

Abstract
RNase H plays an essential role in the replication of human immunodeficiency virus type 1 (HIV-1). Therefore, it is a promising target for drug development. However, the identification of HIV-1 RNase H inhibitors (RHIs) has been hampered by the open morphology of its active site, the limited number of available RNase H crystal structures in complex with inhibitors, and the fact that, due to the high concentrations of Mg2+ needed for protein stability, HIV-1 RNase H is not suitable for nuclear magnetic resonance (NMR) inhibitor studies. We recently showed that the RNase H domains of HIV-1 and prototype foamy virus (PFV) reverse transcriptases (RTs) exhibit a high degree of structural similarity. Thus, we examined whether PFV RNase H can serve as an HIV-1 RNase H model for inhibitor interaction studies. Five HIV-1 RHIs inhibited PFV RNase H activity at low-micromolar concentrations similar to those of HIV-1 RNase H, suggesting pocket similarity of the RNase H domains. NMR titration experiments with the PFV RNase H domain and the RHI RDS1643 (6-[1-(4-fluorophenyl)methyl-1H-pyrrol-2-yl)]-2,4-dioxo-5-hexenoic acid ethyl ester) were performed to determine its binding site. Based on these results and previous data, in silico docking analysis showed a putative RDS1643 binding region that reaches into the PFV RNase H active site. Structural overlays were performed with HIV-1 and PFV RNase H to propose the RDS1643 binding site in HIV-1 RNase H. Our results suggest that this approach can be used to establish PFV RNase H as a model system for HIV-1 RNase H in order to identify putative inhibitor binding sites in HIV-1 RNase H.
INTRODUCTION
In foamy viruses (FVs), the mature reverse transcriptase (RT) harbors the protease (PR) at its N terminus, leading to a multifunctional enzyme that carries out the functions of PR and RT, including the activities of RT-associated RNA-dependent DNA polymerase (RDDP) and RNase H (1–3). In contrast to the human immunodeficiency virus type 1 (HIV-1) RT heterodimer (p66/p51), prototype FV (PFV) PR-RT has been shown to be monomeric and able to catalyze all RT-related functions (3), while PR activation requires dimer formation of the PR domain via the binding of PR-RT to a specific element on the viral RNA, called the protease-activating RNA motif (PARM) (4, 5). Hence, for the retrotranscription functions, PFV PR-RT differs from alpharetroviral and lentiviral RTs (including HIV-1 RT), which are dimeric, and it is similar to the gammaretroviral RTs that are monomeric (6). In addition, the PFV RNase H domain possesses a basic protrusion, including a basic loop and the so-called C-helix, which is important for activity and substrate binding and is absent in the HIV-1 RNase H domain (7, 8). In HIV-1 RT, the basic protrusion is absent; instead, a loop derived from the connection domain of the p66 subunit fulfills a similar function (7, 8). Therefore, in contrast to FV, the free HIV-1 RNase H domain, which lacks the loop, is not enzymatically active (7–11).
In the search for effective drugs to treat HIV-1-infected patients, many HIV-1 RT inhibitors have been identified in the last 25 years (12). They are generally divided into nucleoside analogue RT inhibitors (NRTI), nonnucleoside analogue RT inhibitors (NNRTI), and RNase H inhibitors (RHI) (12–15); however, no RNase H inhibitor is yet in clinical use.
HIV-1 RNase H is an attractive new target for drug discovery, since its enzyme activity is essential for viral replication. During the last 10 years, several classes of HIV-1 RHIs have been identified (12–15). The design of potent RHIs has proven difficult due to the open morphology of the RNase H active site (16) and the limited number of available RNase H crystal structures in complex with inhibitors (17–21). In addition, given the fact that under nuclear magnetic resonance (NMR) conditions, the free HIV-1 RNase H domain is stable only in the presence of 20 to 80 mM MgCl2, it is difficult to perform binding studies with inhibitors that interact with magnesium ions (22–24). Furthermore, since the free HIV-1 RNase H domain is not active, a chimeric HIV-1 RNase H domain harboring the substrate binding loop from Escherichia coli RNase H is often used for inhibitor testing (20, 24–26).
