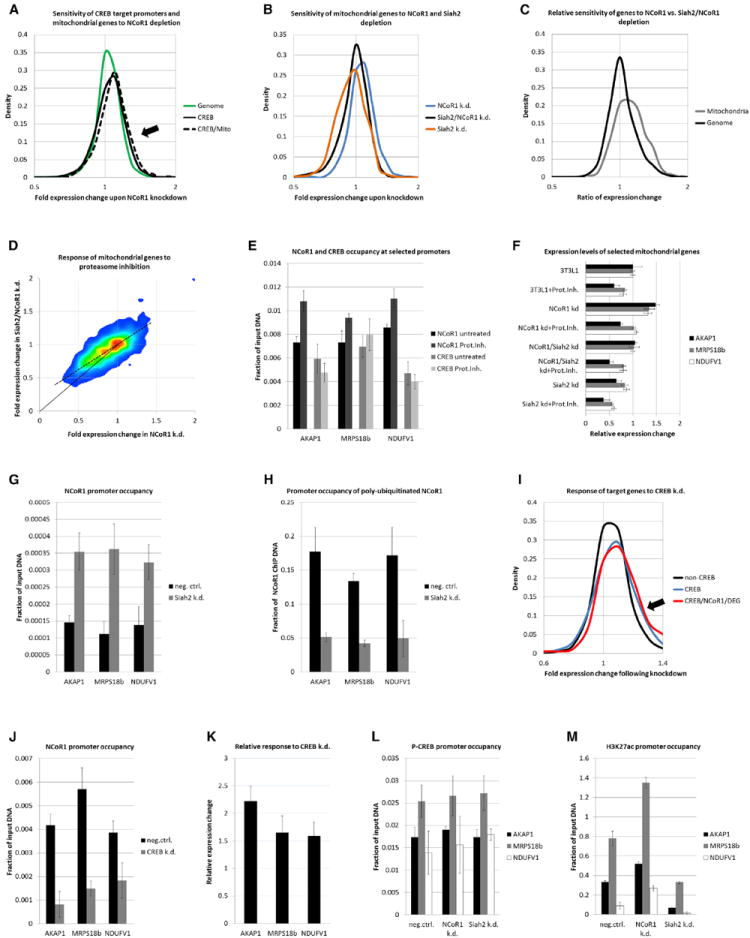
Figure 6. NCoR1 Functionally Restrains Mitochondria.
(A) FACS plots of 3T3-L1 cells transduced with shRNA constructs for NCoR1 and Siah2 or scrambled control (“neg. ctrl.”). The top panel shows staining to assess mitochondrial number, and the bottom panel show staining to quantify Δψm.
(B) Confocal microscopy of scrambled shRNA-transduced 3T3-L1 cells and NCoR1 and Siah2/NCoR1 knockdown cells. Cells were stained with JC-1, which forms red fluorescent aggregates in hyperpolarized mitochondria.
(C) High-resolution microscopy of JC-1-labeled NCoR1 knockdown (top) and Siah2/NCoR1 knockdown cells (bottom).
See also Figure S6.
We verified the electrophysiological results with optical ratiometric analyses using JC-1. This dye is an indicator of mitochondrial function and aggregates upon hyperpolarization, leading to a shift in fluorescence (Collins et al., 2002). Figure 6B depicts the relative increase in J-aggregate formation in NCoR1 knockdown cells. This shift was evident on a population level as well as in the amount of aggregates found within individual hyperpolarized cells (Figure 6C).
Together, these functional tests confirm that NCoR1 and Siah2 adversely influence mitochondrial membrane potential. Stabilization of NCoR1 by proteasome inhibition or Siah2 knockdown presumably reduces ATP production through oxidative phosphorylation.
