

Abstract
This was a phase 1B, dose-ranging, multicenter, pharmacokinetics, and safety study of cyclodextrin-based posaconazole intravenous (i.v.) solution administered through a central line to subjects at high risk for invasive fungal disease (part 1 of a 2-part study [phase 1B/3]). Initially, the safety and tolerability of single-dose posaconazole i.v. 200 mg (n = 10) were compared with those of a placebo (n = 11). Subsequently, 2 doses were evaluated, posaconazole i.v. 200 mg once daily (q.d.) (n = 21) and 300 mg q.d. (n = 24). The subjects received twice-daily (b.i.d.) posaconazole i.v. on day 1, followed by 13 days of posaconazole i.v. q.d., then 14 days of posaconazole oral suspension 400 mg b.i.d. The steady-state (day 14) exposure target (average concentration [areas under concentration-time curve {AUCs}/24 h, average concentrations at steady state {Cavgs}], of ≥500 to ≤2,500 ng/ml in ≥90% of the subjects) was achieved by 94% of the subjects for 200 mg posaconazole q.d. and by 95% of subjects for 300 mg posaconazole q.d. The desired exposure target (mean steady-state Cavg, ∼1,200 ng/ml) was 1,180 ng/ml in the 200-mg dosing cohort and was exceeded in the 300-mg dosing cohort (1,430 ng/ml). Posaconazole i.v. was well tolerated. Posaconazole i.v. 300 mg q.d. was selected for the phase 3 study segment. (This study has been registered at ClinicalTrials.gov under registration no. NCT01075984.)
INTRODUCTION
Posaconazole oral suspension (Noxafil; Merck & Co., Inc., Whitehouse Station, NJ) is a marketed extended-spectrum triazole with demonstrated efficacy as antifungal prophylaxis and treatment (1–5). Posaconazole is approved for the prophylaxis of invasive fungal disease (IFD) in allogeneic hematopoietic stem cell transplant recipients with graft-versus-host disease or in those with certain hematologic malignancies and prolonged neutropenia from chemotherapy, for the prophylaxis and treatment of patients with oropharyngeal candidiasis in the United States and Europe, and for the treatment of patients with refractory IFD in Europe (6, 7). The bioavailability of posaconazole oral suspension is significantly enhanced when given with food (8, 9). Therefore, to address this issue, a new tablet formulation of posaconazole has been developed (10). However, a limitation of any oral formulation is that patients at risk for IFD may be unable to take or absorb oral medication (11, 12).
Therefore, an unmet need exists for an intravenous (i.v.) formulation of posaconazole that allows for administration of posaconazole to patients unable to take an oral medication or for whom absorption is a concern. A posaconazole i.v. formulation was recently developed as an aqueous solution containing the solubilizer sulfobutyl ether beta-cyclodextrin. This is the same solubilizer (and in a similar amount) used in i.v. voriconazole, and it is structurally similar to that used in i.v. itraconazole (13, 14).
Data from previous randomized, active-controlled clinical studies with posaconazole oral suspension suggest an exposure-response relationship exists (15). Based on these data, a targeted exposure range (Cavgs of >500 ng/ml but ≤2,500 ng/ml) for posaconazole was selected in this study. This 2-part study (phase 1B/3) was conducted to evaluate the pharmacokinetics (PK) and safety of posaconazole i.v. when given as antifungal prophylaxis to subjects at high risk for IFD. Here we present the results of the dose-finding part 1 (phase 1B).
(This work was presented previously at the 52nd Interscience Conference on Antimicrobial Agents and Chemotherapy, San Francisco, CA, 9 to 12 September 2012.)
MATERIALS AND METHODS
Study design.
This open-label, phase 1B/3, adaptive, dose-ranging, multicenter PK and safety study (ClinicalTrials.gov identifier NCT01075984; study P05220) was conducted in accordance with the principles of good clinical practice and the Declaration of Helsinki; written informed consent was obtained from each subject before any study-related procedures were performed. The study was conducted at 7 sites in Belgium, Germany, and Switzerland. Ethics committee approval was obtained from each study site.
Three sequential dosing cohorts were included (cohorts 0, 1, and 2) (Fig. 1). In all cohorts, posaconazole was infused over 90 min through a central line. Posaconazole i.v. solution is an aqueous injectable solution containing 18 mg/ml posaconazole and 400 mg/ml sulfobutyl ether beta-cyclodextrin, to be diluted with 0.9% sodium chloride or 5% dextrose in water before i.v. administration. Once the safety and tolerability of a single posaconazole i.v. dose were determined, the decision was made to proceed with cohort 1. In cohort 1 (multiple-dose, 200-mg, once-daily cohort), 21 subjects received a loading dose of 200 mg posaconazole i.v. twice daily on day 1, followed by a maintenance dose of 200 mg posaconazole i.v. once daily (days 2 to 14), followed by a posaconazole oral suspension maintenance dose (400 mg) twice daily for 14 days (days 15 to 28). In cohort 2 (multiple-dose, 300-mg, once-daily cohort), 24 subjects received a loading i.v. dose of 300 mg posaconazole i.v. twice daily on day 1, followed by a maintenance i.v. dose of 300 mg posaconazole i.v. once daily (days 2 to 14), followed by a posaconazole oral suspension maintenance dose (400 mg) twice daily for 14 days (days 15 to 28). In all cohorts, posaconazole i.v. was administered through a central venous line because administration of multiple doses of posaconazole i.v. to healthy volunteers through a peripheral i.v. line was associated with infusion site reactions (single doses were well tolerated) (16).
FIG 1.