Therefore, we wanted to investigate whether HIV-1 RHIs are also able to inhibit PFV RNase H. In fact, structural alignments of the two RNase H domains reveal that with the exception of the basic protrusion, the overall three-dimensional structures of HIV-1 and PFV RNase H are highly homologous (8, 12, 27). If PFV RNase H were inhibited in a similar fashion, it could serve as an HIV-1 RNase H model for inhibitor interaction studies. Moreover, we have shown that FV RNase H is soluble and stable at 6 mM MgCl2 (7, 8). This makes the RNase H suitable for inhibitor binding studies by NMR spectroscopy, especially since several RHIs are Mg2+ ion binding compounds, i.e., diketo acids (28).
We first chose five different HIV-1 RHIs that inhibit HIV-1 RT by binding at different RT pockets (Fig. 1) and the NNRTI efavirenz as a control to test their effects on FV PR-RT in enzyme activity assays. Due to its relatively high solubility in water, the inhibitor RDS1643 that impaired FV RNase H activity appeared to be suitable for NMR spectroscopy. NMR titration experiments identified its putative binding site in the PFV RNase H domain. Sequence and structure alignments with HIV-1 RNase H and docking experiments were used to reveal the corresponding binding site in HIV-1 RNase H. Our data suggest that PFV RNase H can be used as a model for HIV-1 RNase H inhibitor binding.
FIG 1.
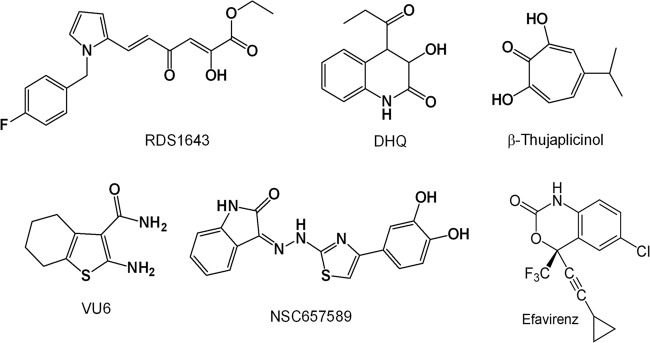
Chemical structures of HIV-1 RHIs and the NNRTI efavirenz.
MATERIALS AND METHODS
Protein purifications.
The purification of PFV PR-RT and 15N-labeled PFV RNase H was performed as described previously (3, 7).
RNase H polymerase-independent cleavage assay.
The PFV RT-associated RNase H activity was measured in a 100-μl reaction volume containing 50 mM Tris HCl (pH 8.1), 6 mM MgCl2, 1 mM dithiothreitol (DTT), 80 mM KCl, 0.25 μM hybrid RNA/DNA (5′-GTTTTCTTTTCCCCCCTGAC-3′-fluorescein, 5′-CAAAAGAAAAGGGGGGACUG-3′-dabcyl), and 2 nM PFV RT. The reaction mixture was incubated for 1 h at 37°C, the reaction was stopped by adding EDTA, and the products were measured with a Victor3 (PerkinElmer) plate reader at 490/528 nm.
DNA polymerase assay.
The PFV RT-associated RDDP activity was measured in a 50-μl volume containing 60 mM Tris-HCl (pH 8.1), 8 mM MgCl2, 60 mM KCl, 13 mM DTT, 100 μM dTTP, 5 nM PFV RT, and 2.5 μM poly(A)-oligo(dT) (EnzChek Invitrogen). The reaction mixture was incubated for 30 min at 37°C. The enzymatic reaction was stopped by adding EDTA. The reaction products were detected by the addition of PicoGreen and measured with a Victor3 (PerkinElmer) plate reader at 502/523 nm.
Compounds.
RDS1643 was kindly provided by Simona Di Santo, University of Rome La Sapienza, Italy; 2-amino-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxamide (VU6) and 2,7-dihydroxy-4-isopropyl-cyclohepta-2,4,6-triene (β-thujaplicinol) were kindly provided by Stuart Le Grice, National Cancer Institute (NCI), USA; (Z)-3-(2-(4-(3,4-dihydroxyphenyl)thiazol-2-yl)hydrazono)indolin-2-one (NSC657589) was obtained from the NCI repository; and 3-hydroxy-4-propionyl-3,4-dihydroquinolin-2(1H)-one (DHQ) was kindly provided by Philippe Cotelle, University of Lille, France. Efavirenz was obtained from Elias Maccioni, University of Caligari, Italy.