Synchronization between Mitochondrial Activity and the NCoR1/Siah2 Axis
Metabolic adaptation requires communication between mitochondria and the nucleus because the organelle only carries 13 protein-coding genes (Ryan and Hoogenraad, 2007). We challenged mitochondria by depolarization with the uncoupling ionophore FCCP and observed induction of mitochondrial genes (Figure 7A, red gate, and S7A; p = 2.8 × 10−20, Fisher’s exact test). Genes that were stimulated by FCCP were repressed in Siah2 knockdown cells (p < 4.51 × 10−308 and p = 1.1 × 10−37 for upregulation of CREB target genes by FCCP, data not shown). We also observed the expected opposite effect of FCCP and proteasome inhibition on mitochondrial genes (Figure 7B; p = 6.31 × 10−40 for repression by lactacystin, p = 3.91 × 10−4 for induction by FCCP, and there is no significant effect for combined treatment). At least part of the activation following depolarization was achieved by Siah2-dependent removal of NCoR1 from target promoters (Figure 7C; p < 0.027 for removal following FCCP, and p < 0.01 for upregulation of endogenous NCoR1 in Siah2 knockdown cells; two-sided Student’s t test). Lack of Siah2 reduced the effect of FCCP on gene induction (Figure 7D; p < 0.015, two-sided Student’s t test). Siah2 itself was robustly upregulated by mitochondrial depolarization, possibly paving the way for accelerated NCoR1 removal (Figure 7D). The antagonism between proteasome inhibition and mitochondrial uncoupling was also observed at the level of H3K27 acetylation (Figure 7E; p < 0.05, two-sided Student’s t test).
Figure 7. Mitochondrial Dysfunction Accelerates NCoR1 Degradation.
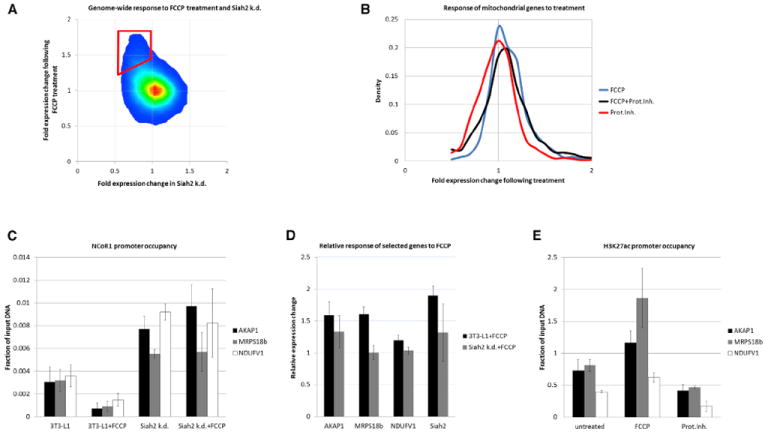
(A) Genome-wide expression changes following treatment with FCCP or Siah2 depletion. The red gate signifies genes that are upregulated by FCCP and downregulated by Siah2 knockdown.
(B) Mitochondrial genes (1,274) are stimulated by treatment with FCCP (blue curve) and repressed by lactacystin (red curve; p < 0.0004) but are not significantly altered by combined treatment.
(C) ChIP of endogenous NCoR1 shows elimination from promoters following FCCP treatment (analysis by qPCR; p < 0.027). Promoter occupancy in Siah2 knockdown cells is higher (p < 0.01), and FCCP does not remove NCoR1 from promoters in these cells.
(D) Upregulation of AKAP1, MRPS18b, and NDUFV1 following FCCP treatment (p < 0.02). No significant upregulation is seen in Siah2 knockdown cells.
(E) Acetylation levels of H3K27 at indicated promoters following FCCP or lactacystin treatment. FCCP increases and lactacystin decreases acetylation (p < 0.05). All bars represent mean ± SD.
See also Figure S7.