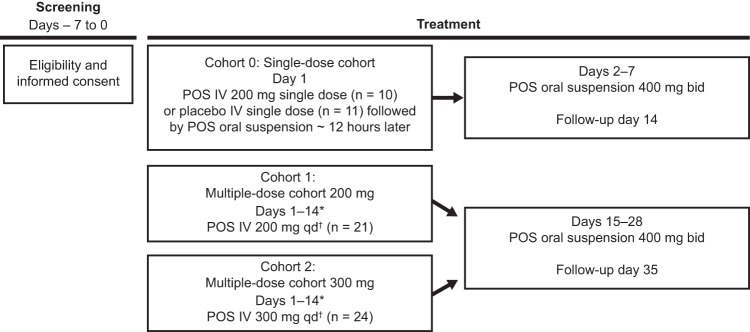
Study design, phase 1B. Each cohort was completed before subjects in the subsequent cohort were dosed. Safety was assessed throughout the study. *, PK samples for analysis of posaconazole were taken on days 1 and 14 at 0 h (predose), 1 h after start of infusion, immediately at the end of infusion, approximately 15 min after the end of infusion, and approximately 4, 8, 12, and 24 h after start of infusion. In cohorts 1 and 2, intravenous posaconazole (200 or 300 mg) was given twice daily as a loading dose on day 1. †, twice daily loading dose on day 1. bid, twice daily; IV, intravenous; POS, posaconazole; qd, once daily.
Subjects.
Adult subjects (≥18 years old) were enrolled after the start of chemotherapy for acute myelogenous leukemia, myelodysplastic syndrome, or secondary leukemia. Subjects had (or, in the opinion of the investigators, were likely to develop within 5 days) neutropenia (absolute neutrophil counts of <500/mm3 [0.5 × 109/liter]) that was likely to last ≥7 days. Subjects were required to have a central venous catheter in place as part of the standard of care for the underlying disease. Subjects were male or female, of any race, with weight of >34 kg. Subjects were excluded if they had known or suspected IFD, a history of type 1 hypersensitivity or idiosyncratic reactions to azoles, moderate or severe liver dysfunction (defined as aspartate aminotransferase [AST] or alanine aminotransferase [ALT] levels of >3× the upper limit of normal [ULN] and a total bilirubin level of >2× ULN), a prolonged corrected QT (QTc) interval (>500 ms), or a creatinine clearance rate of <50 ml/min. Subjects were excluded if they had taken oral posaconazole within 10 days before study enrollment or systemic antifungal therapy within 30 days of study enrollment for reasons other than antifungal prophylaxis. Concomitant medications were monitored throughout the study.
PK sampling.
Blood samples were collected for PK analysis on days 1 and 14 (steady state) at 0 h (predose), 1 h after the start of infusion, immediately at the end of infusion, approximately 15 min after the end of infusion, and approximately 4, 8, 12, and 24 h after the start of infusion (12-hour sample on day 1 taken before the second dose, 24-hour sample on day 1 equivalent to the 12-hour sample after the second dose). Blood samples were also collected during oral suspension dosing (days 15 to 28; results not presented). Plasma samples (2 duplicate sets) were immediately frozen to at least −20°C and were maintained frozen until analyzed. Plasma samples were assayed for posaconazole using a validated liquid chromatography coupled to tandem mass spectrometry detection method (17) with a calibration range of 5 to 5,000 ng/ml.
PK evaluations.
The primary PK parameter of interest was steady-state (day 14) average concentration (Cavg; calculated as the area under the concentration-time curve [AUC]/24 h). Cavg was selected because exposure-response analyses of previous randomized, active-controlled clinical studies with posaconazole oral suspension suggested an inverse association between Cavg and clinical failure rate (15). Other parameters assessed included the AUC during the dosing interval (dosing interval was 12 h on day 1 [AUC over 0 to 12 h {AUC0–12 h}] and 24 h on day 14 [AUC over 0 to 24 h {AUC0–24 h}]), maximum plasma concentration (Cmax) on days 1 and 14, time to Cmax (Tmax) on days 1 and 14, and minimum plasma concentration (Cmin). Posaconazole PK parameters were calculated using the software Phoenix Build 6.3.0, WinNonLin 6.3, and Connect 1.3 (Certara, St. Louis, MO). Cmax and Tmax were generated by WinNonLin from each subject's plasma concentration-time data. All AUC parameters were calculated using the linear trapezoidal method for ascending concentrations and the log trapezoidal method for descending concentrations (linear up/log down).
Safety.
Safety assessments included adverse event (AE) and serious AE (SAE) reports (including catheter-related events and infusion site reactions), vital signs, clinical laboratory tests, and electrocardiograms. Safety was assessed throughout the study (during i.v. and oral suspension dosing) until the follow-up visit (7 days after the last dose of study drug). Patients were assessed for AEs daily throughout the study. Electrocardiography was performed at baseline, on the morning of i.v. administration, and on day 4 during oral administration. Hematology and serum chemistries were assessed at baseline, before i.v. administration, on days 2, 4, and 6 during oral administration, at the end of treatment, and 7 days after the end of treatment. Safety analyses were descriptive, summarized by dose level, and performed on all treated subjects. An independent data monitoring committee reviewed the safety data after each cohort was completed to assess whether the next cohort should proceed according to predefined safety criteria (including no subjects with QT effect [QTc >500 ms] or hepatic effect [AST/ALT levels of >3× ULN or bilirubin level of >2× ULN without underlying hepatic disorder]).
Breakthrough IFD.
Proven, probable, and possible cases of IFD were categorized by the investigators according to the 2008 European Organization for Research and Treatment of Cancer Mycoses Study Group criteria (18).
Statistical analysis.
The determination of sample size was based on the targeted number of subjects for evaluation of PK and safety and on the anticipated discontinuation rate. A recently completed trial with a similar population showed a day-28 subject discontinuation rate of approximately 35%. Therefore, in order to have approximately 30 PK-evaluable subjects in part 1 reach steady state, the plan was to enroll approximately 50 subjects in part 1 of this study.