NMR analyses.
Standard nuclear magnetic resonance (NMR) heteronuclear single quantum coherence (HSQC) experiments were recorded using 50 to 80 μM 15N-labeled PFV RNase H in 5 mM sodium phosphate (pH 7.0), 100 mM NaCl, 6 mM MgCl2, 0.5 mM DTT, 10% (vol/vol) D2O, and 6% deuterated dimethyl sulfoxide (DMSO) at 25°C on Bruker Avance 700 and 800 MHz spectrometers partially equipped with a cryogenically cooled probe. In-house protocols were used to process the NMR data, and the program NMRView was utilized for analysis (B.A. Johnson; Merck, Whitehouse Station, NJ, USA). The inhibitors were dissolved in 100% deuterated DMSO to a final concentration of 100 mM and added at the concentrations indicated in Results and Discussion. The control HSQC spectra of PFV RNase H at various concentrations of deuterated DMSO were recorded for each titration step. The final DMSO concentrations for RDS1643 did not exceed 11%.
Changes in the chemical shifts were expressed by calculating the weighted geometric average (equation 1, c15N = 0.1, of chemical shifts of 1H and 15N spins). A chemical shift change with a weighted geometric average of ≥0.02 ppm was considered significant.
| (1) |
Inhibitor docking.
Docking experiments were performed using the program AutoDock Vina (29). Nonpolar hydrogen atoms were removed from both protein and ligand and saved in a PDBQT format. A grid box was set with 20 × 36 × 28 points in x, y, z direction and −8.5, −18.0, 7.0 grid center for PFV RNase H (Protein Data Bank identification no. [PDB ID] 2LSN). The grid box included the active-site residues and the adjacent residues exhibiting significant chemical shift changes These parameters were saved in a configuration text file (config.txt), followed by autodocking. The results generated were visualized in PyMOL. Among the inhibitor binding orientations obtained, the orientation that fit best to the NMR data was chosen.
RESULTS AND DISCUSSION
Inhibition of PFV PR-RT enzyme activities.
For the analysis of FV PR-RT inhibition, we chose the diketo acid derivative 6-[1-(4-fluorophenyl)methyl-1H-pyrrol-2-yl)]-2,4-dioxo-5-hexenoic acid ethyl ester (RDS1643) (28), 3-hydroxy-4-propionyl-3,4-dihydroquinolin-2(1H)-one (DHQ) (30), and the natural product 2,7-dihydroxy-4-isopropyl-cyclohepta-2,4,6-triene (β-thujaplicinol) (31), which were reported to inhibit HIV-1 RNase H by binding the two divalent Mg2+ ions in the catalytic site. We also chose the vinylogous urea derivative 2-amino-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxamide (VU6), which was reported to inhibit HIV-1 RNase H by interacting with a pocket located at the junction between the p51 subunit thumb subdomain and the p66 RNase H domain, possibly affecting the conformation of the adjacent p66 RNase H domain (32), and the hydrazonoindolin-2-one derivative, (Z)-3-(2-(4-(3,4-dihydroxyphenyl)thiazol-2-yl)hydrazono)indolin-2-one (NSC657589), which was shown to inhibit both HIV-1 RT-associated RDDP and RNase H activities, possibly by binding to a site located in a region between the polymerase catalytic aspartate triad (Asp110, Asp185, Asp186) and the NNRTI pocket, hence contiguous to the NNRTI pocket but different from it (33). Figure 1 shows the chemical structures of the inhibitors used in this study. When these HIV-1 RHIs were tested on PFV PR-RT in biochemical assays using the methods previously described for HIV-1 RT-associated RNase H and polymerase functions (34–36) and the reaction conditions previously optimized for PFV PR-RT (3), we observed that all selected RHIs inhibited the PFV PR-RT (Table 1 and Fig. 2). In particular, the metal binding catalytic site binder RHIs, RDS1643, DHQ, and β-thujaplicinol, inhibited the PFV PR-RT RNase H activity, with 50% inhibitory concentrations (IC50s) comparable to the ones obtained for HIV-1 RT, with the exception of DHQ, which was 2.5-fold-less potent on PFV PR-RT than on HIV-1 RT. Unexpectedly, while RDS1643 and DHQ have been reported to be selective for the HIV-1 RNase H function (Table 1), they also inhibited the PFV PR-RT-associated RDDP activity with the same potency shown for the RNase H function (Fig. 2). In contrast, β-thujaplicinol was inactive on the associated RDDP activities of both enzymes (Table 1 and Fig. 2). On one hand, these results may indicate that RDS1643 and DHQ also bind the Mg2+ ions in the PFV PR-RT DNA polymerase (DP) site, while β-thujaplicinol is not able to act on the DP site; however, in all the retroviral RTs crystallized so far, the geometrical characteristics of the two sites are quite different, and this hypothesis is not very likely to be accurate. It is more plausible that the binding of RDS1643 and DHQ at the PFV RNase H active site may reduce the PFV PR-RT flexibility and, with a long-range effect, it may also affect its RDDP activity. Moreover, RDS1643 probably contacts the nucleic acid in the RNase H active site and thus might hamper polymerization activity due to a change of primer position in the polymerase active site. Possibly, given the structure of β-thujaplicinol, its binding to the PFV RNase H site does not involve the amino acid residues interacting with RDS1643 and DHQ, so that a long-range effect cannot take place.
TABLE 1.
PFV PR-RT inhibition by HIV-1 RT RHIs
| Compound | Mean ± SD IC50 (μM) fora: |
|||
|---|---|---|---|---|
| PFV PR-RT |
HIV-1 RT |
|||
| RNase H | RDDP | RNase H | RDDP | |
| RDS1643 | 7.6 ± 2.0 | 5.2 ± 0.5 | 8.0 ± 1.5 | >100 |
| DHQ | 41.0 ± 8.0 | 41.0 ± 12.0 | 16.0 ± 4.0 | >100 |
| β-Thujaplicinol | 0.3 ± 0.1 | >100 | 0.22 ± 0.03 | >100 |
| VU6 | 6.5 ± 0.4 | 15.2 ± 1.8 | 18.7 ± 4.8 | >100 |
| NSC657589 | 1.7 ± 0.3 | 2.0 ± 0.5 | 4.4 ± 1.3 | 7.4 ± 1.7 |
| Efavirenz | >20 | >20 | >20 | 0.015 ± 0.005 |
Compound concentration required to inhibit RT-associated RNase H activity by 50% and compound concentration required to reduce RT-associated RDDP activity by 50%, respectively. The results were calculated from ≥3 independent experiments.
FIG 2.

Dose-dependent PFV PR-RT inhibition by HIV-1 RHIs. PFV PR-RT-associated RNase H (filled circles) and RDDP (open circles) activities were assayed at 37°C as described for 1 h (RNase H) and 30 min (RDDP), in the absence and in the presence of different concentrations of RDS1643 (A), VU6 (B), β-thujaplicinol (C), and NSC657589 (D). The results represent the average and standard deviation values of three independent experiments.
Next, we assayed the vinylogous urea derivative VU6, showing that it inhibited the PFV PR-RT-associated RNase H function 2.9-fold-more potently than did HIV-1 RT. Interestingly, VU6 was also able to inhibit PFV PR-RT-associated RDDP activity, while it was inactive on the HIV-1 RT RDDP (Table 1). Of note, the inhibition of PFV RDDP and RNase H activities occurs at comparable compound concentrations (Table 1), suggesting a unique inhibitor binding site exerting an effect on both activities. Since vinylogous urea derivatives have been proposed to bind to a pocket located at the junction between the p51 subunit thumb subdomain and the p66 RNase H domain of HIV-1 RT and, in particular, to interact with an α-helix adjacent to the RNase H domain in the p51 subunit (32, 37), we speculate that even though HIV-1 RT is a heterodimer and PFV PR-RT is a monomer, this pocket is present in both proteins. Therefore, VU6 inhibition of both PFV PR-RT RNase H and RDDP functions reinforces the hypothesis that the binding of RHIs to PFV PR-RT may decrease its flexibility, leading to a complete loss of its catalytic activities.