DISCUSSION
Dynamic regulation of gene expression requires the binding as well as the removal of transcription factors. To investigate the latter, we established a method to localize and quantify genomic sites of protein turnover. We focused on ubiquitination as the initial “kiss of death” to identify DNA elements that correlate with proteolysis. Our data suggest that DNA-associated degradation occurs at genes with high activity, and we propose that the majority of DNA-linked proteolysis affects transcriptional regulators, in line with previous work (Salghetti et al., 2000). Furthermore, we could exclude RNA polymerase II itself as a major substrate of degradation (data not shown).
Our data demonstrate that protein turnover is enriched at CREB-occupied promoters. We propose that this is caused by degradation of the associated corepressor NCoR1. Proteolysis of NCoR1 is a well-documented mechanism that controls the switch from repression to activation in nuclear hormone receptor target genes (Perissi et al., 2010). NCoR1 depletion increases insulin signaling, metabolic efficiency, and muscle size, and NCoR1 inhibition may provide treatment options for type II diabetes and sarcopenia (Li et al., 2011; Yamamoto et al., 2011). CREB activation and NCoR1 depletion both stimulate mitochondrial activity and biogenesis in muscle cells (Wu et al., 2006; Yamamoto et al., 2011). In addition, the kinase AKT can simultaneously activate CREB (Du and Montminy, 1998) and inactivate NCoR1 (Perissi et al., 2010), suggesting antagonism between both. Consistent with this notion, we provide here evidence for an immediate repression of CREB by NCoR1. Both proteins physically interact either directly or via cofactors at promoters that are predominantly TATA-less (Conkright et al., 2003) (Figure S7B). CREB-bound genes that are repressed by the proteasome are induced by the cAMP-generating drug forskolin (Figures S7C and S7D), suggesting they are not fully activated at steady state. Combined, these results support a model by which CREB activity can be modulated at three levels: repression by NCoR1, derepression by elimination of NCoR1, and full activation by removal of NCoR1 and phosphorylation of CREB. It remains to be determined whether derepression and activation occur independently under physiological conditions.
Ubiquitination of NCoR1 is triggered by the ligase Siah2 (Zhang et al., 1998), but substrate recognition requires additional factors such as F-box-like/WD40-containing proteins (Perissi et al., 2004). Siah2 also directly regulates mitochondrial proteins under hypoxic conditions (Carlucci et al., 2008; Kim et al., 2011a). We grew cells under ambient oxygen, and our experimental design probed for the expression of nuclear genes as primary readout. The fact that a combined knockdown with NCoR1 rescued the effects of Siah2 deficiency alone suggests that the main impact of Siah2 on mitochondrial activity under normal oxygen occurs in the nucleus. In addition, Siah2 regulates NCoR1 abundance at target promoters, and we provide evidence for antagonistic control of H3K27 acetylation at mitochondrial genes by these two factors (Figures 5B, 5F, 5G, and 5M). However, it is intriguing that hypoxia, as well as mitochondrial dysfunction under normoxic conditions (FCCP treatment), induces Siah2 (Nakayama et al., 2004). Under low oxygen, the E3 ligase attenuates oxidative phosphorylation by eliminating mitochondrial proteins and stabilizing the HIF pathway, an obvious response to the environment. Under normoxic conditions, we observed an opposite effect, in which Siah2 stimulates mitochondrial activity. Binding of Siah2 to substrate is mediated through WD40 domain-containing proteins. Genes encoding these modules are among the most upregulated following FCCP treatment (p = 1.6 × 10−7, Fisher’s exact test; data not shown). It is therefore conceivable that Siah2 could adjust its function by recruiting different recognition modules and promoting degradation of distinct substrates in dependence of the metabolic milieu. Under this assumption, one might expect that hypoxia induces stabilization of NCoR1, despite upregulation of Siah2. Also, Siah2 is phosphorylated under hypoxia, which facilitates its nuclear export (Khurana et al., 2006).