Posaconazole plasma concentrations and PK parameters were listed and summarized by treatment group using descriptive statistics and graphics for the multiple-dose cohorts 1 and 2. The desired exposure target was a mean steady-state Cavg of approximately 1,200 ng/ml (or AUC0–24 h of 28,800 ng · h/ml), with at least 90% of subjects in the PK-evaluable dosing cohort having Cavg of ≥500 ng/ml and ≤2,500 ng/ml (or AUC0–24 h of ≥12,000 ng · h/ml and ≤60,000 h · ng/ml). For the multiple-dose cohorts (cohorts 1 and 2), PK criteria had to be fulfilled for each dose before dosing could proceed in the next cohort. For the study to proceed to cohort 2, no subject in cohort 1 could have a steady-state plasma Cavg of ≥3,750 ng/ml or a steady-state AUC of >90,000 ng · h/ml. For the study to proceed to cohort 3 with either dose (phase 3 of the study; not presented here), the desired exposure target had to be met, and no subject at the selected dose could have a steady-state plasma Cavg of >3,650 ng/ml or a steady-state AUC of >87,600 ng · h/ml.
RESULTS
Subjects.
Sixty-six subjects were enrolled into the phase 1B portion of the study, 21 subjects (10 subjects receiving posaconazole i.v. solution and 11 subjects receiving placebo) in the single-dose cohort (cohort 0), 21 subjects in the 200-mg multiple-dose cohort (cohort 1), and 24 subjects in the 300-mg multiple-dose cohort (cohort 2). Table 1 shows subject demographics and baseline characteristics. In the single-dose cohort (cohort 0), 20 of 21 (95%) subjects completed the study through day 7 (last day of posaconazole oral suspension), and 1 subject (placebo group) was discontinued for an AE (nausea) after receiving the first oral dose of posaconazole suspension. In the 200-mg multiple-dose cohort (cohort 1), 14 of 21 (67%) subjects completed i.v. treatment (days 1 to 14) and oral treatment (days 15 to 28) and 7 subjects were discontinued (3 subjects for AEs/treatment failure during i.v. dosing and 1 subject for an SAE during oral suspension dosing, 2 subjects did not want to continue because of reasons unrelated to the study treatment [1 during i.v. dosing and 1 during oral dosing], and 1 subject was noncompliant with the study protocol during oral dosing). In the 300-mg multiple-dose cohort (cohort 2), 17 of 24 (71%) subjects completed i.v. treatment (days 1 to 14) and oral treatment (days 15 to 28); 7 subjects discontinued because of AEs (5 during i.v. dosing and 2 during oral suspension dosing).
TABLE 1.
Subject demographics
| Demographic characteristic | Cohort 0: single dosea |
Cohort 1: POS 200 mg i.v., multiple doses (n = 21) | Cohort 2: POS 300 mg i.v., multiple doses (n = 24) | |
|---|---|---|---|---|
| Placebo i.v. (n = 11) | POS 200 mg i.v. (n = 10) | |||
| Age (yr) | ||||
| Mean (SD) | 59.5 (12.3) | 56.8 (17.4) | 49.1 (14.7) | 52.4 (13.4) |
| ≥65 yr (n [%]) | 4 (36) | 4 (40) | 2 (10) | 4 (17) |
| Sex (n [%]) | ||||
| Male | 5 (45) | 5 (50) | 13 (62) | 13 (54) |
| Female | 6 (55) | 5 (50) | 8 (38) | 11 (46) |
| Race (n [%]) | ||||
| White | 11 (100) | 10 (100) | 21 (100) | 24 (100) |
| Ethnicity (n [%]) | ||||
| Hispanic or Latino | 0 | 0 | 0 | 0 |
| Neither Hispanic nor Latino | 11 (100) | 10 (100) | 21 (100) | 24 (100) |
| Wt (mean [SD]) (kg) | 73.8 (8.6) | 79.3 (18.1) | 77.2 (11.4) | 76.9 (13.8) |
| Ht (mean [SD]) (cm) | 169.3 (8.3) | 171.3 (12.5) | 173.6 (9.5) | 171.9 (8.1) |
| Primary diagnosis (n [%]) | ||||
| AMLb | 10 (90.9) | 10 (100) | 20 (95.2) | 22 (91.7) |
| MDSc | 1 (9.1) | 0 | 1 (4.8) | 2 (8.3) |
i.v., intravenous; POS, posaconazole.
AML, acute myelogenous leukemia.
MDS, myelodysplastic syndrome.
PK results.
PK parameters are log-normally distributed and are presented as means (with coefficient of variation percentages [CV%] in parentheses). For the PK analysis on day 1, 20 of 21 subjects in the 200-mg multiple-dose cohort (cohort 1) and 22 of 24 subjects in the 300-mg multiple-dose cohort (cohort 2) were considered evaluable. For the PK analysis at steady state (day 14), 15 of 21 subjects in the 200-mg cohort (cohort 1) were considered evaluable. In the 300-mg cohort (cohort 2) steady-state (day 14) PK analysis, 19 of 24 subjects were considered evaluable.
PK parameters for the multiple-dose cohorts are summarized in Table 2. On day 1, posaconazole i.v. 200 mg attained a median Tmax of 1.48 h (range, 1.0 to 4.0 h), a mean (CV%) Cmax of 990 (47) ng/ml, and a mean (CV%) exposure (AUC0–12 h) of 5,390 (29) ng · h/ml. Posaconazole i.v. 300 mg attained a median Tmax of 1.54 h (range, 1.0 to 2.0 h), a mean (CV%) Cmax of 1,590 (61) ng/ml and a mean (CV%) exposure (AUC0–12 h) of 8,240 (26) ng · h/ml. Mean (standard deviation) plasma concentration profiles for days 1 and 14 are shown in Fig. 2A for the 200-mg multiple-dose cohort (cohort 1) and in Fig. 2B for the 300-mg multiple-dose cohort (cohort 2).
TABLE 2.