Subsequently, we tested the hydrazonoindolin-2-one derivative NSC657589, which has been reported to inhibit both HIV-1 RT-associated functions (33). Our data reveal that it inhibited both PFV PR-RT RNase H and RDDP functions. The IC50s observed for PFV PR-RT inhibition by NSC657589 were 2- to 3-fold lower than those observed for HIV-1 RT inhibition. NSC657589 has been proposed to inhibit HIV-1 RT by binding a site close to but distinct from the NNRTI binding pocket (33). Therefore, our results suggest that either this pocket is present also in PFV PR-RT or that the NSC657589 activity on HIV-1 RT is due to the binding to a different site, possibly close to the RNase H binding pocket.
In addition, we wanted to test the NNRTI efavirenz on PFV PR-RT functions, since it is known that the NNRTI binding pocket is present in HIV-1 RT only and not in the highly homologous HIV-2 RT (38). In HIV-1 RT, two tyrosines, Y181 and Y188, close to the (underlined) active-site motif Y183MDD (VIY181QYMDDLY188V), as well as the substitution K103N, are involved in efavirenz resistance (38). Sequence comparisons show that in PFV RT, only the tyrosine corresponding to Y188 is present (NVQVYVDDIY317L); however, due to the low sequence identity (ca. 26%), it is difficult to judge whether the lysine corresponding to K103 is also present in PFV RT. Our results showed that efavirenz was not able to inhibit either PFV PR-RT function (Table 1), confirming that the NNRTI binding pocket is not present in PFV PR-RT and that the hydrazonoindolin-2-one derivative NSC657589 binds to a different site from that of the NNRTI binding pocket.
NMR titration experiments with the free PFV RNase H domain.
To learn more about RNase H inhibitor binding, we tested the binding of several of the inhibitors, namely, RDS1643, NSC657589, and VU6, to the free PFV RNase H domain in NMR titration experiments. An NMR spectrum correlating the resonance frequencies of chemical shifts of amide protons directly bonded to 15N-labeled nitrogen atoms (2D [1H-15N] heteronuclear single quantum correlation [HSQC]) allows the individual detection of peptide backbone signals. Each signal in the spectrum represents a single amino acid of the peptide chain and can be assigned to an individual residue. Changes in the chemical environment of a magnetically active atom, e.g., binding of an inhibitor or ligand, led to changes in the chemical shifts of the signal. Thus, HSQC spectra can be used to investigate inhibitor binding.
We previously solved the structure of PFV RNase H by NMR spectroscopy (PDB ID 2LSN) (7, 8). Thus, NMR assignment and structural information can be used for inhibitor titration experiments. However, measurements using the RNase H inhibitors and PFV RNase H were partially hampered by the low solubility of the inhibitors in aqueous solutions and the high inhibitor concentrations needed in NMR spectroscopy, since concentrations of 15N-labeled enzyme of around 50 to 100 μM are necessary to obtain analyzable signals. In fact, the addition of NSC657589 and VU6 to 15N-labeled PFV RNase H resulted in partial precipitation of the inhibitors. Final concentrations of about 2 mM NSC657589 (ca. 32-fold excess) or 12 mM VU6 (ca. 194-fold excess) were added to 15N-labeled PFV RNase H. However, due to the partial precipitation of inhibitors and possibly enzymes even at lower inhibitor concentrations during the measurements, the spectra could not be evaluated (data not shown).
In contrast, the addition of the inhibitor RDS1643 (49-fold excess) gave rise to relevant chemical shift changes of certain residues in the HSQC spectrum (Fig. 3; see also Fig. S1 in the supplemental material). A chemical shift change of ≥0.02 ppm was considered relevant (Fig. 3A). Significant chemical shift changes upon the addition of inhibitors were observed with residues T598, A614, G617, T641, I647, A648, T668, Y672, A674, W703, K722, H732, and T733. The overlays of the HSQC spectra of residues T641 and W703 recorded at various inhibitor concentrations are illustrated in Fig. 3B. The data confirm that RDS1643 binds to the RNase H domain and suggest that the effects on polymerization are indirect. However, we cannot exclude a second binding site in the polymerase domain.
FIG 3.