Our inability to detect binding of endogenous NCoR1 to CREB in the absence of crosslinking may reflect the short duration of this interaction at steady state. Further studies are needed to examine how different physiological conditions impact the NCoR1/CREB complex and how Siah2 achieves specificity for the removal of NCoR1 in a subset of genes, a phenomenon that was recently exemplified by the role of this E3 ligase in gene-selective androgen receptor regulation (Qi et al., 2013).
As an adaptor molecule, NCoR1 is involved in HDAC recruitment (You et al., 2013) and other mechanisms of transcriptional control (Zhou et al., 2008). It is surprising that a cell would invest in the continuous destruction of a large protein such as NCoR1, whose synthesis is so ATP consuming. This costly mechanism may provide a suitable feedback between the nucleus and mitochondria when energy deficiency translates into diminished corepressor levels, which then releases mitochondrial genes to increase ATP production.
The examination of degradation sites for transcription factor motifs revealed more than 80 candidates, including nuclear hormone receptors (Perissi et al., 2010). The most prominent motifs within this family belong to the glucocorticoid receptor GCR/NR3C1, the thyroid hormone receptor THRA, and the retinoic acid receptor RARG (p < 0.0006, Z test). Therefore, our results do not contradict earlier reports on proteasomal regulation of nuclear hormone receptors.
NCoR1 has been shown to repress several transcription factors (Ghisletti et al., 2009), in part by recruitment through non-DNA-bound nuclear hormone receptors (Pascual et al., 2005). It is therefore conceivable that a nuclear hormone receptor forms a complex with CREB and NCoR1. However, we have no data to support such a connection because NCoR1 does not require the receptor interacting domain for association with CREB (Figure 4H).
Our method favors the detection of DNA regions with dynamic protein turnover. It is very well possible that the majority of NCoR1 at steady state binds to nuclear hormone receptors, but it is the particularly short-lived interaction with CREB that we identified. There are intriguing parallels between nuclear hormone receptors and CREB. Both recruit coactivators upon stimulation and are involved in homeostasis and metabolic adaptation (Altarejos and Montminy, 2011). The parallel that has been less explored is that of a corepressor. We propose that NCoR1 dampens CREB target genes, and its constant elimination by the ubiquitin-proteasome system is required to maintain gene expression. Given the rapid and reversible nature of CREB activation and its role as an integrator of environmental signals (Altarejos and Montminy, 2011), it would be consistent that NCoR1 degradation is particularly high at metabolic CREB target promoters.
In summary, we present a functional annotation of DNA-linked protein degradation, offering mechanistic insights into how the UPS regulates global gene expression. Our methodology provides a framework for future studies to dissect the precise role of individual enzymes of the UPS in DNA-associated degradation and to investigate differences between various cell types.
EXPERIMENTAL PROCEDURES
Full descriptions of experimental procedures as well as gene expression values, ChIP-on-chip results, gene annotations, and ChIP-seq maps are included in the Extended Experimental Procedures and in Tables S1 and S2.
Cell Culture
3T3-L1 cells were grown in DMEM/10% FCS (37°C, 5% CO2). The proteasome was inhibited with lactacystin for 3 (ChIPs) or 6 hr (expression analyses).
Gene Expression
Expression was measured with Affymetrix Mouse Gene 1.0 ST arrays. Libraries were constructed using oligo-dT in combination with random hexamer priming. SYBR-Green based RT-qPCR was performed with transcript-specific reverse transcription.
ChIP-Seq and Data Analyses
We used M2 anti-FLAG or protein-specific antibodies for ChIP. DNA was prepared according to Illumina’s protocols for single-end sequencing (HiSeq 2000) and mapped to genome mm9. ChIP-seq peaks were called with MACS1.4, and motifs were detected with SeqPos (2 kb window). GO analyses were conducted with DAVID (Huang da et al., 2009). Chromatin states are based on GSE20752 (Mikkelsen et al., 2010), and the genomic distribution of NCoR1 is based on ERR103444 (Raghav et al., 2012). Unless indicated otherwise, all statistical tests are based on the two-sided Wilcoxon rank-sum test with Bonferroni correction.