PK parameter values after twice-daily dosing of POS i.v. (day 1) and multiple doses of POS i.v. (day 14) administered to subjects at high risk for IFDa
| POS day of administration, cohort, and dosageb | No. of subjects | Cmax (mean [CV%c]) (ng/ml) | Tmax (median [range]) (h) | AUCd (mean [CV%]) (ng · h/ml) | Cavge (mean [CV%]) (ng/ml) | Cmin (mean [CV%]) (ng/ml) | Accumulation ratio (mean [CV%])f | Cavg of ≥500 and ≤2,500 ng/ml (%) |
|---|---|---|---|---|---|---|---|---|
| Day 1 | ||||||||
| Cohort 1: 200 mg b.i.d. | 20 | 990 (47) | 1.48 (1.0–4.0) | 5,390 (29) | NA | NA | NA | NA |
| Cohort 2: 300 mg b.i.d. | 22 | 1,590 (61) | 1.54 (1.0–2.0) | 8,240 (26) | NA | NA | NA | NA |
| Day 14 | ||||||||
| Cohort 1: 200 mg q.d. | 15 | 1,950 (50) | 1.00 (1.0–4.0) | 28,200 (51) | 1,180 (51) | 958 (63) | 3.6 (44) | 94 |
| Cohort 2: 300 mg q.d. | 19 | 2,610 (39) | 1.50 (0.98–4.0) | 34,300 (42) | 1,430 (42) | 1,068 (50) | 2.8 (31) | 95 |
i.v., intravenous; IFD, invasive fungal disease; POS, posaconazole; PK, pharmacokinetics; NA, not applicable.
b.i.d., twice daily; q.d., once daily.
CV, coefficient of variation; Cmax, maximum observed concentration; Tmax, time to Cmax; AUC, area under concentration-time curve; Cavg, average concentration at steady state; Cmin, minimum plasma concentration.
AUC from 0 to 12 h (AUC0–12 h) presented for day 1; AUC0–24 h presented for day 14.
Cavg = AUC0–24 h/dose interval, based on Cmax.
Accumulation ratio based on AUC0–24 h.
FIG 2.

Mean (standard deviation) plasma concentration profiles (days 1 and 14). (A) Cohort 1: intravenous posaconazole 200 mg daily (after 200 mg twice daily on day 1) administered to subjects at high risk for IFD. (B) Cohort 2: intravenous posaconazole 300 mg daily (after 300 mg twice daily on day 1) administered to subjects at high risk for IFD. IV, intravenous.
At day 14, for the 200-mg cohort (cohort 1), based on data in 15 PK-evaluable subjects (day 14), median Tmax was 1.0 h (range, 1.0 to 4.0 h), mean (CV%) Cmax was 1,950 (50) ng/ml, mean (CV%) exposure (AUC0–24 h) was 28,200 (51) ng · h/ml, and mean (CV%) Cmin was 958 (63) ng/ml. Mean accumulation ratio (CV%) based on AUC0–24 was 3.6 (44). Fourteen of 15 (94%) subjects attained a steady-state (day 14) Cavg of ≥500 and ≤2,500 ng/ml, and 1 (6%) subject attained a steady-state Cavg of >2,500 ng/ml but <3,650 ng/ml. Applying the same criteria to Cmin revealed that 3 of 15 (20%) subjects attained a steady-state (day 14) Cmin of <500 ng/ml, 11 of 15 (73%) subjects attained a steady-state (day 14) Cmin of ≥500 and ≤2,500 ng/ml, and 1 (6%) subject attained a steady-state Cmin of >2,500 ng/ml but <3,650 ng/ml. The mean (CV%) steady-state Cavg was 1,180 (51) ng/ml (prespecified Cavg target, 1,200 ng/ml). No subject in cohort 1 had a steady-state Cavg plasma concentration of ≥3,750 ng/ml or a steady-state AUC of >90,000 ng · h/ml; therefore, dosing was allowed to proceed for cohort 2.
For the 300-mg cohort (cohort 2), based on data in 19 PK-evaluable subjects (day 14), median Tmax was 1.5 h (range, 0.98 to 4.0 h), mean (CV%) Cmax was 2,610 (39) ng/ml, mean (CV%) exposure (AUC0–24 h) was 33,800 (42) ng · h/ml, and mean (CV%) Cmin was 1,046 (50) ng/ml. The mean accumulation ratio (CV%) based on AUC0–24 was 2.8 (31). Eighteen of 19 (95%) subjects attained steady-state (day 14) Cavgs of ≥500 and ≤2,500 ng/ml, and 1 of 19 (5%) subjects attained a steady-state Cavg of >2,500 ng/ml but ≤3,650 ng/ml. The mean (CV%) steady-state Cavg was 1,430 (42) ng/ml, which was higher than the prespecified Cavg target of 1,200 ng/ml. Applying the same criteria to Cmins revealed that 4 of 19 (21%) subjects attained steady-state (day 14) Cmins of <500 ng/ml, 15 of 19 (79%) subjects attained steady-state (day 14) Cmins of ≥500 and ≤2,500 ng/ml, and 0 (0%) subjects attained steady-state Cavgs of >2,500 ng/ml but <3,650 ng/ml. No subject in cohort 2 had a steady-state Cavg plasma concentration of >3,650 ng/ml or a steady-state AUC of >87,600 ng · h/ml; therefore, the phase 3 portion of the study was allowed to proceed. Exposure increases between the 200- and 300-mg dose levels were slightly less than dose proportional but, based on the totality of clinical experience, the PK of posaconazole i.v. have been observed to be generally dose proportional in the clinically relevant dose range of 200 to 300 mg. In posaconazole i.v. (200-mg and 300-mg) cohorts, >90% of subjects achieved Cavgs of ≥500 ng/ml and ≤2,500 ng/ml.
Safety.
In the single-dose cohort (cohort 0), all subjects (100% [10/10] posaconazole i.v.; 100% [11/11] placebo i.v.) experienced at least 1 treatment-emergent AE. One AE (severe nausea in the placebo group) led to study drug discontinuation. Six (60%) posaconazole i.v. subjects and 3 (27%) placebo i.v. subjects reported AEs considered at least possibly related to study treatment, as determined by the investigator. The most commonly reported treatment-related AEs in the posaconazole i.v. group in cohort 0 were constipation, hypokalemia (1 subject with 3.5 meq/liter and 1 subject with 3.4 meq/liter), and nausea (each reported in 2 subjects [20%]). All other treatment-related AEs were reported by 1 subject each (10%). One SAE (grade 4 hyperbilirubinemia in the placebo group) was considered possibly related to treatment. No catheter-related or local infusion site events were considered related to posaconazole i.v. treatment. Two deaths were reported in cohort 0 (1 in the posaconazole i.v. group and 1 in the placebo group, both caused by sepsis); neither was considered by the investigator to be treatment related.