Backbone chemical shift changes upon inhibitor binding. (A) Changes of chemical shifts of PFV RNase H after adding 3.9 mM RDS1643 as a function of the primary sequence. Normalized chemical shift changes of >0.02 ppm, indicated by a horizontal gray line, were considered significant. Residues that were not assigned are marked by an x. (B) [1H-15N]HSQC overlays of the T641 (left panel) and W703 (right panel) amide cross peaks at each inhibitor titration step shown in different colors (black, 0 mM; blue, 1.0 mM; green, 2.0 mM; cyan, 3.0 mM; red, 3.9 mM).
The residues exhibiting significant chemical shift changes upon the addition of inhibitors are highlighted in the three-dimensional structure of PFV RNase H (Fig. 4A). Among the residues leading to significant chemical shift changes, T598, A614, G617, A648, T668, and A674 are not surface exposed, while residues, T641, I647, Y672, and W703 are spatially close together and oriented toward the protein surface (Fig. 4B).
FIG 4.

Identification of the putative inhibitor binding site on the structure of PFV RNase H. The normalized chemical shift changes (weighted geometric average of 1HN and 15N chemical shift changes) are shown in the PFV RNase H. Amino acids showing significant chemical shift changes at an RDS1643 concentration of 3.9 mM are highlighted. Changes of ≥0.02 ppm are illustrated in pink, changes of >0.03 ppm are shown in violet, and changes of >0.04 ppm are shown in red. The active-site residues are represented in green. (A) Ribbon presentation of PFV RNase H (PDB ID 2LSN). (B) Surface presentation of PFV RNase H highlighting the surface-exposed residues with significant chemical shift changes. (C) Surface presentation of PFV RNase H with an RNA/DNA hybrid. Based on the structure of the human RNase H-substrate complex (PDB ID 2QK9), a structural alignment of the PFV and human RNase H was performed to model the RNA/DNA substrate of the human RNase H onto PFV RNase H (root mean square deviation [RMSD], 2.35 Å) (8). The RNA strand is shown in dark blue and the DNA strand in light blue. (D) Inhibitor modeling. RDS1643 was modeled onto the structure of the PFV RNase H using the modeling program AutoDock Vina (29).
Residues T598, I647, A648, and T668 are adjacent to the active-site residues D599, E646, and D669, respectively (Fig. 4A). W703 is in the alpha-helix D, which follows the basic loop that has been shown to be involved in nucleic acid substrate binding (8). T641 is located at the beginning of the alpha-helix A, which also harbors the active-site residue E646.
These data indicate that the inhibitor binding pocket is located next to the active site and might be formed by the surface-exposed residues T641, I647, Y672, and W703. The residues K722, H732, and T733 located at the beginning of the alpha-helix E and the adjacent N-terminal loop are also surface exposed. However, they exhibited only small chemical shift changes (<0.03 ppm), which might be due to minor direct or indirect conformational changes induced by the binding of the inhibitor.
Most of the other residues shown in Fig. 4A are located in the interior of the protein (Fig. 4B) and therefore do not come into consideration for direct inhibitor contact. Rather, the binding of the inhibitor can lead to indirect changes in the local structure of the protein by changing side-chain orientations.
To show how the nucleic acid substrate is bound in relation to the putative inhibitor binding pocket, we modeled the RNA/DNA substrate of the human RNase H-RNA-DNA complex (PDB ID 2QK9) onto the PFV RNase H (8). For clarity, only the nucleic acid without the human RNase H is presented (Fig. 4C). It has been shown by UV/visible light (Vis) spectroscopy and isothermal titration calorimetry that RDS1643 and other diketo acids (DKAs) bind Mg2+ or Mn2+ (26, 28) and thus can interact with the metal ions in the active site of the RNase H. This is consistent with our observation of chemical shift changes in the NMR titration experiment close or next to the active-site residues. Overall, these data suggest that RDS1643 can bind Mg2+ in the RNase H active site and that it can make additional interactions with residues next to it to stabilize its binding.
To further verify this mode of interaction, we performed docking calculations with RDS1643 and PFV RNase H using the program AutoDock Vina (29). Based on our NMR data, we limited the potential binding site to the active site and the area that includes residues T641, I647, Y672, and W703. Figure 4D depicts a possible binding mode of the inhibitor that is in good agreement with our results. The keto groups of RDS1643 reach into the RNase H active site and thus may be able to bind the metal ions, while the hydrophobic part of the molecule is close to Y672 and W703.