Supplementary Material
Acknowledgments
We thank Carlota Dao, Laura Prickett-Rice, Kat Folz-Donahue, Meredith Weglarz, Katherine Kulig, Christa Buecker, Hsu-Hsin Chen, Mark Borowsky, Caroline Woo, Youn-Kyoung Lee, Alison Brown, and Vance Morgan for support. T.H. is a fellow of the German National Academic Foundation. A.C. appreciates mentorship by Daniel Finley, Robert Kingston, Derrick Rossi, and Ron Kohanski (NIH/NIA). A.C. is an Irvington Fellow of the Cancer Research Institute and is supported by the Margaret Dammann Eisner Foundation and by National Institute on Aging grant K01-AG036744. D.T.S. is supported by NIH grant DK050234. We thank the reviewers for constructive suggestions and apologize for references we could not include.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Extended Experimental Procedures, seven figures, and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2013.11.016.
ACCESSION NUMBERS
Sequencing data are available at the Gene Expression Omnibus (GSE33821) under accession numbers GSM838021, GSM838022, GSM838023, GSM838024, GSM841627, GSM1095377, GSM1095378, GSM1095379, GSM1095380, GSM1095381, GSM1095382, and GSM1095383.
References
- Altarejos JY, Montminy M. CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat Rev Mol Cell Biol. 2011;12:141–151. doi: 10.1038/nrm3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auld KL, Brown CR, Casolari JM, Komili S, Silver PA. Genomic association of the proteasome demonstrates overlapping gene regulatory activity with transcription factor substrates. Mol Cell. 2006;21:861–871. doi: 10.1016/j.molcel.2006.02.020. [DOI] [PubMed] [Google Scholar]
- Blendy JA, Kaestner KH, Schmid W, Gass P, Schutz G. Targeting of the CREB gene leads to up-regulation of a novel CREB mRNA isoform. EMBO J. 1996;15:1098–1106. [PMC free article] [PubMed] [Google Scholar]
- Carlucci A, Adornetto A, Scorziello A, Viggiano D, Foca M, Cuomo O, Annunziato L, Gottesman M, Feliciello A. Proteolysis of AKAP121 regulates mitochondrial activity during cellular hypoxia and brain ischaemia. EMBO J. 2008;27:1073–1084. doi: 10.1038/emboj.2008.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A. Intracellular protein degradation: from a vague idea thru the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Biochim Biophys Acta. 2012;1824:3–13. doi: 10.1016/j.bbapap.2011.03.007. [DOI] [PubMed] [Google Scholar]
- Collins TJ, Berridge MJ, Lipp P, Bootman MD. Mitochondria are morphologically and functionally heterogeneous within cells. EMBO J. 2002;21:1616–1627. doi: 10.1093/emboj/21.7.1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conkright MD, Guzmán E, Flechner L, Su AI, Hogenesch JB, Montminy M. Genome-wide analysis of CREB target genes reveals a core promoter requirement for cAMP responsiveness. Mol Cell. 2003;11:1101–1108. doi: 10.1016/s1097-2765(03)00134-5. [DOI] [PubMed] [Google Scholar]
- Du K, Montminy M. CREB is a regulatory target for the protein kinase Akt/PKB. J Biol Chem. 1998;273:32377–32379. doi: 10.1074/jbc.273.49.32377. [DOI] [PubMed] [Google Scholar]