AEs from the multiple-dose cohorts are summarized in Table 3; the safety data incorporate 14 days of posaconazole i.v. dosing (200 or 300 mg) plus 14 days of twice-daily posaconazole oral suspension (400 mg twice daily). AEs leading to discontinuation were reported in 4 of 21 (19%) subjects in the 200-mg once-daily cohort (cohort 1) (3 during i.v. dosing and 1 during oral dosing) and in 7 of 24 (29%) subjects in the 300-mg once-daily cohort (cohort 2) (5 during i.v. dosing and 2 during oral dosing). AEs that led to discontinuation during i.v. dosing in cohort 1 were candidemia (also considered treatment failure), fatigue and nausea, and inflammation at the infusion site (unrelated to study drug), leading to central line removal. AEs that led to discontinuation during i.v. dosing in cohort 2 were bacterial sepsis (with hepatic and renal dysfunction), elevated liver function test (LFT) results, possible pulmonary fungal infection, inflammation around the central catheter, and pulmonary hemorrhage. Treatment-related AEs were reported in 3 (14%) subjects in cohort 1 and in 8 (33%) subjects in cohort 2. Rash and nausea were the only treatment-related AEs reported by more than 1 subject; treatment-related rash was reported by 3 subjects overall (1 subject in cohort 1 and 2 subjects in cohort 2); treatment-related nausea was reported by 1 subject in each cohort. One death was reported in cohort 1 (caused by bilateral pulmonary artery embolism and intrapulmonary hemorrhage in a patient with disseminated aspergillosis), and 3 deaths were reported in cohort 2 (1 caused by acute renal failure and 2 by sepsis); none was considered treatment related. No catheter-related events were considered related to treatment in either cohort. No AEs related to renal function were reported for subjects in cohort 0 or cohort 1. One subject in cohort 2 had renal failure considered unrelated to study treatment (acute renal failure attributed by the investigator to Escherichia coli sepsis). The most commonly reported AEs (≥20% of subjects in any cohort) are presented in Table 4.
TABLE 3.
AEs after multiple doses of POS i.v. 200/300 mg followed by multiple doses of twice-daily POS oral suspension 400 mg (cohorts 1 and 2)a
| AE type | No. (%) of subjects with indicated AEb |
|
|---|---|---|
| Cohort 1: POS i.v. 200 mg q.d.c (n = 21) | Cohort 2: POS i.v. 300 mg q.d. (n = 24) | |
| Any treatment-emergent AE | 20 (95) | 24 (100) |
| Treatment-related AE | 3 (14) | 8 (33) |
| SAEd | 4 (19) | 9 (38) |
| Treatment-related SAE | 0 | 1 (4) |
| AE leading to study drug discontinuation | 4 (19) | 7 (29) |
| Discontinuation during POS i.v. | 3 | 5 |
| Discontinuation during POS oral suspension | 1 | 2 |
| SAE leading to study drug discontinuation | 1 (5) | 2 (8) |
| Death | 1 (5) | 3 (13) |
AE, adverse event; POS, posaconazole; i.v., intravenous.
All subjects received POS i.v. 200 or 300 mg on days 1 to 14 followed by 14 days of twice-daily POS oral suspension 400 mg.
q.d., once daily
SAE, serious adverse event.
TABLE 4.
Most commonly reported (≥20% subjects in either cohort) AEs after multiple doses of POS i.v. followed by multiple doses of twice-daily POS oral suspensiona
| AE | No. (%) of subjects with indicated adverse event |
|||
|---|---|---|---|---|
| Cohort 1: POS i.v. 200 mg q.d.b (n = 21) |
Cohort 2: POS i.v. 300 mg q.d. (n = 24) |
|||
| i.v. phasec | Overalld | i.v. phasec | Overalld | |
| Nausea | 6 (29) | 9 (43) | 4 (17) | 7 (29) |
| Diarrhea | 5 (24) | 6 (29) | 9 (38) | 10 (42) |
| Mucosal inflammation | 2 (10) | 2 (10) | 8 (33) | 8 (33) |
| Pyrexia | 5 (24) | 6 (29) | 6 (25) | 8 (33) |
| Rash | 2 (10) | 2 (10) | 6 (25) | 8 (33) |
| Febrile neutropenia | 5 (24) | 6 (29) | 7 (29) | 7 (29) |
| Cough | 5 (24) | 6 (29) | 1 (4) | 1 (4) |
| Headache | 2 (10) | 2 (10) | 6 (25) | 6 (25) |
| Vomiting | 1 (5) | 2 (10) | 3 (13) | 5 (21) |
| Abdominal pain | 3 (14) | 3 (14) | 3 (13) | 5 (21) |
| Hemorrhoids | 0 | 1 (5) | 5 (21) | 5 (21) |
| Hypokalemia | 3 (14) | 3 (14) | 4 (17) | 5 (21) |
AE, adverse event; POS, posaconazole; i.v., intravenous.
q.d., once daily.
Adverse reactions reported in patients with an onset during the posaconazole i.v. dosing phase of the study.
Adverse reactions reported with an onset at any time during the study in patients who were treated with posaconazole i.v. and oral posaconazole for up to 28 days of posaconazole therapy.
No safety concerns were noted in any of the other parameters, including LFTs and electrocardiogram measurements, and no subjects met prespecified criteria for QT effect. One subject in cohort 1 (with a history of hyperbilirubinemia) had an ALT and/or AST level of ≥3× ULN (alkaline phosphatase [ALK-P] was ≤2× ULN, and total bilirubin was ≥2× ULN). Liver function improved while the subject continued to receive posaconazole; therefore, the LFT abnormalities were not considered treatment related. Three subjects in cohort 2 had an ALT and/or AST level of ≥3× ULN (ALK-P was ≤2× ULN, and total bilirubin was ≥2× ULN); however, each of these occurred in the setting of sepsis, and the LFT abnormalities were considered attributable to underlying disease.
Breakthrough IFD.
One case of probable/proven breakthrough IFD was reported in each of the multiple-dose cohorts (5% and 4% in cohorts 1 and 2, respectively); candidemia was reported 3 days after the start of i.v. treatment in cohort 1 (this patient was also found to have disseminated aspergillosis at autopsy), and pulmonary mycosis was reported on day 21 in cohort 2. Cavg was not calculated for the subject with candidemia in cohort 1 because the subject received only 5 days of i.v. therapy; the Cavg value during the i.v. treatment phase was 1,370 ng/ml for the subject with pulmonary mycosis in cohort 2. Four possible cases of IFD were reported. All proven, possible, and probable cases of IFD are listed in Table S1 in the supplemental material.
DISCUSSION
An i.v. formulation of posaconazole has been developed to ensure adequate exposure among patients at high risk for IFD who are unable to tolerate oral medication or for whom absorption of an oral medication is a concern. The aim of this study was to evaluate the PK, safety, and tolerability of posaconazole i.v. solution in subjects at risk for IFD and to identify a dose of posaconazole i.v. solution that attained a prespecified exposure target for further study in a larger and more diverse population in phase 3 (part 2) of the study. This is the first report on the use of posaconazole i.v. solution in patients at risk for IFD.
The correlation between plasma concentrations and efficacy or toxicity of any antifungal agent stems from experimental models of fungal infection and from pharmacodynamic and PK assessments of preclinical and clinical data (19). Although an absolute threshold plasma level for the prevention of breakthrough IFD has yet to be clearly defined for posaconazole (20, 21), posaconazole plasma levels are important for maintaining efficacy in prophylaxis and treatment. A positive association between posaconazole exposure and response was previously reported in a nonrandomized trial of posaconazole salvage treatment for invasive aspergillosis (4) and in a recent retrospective analysis conducted to investigate relationships between posaconazole concentration and clinical outcomes (22). Evidence suggests a positive exposure-response relationship with posaconazole. Further investigation will provide additional support in defining the target concentrations to achieve optimal efficacy with prophylaxis and treatment of patients with IFD (15, 23).
In this study, the lower end of the target exposure range (Cavg >500 ng/ml) was based on the exposure-response analysis (4) and was chosen because posaconazole 500 ng/ml is the MIC90 of most clinically important Aspergillus species (24). A steady-state target posaconazole plasma level of 500 ng/ml has previously been used in a trial of patients with compromised gastrointestinal function who were at high risk for IFD (19). The target in this study (Cavg >500 ng/ml in >90% of subjects) was higher than previously seen with the oral suspension (25). The upper end of the target exposure range (Cavg ≤2,500 ng/ml) was selected to be within the range at which posaconazole exposure response and safety have been characterized for most subjects in earlier clinical studies for the prophylaxis and treatment of refractory IFD.
In this study, posaconazole i.v. solution 300 mg once daily (twice daily on day 1 to enable more rapid achievement of steady state [i.e., generally between 6 and 8 days of dosing {data on file}]) attained the prespecified PK exposure target in ≥90% of subjects at high risk for IFD; 18 of 19 (95%) subjects attained steady-state Cavg ≥500 and ≤2,500 ng/ml. Posaconazole i.v. 300 mg achieved exposures in the upper quartiles previously studied, with a mean Cavg of 1,430 ng/ml and an AUC0–24 h of 34,300 ng · h/ml. Exposures achieved in the present study are within the upper quartiles of exposures achieved in previous studies assessing posaconazole prophylactic efficacy (1, 2). These exposures also reduce the risk for failure of antifungal prophylaxis. The posaconazole i.v. 200-mg dose resulted in exposures slightly below the prespecified PK exposure targets, whereas the 300-mg dose met the Cavg and the AUC0–24 h targets and resulted in higher steady-state Cmins than the 200-mg dose. Therefore, the posaconazole i.v. 300-mg dose was selected as the dose to use in proceeding to cohort 3 (phase 3).
No unexpected safety concerns compared with the previous oral use of i.v. administration were identified during our study. Posaconazole i.v. was safe when administered through a central line and showed a safety profile similar to that previously reported for posaconazole oral suspension administered to subjects at high risk for IFD (1, 2). No catheter-related AEs were considered related to treatment; central-line administration appears to mitigate peripheral i.v. intolerability such as thrombophlebitis and infusion site reactions (16). Overall, 2 of 45 (4%) subjects reported probable or proven breakthrough IFD, and 4 (9%) patients had possible IFD.
Posaconazole is metabolized primarily by UDP glucuronidation, is a substrate of p-glycoprotein efflux, and is a strong inhibitor of CYP3A4 (6). Plasma concentrations of drugs predominantly metabolized by CYP3A4 (such as sirolimus, tacrolimus, cyclosporine, simvastatin, ergot alkaloids, and benzodiazepines) may be increased by posaconazole (6).
Posaconazole i.v. solution achieves target exposure rapidly with once-daily dosing (after a twice-daily dose on day 1). This new formulation allows the administration of posaconazole to patients unable to take an oral medication and to patients who are at high risk for IFD but for whom oral absorption is a concern.
Summary and conclusions.
Posaconazole i.v. solution was well tolerated in subjects at high risk for IFD. Further, PK analysis demonstrated that posaconazole 300 mg i.v. achieved exposures in the range of the upper quartiles known to be associated with a positive response relationship. Based on the combined PK and safety findings, posaconazole i.v. 300 mg once daily was selected as the dose to use in the phase 3 study for further investigation in a larger, more diverse patient population.