- Geng F, Tansey WP. Similar temporal and spatial recruitment of native 19S and 20S proteasome subunits to transcriptionally active chromatin. Proc Natl Acad Sci USA. 2012;109:6060–6065. doi: 10.1073/pnas.1200854109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghisletti S, Huang W, Jepsen K, Benner C, Hardiman G, Rosenfeld MG, Glass CK. Cooperative NCoR/SMRT interactions establish a corepressor-based strategy for integration of inflammatory and anti-inflammatory signaling pathways. Genes Dev. 2009;23:681–693. doi: 10.1101/gad.1773109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Khurana A, Nakayama K, Williams S, Davis RJ, Mustelin T, Ronai Z. Regulation of the ring finger E3 ligase Siah2 by p38 MAPK. J Biol Chem. 2006;281:35316–35326. doi: 10.1074/jbc.M606568200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Scimia MC, Wilkinson D, Trelles RD, Wood MR, Bowtell D, Dillin A, Mercola M, Ronai ZA. Fine-tuning of Drp1/Fis1 availability by AKAP121/Siah2 regulates mitochondrial adaptation to hypoxia. Mol Cell. 2011a;44:532–544. doi: 10.1016/j.molcel.2011.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, Sowa ME, Rad R, Rush J, Comb MJ, et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell. 2011b;44:325–340. doi: 10.1016/j.molcel.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Kriegenburg F, Seeger M, Saeki Y, Tanaka K, Lauridsen AM, Hartmann-Petersen R, Hendil KB. Mammalian 26S proteasomes remain intact during protein degradation. Cell. 2008;135:355–365. doi: 10.1016/j.cell.2008.08.032. [DOI] [PubMed] [Google Scholar]
- Li P, Fan W, Xu J, Lu M, Yamamoto H, Auwerx J, Sears DD, Talukdar S, Oh D, Chen A, et al. Adipocyte NCoR knockout decreases PPARγ phosphorylation and enhances PPARγ activity and insulin sensitivity. Cell. 2011;147:815–826. doi: 10.1016/j.cell.2011.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipford JR, Deshaies RJ. Diverse roles for ubiquitin-dependent proteolysis in transcriptional activation. Nat Cell Biol. 2003;5:845–850. doi: 10.1038/ncb1003-845. [DOI] [PubMed] [Google Scholar]
- Mikkelsen TS, Xu Z, Zhang X, Wang L, Gimble JM, Lander ES, Rosen ED. Comparative epigenomic analysis of murine and human adipogenesis. Cell. 2010;143:156–169. doi: 10.1016/j.cell.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama K, Frew IJ, Hagensen M, Skals M, Habelhah H, Bhoumik A, Kadoya T, Erdjument-Bromage H, Tempst P, Frappell PB, et al. Siah2 regulates stability of prolyl-hydroxylases, controls HIF1alpha abundance, and modulates physiological responses to hypoxia. Cell. 2004;117:941–952. doi: 10.1016/j.cell.2004.06.001. [DOI] [PubMed] [Google Scholar]
- Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, Glass CK. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature. 2005;437:759–763. doi: 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perissi V, Aggarwal A, Glass CK, Rose DW, Rosenfeld MG. A corepressor/coactivator exchange complex required for transcriptional activation by nuclear receptors and other regulated transcription factors. Cell. 2004;116:511–526. doi: 10.1016/s0092-8674(04)00133-3. [DOI] [PubMed] [Google Scholar]
- Perissi V, Jepsen K, Glass CK, Rosenfeld MG. Deconstructing repression: evolving models of co-repressor action. Nat Rev Genet. 2010;11:109–123. doi: 10.1038/nrg2736. [DOI] [PubMed] [Google Scholar]
- Perry SW, Norman JP, Barbieri J, Brown EB, Gelbard HA. Mitochondrial membrane potential probes and the proton gradient: a practical usage guide. Biotechniques. 2011;50:98–115. doi: 10.2144/000113610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi J, Tripathi M, Mishra R, Sahgal N, Fazli L, Ettinger S, Placzek WJ, Claps G, Chung LW, Bowtell D, et al. The E3 ubiquitin ligase Siah2 contributes to castration-resistant prostate cancer by regulation of androgen receptor transcriptional activity. Cancer Cell. 2013;23:332–346. doi: 10.1016/j.ccr.2013.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghav SK, Waszak SM, Krier I, Gubelmann C, Isakova A, Mikkelsen TS, Deplancke B. Integrative genomics identifies the corepressor SMRT as a gatekeeper of adipogenesis through the transcription factors C/EBPβ and KAISO. Mol Cell. 2012;46:335–350. doi: 10.1016/j.molcel.2012.03.017. [DOI] [PubMed] [Google Scholar]