Supplementary Material
ACKNOWLEDGMENTS
We thank the additional investigators involved in the phase 1B portion of the trial, Helmut Ostermann (University of Munich, Germany), Urs Schanz (University Hospital Zurich, Switzerland), and Stefan Zimmerli (University of Bern, Switzerland). We also thank Brenda Paton for her contributions to the manuscript.
Medical writing and editorial assistance were provided by Sheena Hunt, PhD, Denise Balog, PharmD, and David Gibson, PhD, CMPP, of ApotheCom. This assistance was funded by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Whitehouse Station, NJ, USA. This study was supported by Merck, Sharp & Dohme Corp.
All authors made substantial contributions to the work by critically reviewing and revising the manuscript, approving the final manuscript for submission, and agreeing to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. The authors also made the following substantial contributions: J.M. collected data and provided study materials or patients. O.A.C. collected data, supervised the research group, provided study materials or data, and provided administrative or logistic support. A.J.U. collected data and interpreted the results. W.J.H. collected data and provided study materials or patients. G.K. conceived of and designed the study, interpreted the data, and supervised the research group. H.P. designed the study and supervised the research group. M.C. performed pharmacokinetic analysis. N.K. designed the study, analyzed the data, and supervised the research group. H.W. provided technical help, collected data, supervised the research group, provided study materials or patients, analyzed the data, and provided administrative or logistic support. M.N.R. provided technical help, supervised the research group, and provided administrative or logistic support.
Conflicts of interest: J.M., consultant for Merck, Basilea, F2G, Gilead, Pfizer, Amgen, GSK, Astellas, Viropharma, and Bio-Rad and on speakers' bureaus for Merck, Gilead, Pfizer, Astellas, and Bio-Rad; O.A.C., consulted for 3M, Astellas, Basilea, Cubist, F2G, Gilead, GSK, Merck, Optimer, Pfizer, and Sanofi Pasteur, received research grants from 3M, Actelion, Astellas, Basilea, Bayer, Celgene, Cubist, F2G, Genzyme, Gilead, GSK, Merck, Miltenyi, Optimer, Pfizer, Quintiles, and Viropharma, and on speakers' bureaus for Astellas, Gilead, Merck/MSD, and Pfizer; A.J.U., received a research grant from Merck, consulted and on speakers' bureaus for Astellas, Merck, Pfizer, and Gilead, received travel support from Merck, Pfizer, and Astellas, and gave educational presentations for Gilead; W.J.H., received research grants from Astellas, Basilea, Gilead, Merck, and Pfizer, consulted for Merck, and on speakers' bureaus for Astellas, Bristol-Myers Squibb, Essex/Schering-Plough, Gilead, Merck, and Pfizer. G.K. and H.P. were Merck employees at the time of the study and own stock in Merck. M.C., N.K., H.W., and M.N.R. are Merck employees and own stock in Merck.
Footnotes
Published ahead of print 14 April 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02686-13.
REFERENCES
- 1.Cornely OA, Maertens J, Winston DJ, Perfect J, Ullmann AJ, Walsh TJ, Helfgott D, Holowiecki J, Stockelberg D, Goh YT, Petrini M, Hardalo C, Suresh R, Angulo-Gonzalez D. 2007. Posaconazole vs. fluconazole or itraconazole prophylaxis in patients with neutropenia. N. Engl. J. Med. 356:348–359. 10.1056/NEJMoa061094 [DOI] [PubMed] [Google Scholar]
- 2.Ullmann AJ, Lipton JH, Vesole DH, Chandrasekar P, Langston A, Tarantolo SR, Greinix H, Morais de Azevedo W, Reddy V, Boparai N, Pedicone L, Patino H, Durrant S. 2007. Posaconazole or fluconazole for prophylaxis in severe graft-versus-host disease. N. Engl. J. Med. 356:335–347. 10.1056/NEJMoa061098 [DOI] [PubMed] [Google Scholar]
- 3.Keating GM. 2005. Posaconazole. Drugs 65:1553–1567. 10.2165/00003495-200565110-00007 [DOI] [PubMed] [Google Scholar]
- 4.Walsh TJ, Raad I, Patterson TF, Chandrasekar P, Donowitz GR, Graybill R, Greene RE, Hachem R, Hadley S, Herbrecht R, Langston A, Louie A, Ribaud P, Segal BH, Stevens DA, van Burik JA, White CS, Corcoran G, Gogate J, Krishna G, Pedicone L, Hardalo C, Perfect JR. 2007. Treatment of invasive aspergillosis with posaconazole in patients who are refractory to or intolerant of conventional therapy: an externally controlled trial. Clin. Infect. Dis. 44:2–12. 10.1086/508774 [DOI] [PubMed] [Google Scholar]
- 5.Raad II, Hachem RY, Herbrecht R, Graybill JR, Hare R, Corcoran G, Kontoyiannis DP. 2006. Posaconazole as salvage treatment of invasive fusariosis in patients with underlying hematologic malignancy and other conditions. Clin. Infect. Dis. 42:1398–1403. 10.1086/503425 [DOI] [PubMed] [Google Scholar]
- 6.Noxafil. 2012. Noxafil (posaconazole) oral suspension; prescribing information. Merck & Co. Inc., Whitehouse Station, NJ [Google Scholar]
- 7.Merck Sharp & Dohme Limited. 2012. Noxafil (posaconazole) oral suspension: summary of product characteristics. Merck Sharp & Dohme Limited, Hertfordshire, United Kingdom [Google Scholar]