- Rubenstein EM, Hochstrasser M. Redundancy and variation in the ubiquitin-mediated proteolytic targeting of a transcription factor. Cell Cycle. 2010;9:4282–4285. doi: 10.4161/cc.9.21.13741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan MT, Hoogenraad NJ. Mitochondrial-nuclear communications. Annu Rev Biochem. 2007;76:701–722. doi: 10.1146/annurev.biochem.76.052305.091720. [DOI] [PubMed] [Google Scholar]
- Sakata E, Bohn S, Mihalache O, Kiss P, Beck F, Nagy I, Nickell S, Tanaka K, Saeki Y, Förster F, Baumeister W. Localization of the proteasomal ubiquitin receptors Rpn10 and Rpn13 by electron cryomicroscopy. Proc Natl Acad Sci USA. 2012;109:1479–1484. doi: 10.1073/pnas.1119394109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salghetti SE, Muratani M, Wijnen H, Futcher B, Tansey WP. Functional overlap of sequences that activate transcription and signal ubiquitin-mediated proteolysis. Proc Natl Acad Sci USA. 2000;97:3118–3123. doi: 10.1073/pnas.050007597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- Wagner SA, Beli P, Weinert BT, Nielsen ML, Cox J, Mann M, Choudhary C. A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol Cell Proteomics. 2011;10 doi: 10.1074/mcp.M111.013284. M111.013284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Huang X, Feng Y, Handschin C, Feng Y, Gullicksen PS, Bare O, Labow M, Spiegelman B, Stevenson SC. Transducer of regulated CREB-binding proteins (TORCs) induce PGC-1alpha transcription and mitochondrial biogenesis in muscle cells. Proc Natl Acad Sci USA. 2006;103:14379–14384. doi: 10.1073/pnas.0606714103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Duong DM, Seyfried NT, Cheng D, Xie Y, Robert J, Rush J, Hochstrasser M, Finley D, Peng J. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell. 2009;137:133–145. doi: 10.1016/j.cell.2009.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Williams EG, Mouchiroud L, Cantó C, Fan W, Downes M, Héligon C, Barish GD, Desvergne B, Evans RM, et al. NCoR1 is a conserved physiological modulator of muscle mass and oxidative function. Cell. 2011;147:827–839. doi: 10.1016/j.cell.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon HG, Chan DW, Reynolds AB, Qin J, Wong J. N-CoR mediates DNA methylation-dependent repression through a methyl CpG binding protein Kaiso. Mol Cell. 2003;12:723–734. doi: 10.1016/j.molcel.2003.08.008. [DOI] [PubMed] [Google Scholar]
- You SH, Lim HW, Sun Z, Broache M, Won KJ, Lazar MA. Nuclear receptor co-repressors are required for the histone-deacetylase activity of HDAC3 in vivo. Nat Struct Mol Biol. 2013;20:182–187. doi: 10.1038/nsmb.2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Guenther MG, Carthew RW, Lazar MA. Proteasomal regulation of nuclear receptor corepressor-mediated repression. Genes Dev. 1998;12:1775–1780. doi: 10.1101/gad.12.12.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Zhu P, Wang J, Pascual G, Ohgi KA, Lozach J, Glass CK, Rosenfeld MG. Histone H2A monoubiquitination represses transcription by inhibiting RNA polymerase II transcriptional elongation. Mol Cell. 2008;29:69–80. doi: 10.1016/j.molcel.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.