- 8.Courtney R, Wexler D, Radwanski E, Lim J, Laughlin M. 2004. Effect of food on the relative bioavailability of two oral formulations of posaconazole in healthy adults. Br. J. Clin. Pharmacol. 57:218–222. 10.1046/j.1365-2125.2003.01977.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krishna G, Moton A, Ma L, Medlock MM, McLeod J. 2009. The pharmacokinetics and absorption of posaconazole oral suspension under various gastric conditions in healthy volunteers. Antimicrob. Agents Chemother. 53:958–966. 10.1128/AAC.01034-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krishna G, Ma L, Martinho M, O'Mara E. 2012. A single dose phase I study to evaluate the pharmacokinetics of posaconazole new tablet and capsule formulations relative to oral suspension. Antimicrob. Agents Chemother. 56:4196–4201. 10.1128/AAC.00222-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pille S, Bohmer D. 1998. Options for artificial nutrition of cancer patients. Strahlenther. Onkol. 174:52–55 [PubMed] [Google Scholar]
- 12.Sansone-Parsons A, Krishna G, Calzetta A, Wexler D, Kantesaria B, Rosenberg MA, Saltzman MA. 2006. Effect of a nutritional supplement on posaconazole pharmacokinetics following oral administration to healthy volunteers. Antimicrob. Agents Chemother. 50:1881–1883. 10.1128/AAC.50.5.1881-1883.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pfizer Inc. 2011. VFEND IV (voriconazole) for injection, VFEND tablets (voriconazole), VFEND (voriconazole) for oral suspension: prescribing information. Pfizer, Inc., New York, NY [Google Scholar]
- 14.Janssen-Cilag Limited. 2013. Sporanox IV (itraconazole) 10 mg/ml concentrate and solvent for solution for infusion: summary of product characteristics. Janssen-Cilag Limited, Buckinghamshire, United Kingdom [Google Scholar]
- 15.Jang SH, Colangelo PM, Gobburu JV. 2010. Exposure-response of posaconazole used for prophylaxis against invasive fungal infections: evaluating the need to adjust doses based on drug concentrations in plasma. Clin. Pharmacol. Ther. 88:115–119. 10.1038/clpt.2010.64 [DOI] [PubMed] [Google Scholar]
- 16.van Iersel T, Nassander U, Kersemaekers WM, Xuan F, Caceres M, Waskin H. 2013. Pharmacokinetics (PK) and safety study of posaconazole (POS) i.v. solution via peripheral administration in healthy subjects. Poster. 53rd Intersci. Conf. Antimicrob. Agents Chemother. (ICAAC); American Society for Microbiology, Denver, CO, 10 to 13 September 2013 [Google Scholar]
- 17.Shen JX, Krishna G, Hayes RN. 2007. A sensitive liquid chromatography and mass spectrometry method for the determination of posaconazole in human plasma. J. Pharm. Biomed. Anal. 43:228–236. 10.1016/j.jpba.2006.06.011 [DOI] [PubMed] [Google Scholar]
- 18.De Pauw B, Walsh TJ, Donnelly JP, Stevens DA, Edwards JE, Calandra T, Pappas PG, Maertens J, Lortholary O, Kauffman CA, Denning DW, Patterson TF, Maschmeyer G, Bille J, Dismukes WE, Herbrecht R, Hope WW, Kibbler CC, Kullberg BJ, Marr KA, Muñoz P, Odds FC, Perfect JR, Restrepo A, Ruhnke M, Segal BH, Sobel JD, Sorrell TC, Viscoli C, Wingard JR, Zaoutis T, Bennett JE, European Organization for Research and Treatment of Cancer/Invasive Fungal Infections Cooperative Group, National Institute of Allergy and Infectious Diseases Mycoses Study Group (EORTC/MSG) Consensus Group 2008. Revised definitions of invasive fungal disease from the European Organization for Research and Treatment of Cancer/Invasive Fungal Infections Cooperative Group and the National Institute of Allergy and Infectious Diseases Mycoses Study Group (EORTC/MSG) Consensus Group. Clin. Infect. Dis. 46:1813–1821. 10.1086/588660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cornely OA, Helfgott D, Langston A, Heinz W, Vehreschild JJ, Vehreschild MJ, Krishna G, Ma L, Huyck S, McCarthy MC. 2012. Pharmacokinetics of different dosing strategies of oral posaconazole in patients with compromised gastrointestinal function and who are at high risk for invasive fungal infection. Antimicrob. Agents Chemother. 56:2652–2658. 10.1128/AAC.05937-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krishna G, Martinho M, Chandrasekar P, Ullmann AJ, Patino H. 2007. Pharmacokinetics of oral posaconazole in allogeneic hematopoietic stem cell transplant recipients with graft-versus-host disease. Pharmacotherapy 27:1627–1636. 10.1592/phco.27.12.1627 [DOI] [PubMed] [Google Scholar]
- 21.Cornely OA, Ullmann AJ. 2011. Lack of evidence for exposure-response relationship of posaconazole used for prophylaxis against invasive fungal infections. Clin. Pharmacol. Ther. 89:351–352. 10.1038/clpt.2010.261 [DOI] [PubMed] [Google Scholar]
- 22.Dolton MJ, Ray JE, Chen SC, Ng K, Pont L, McLachlan AJ. 2012. Multicenter study of posaconazole therapeutic drug monitoring: exposure-response relationship and factors affecting concentration. Antimicrob. Agents Chemother. 56:5503–5510. 10.1128/AAC.00802-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dolton MJ, Ray JE, Marriott D, McLachlan AJ. 2012. Posaconazole exposure-response relationship: evaluating the utility of therapeutic drug monitoring. Antimicrob. Agents Chemother. 56:2806–2813. 10.1128/AAC.05900-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sabatelli F, Patel R, Mann PA, Mendrick CA, Norris CC, Hare R, Loebenberg D, Black TA, McNicholas PM. 2006. In vitro activities of posaconazole, fluconazole, itraconazole, voriconazole, and amphotericin B against a large collection of clinically important molds and yeasts. Antimicrob. Agents Chemother. 50:2009–2015. 10.1128/AAC.00163-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krishna G, Tarif MA, Xuan F, Martinho M, Angulo D, Cornely OA. 2008. Pharmacokinetics of oral posaconazole in neutropenic patients receiving chemotherapy for acute myelogenous leukemia or myelodysplastic syndrome. Pharmacotherapy 28:1223–1232. 10.1592/phco.28.10.1223 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.