Structure comparisons.
To elucidate the binding of RDS1643 to HIV-1 RNase H, we performed a secondary structural alignment of HIV-1 RNase H and PFV RNase H (Fig. 5A). In HIV-1 RNase H, residues T473, L479, Y501, and V518 correspond to T641, I647, Y672, and W703 in the inhibitor binding pocket of PFV RNase H. The structural overlay of PFV RNase H with HIV-1 RNase H presented in Fig. 5B implies that in HIV-1 RNase H, the binding of the inhibitor might take place in a similar way. Nevertheless, RDS1643 binding to the RNase H might have different effects on the RDDP activities of the two enzymes, owing to structural differences, i.e., being a heterodimer versus a monomer. Therefore, specific studies on HIV-1 RT are needed to elucidate the RDS1643 mode of RNase H inhibition.
FIG 5.
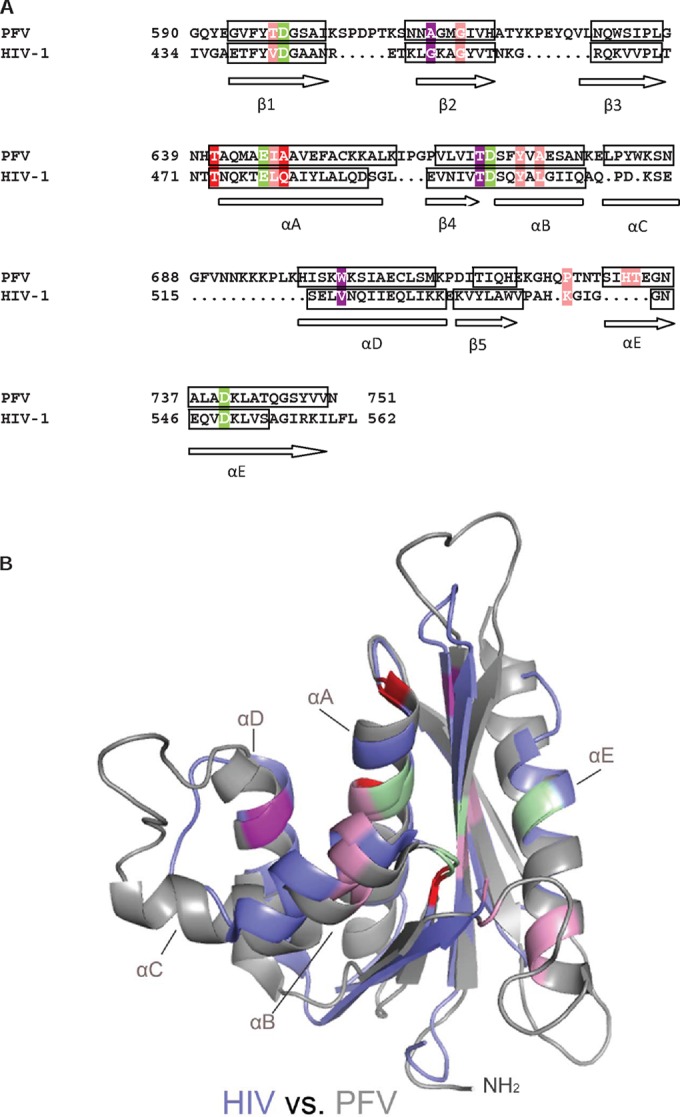
Identification of the putative inhibitor binding pocket in HIV-1 RNase H. (A) Sequence and secondary structure alignment of PFV and HIV-1 RNase to identify the corresponding residues HIV-1 RNase H (PDB ID 1HRH). The active-site residues are highlighted in green. The four residues in HIV-1 RNase H that are equivalent to the ones in PFV RNase H exhibiting chemical shift changes at 3.9 mM RDS1643 are labeled according to the color coding described for Fig. 4A. (B) Structural overlay of the RNases H from PFV (PDB ID 2LSN) (gray) and HIV-1 (PDB ID 1HRH). (blue). Color coding of relevant residues is as described in Fig. 5A.
Despite the utility of PFV RNase H as a model system, we did not attempt to select for resistant virus, since the replication cycles between the two viruses and the time point of reverse transcription in those cycles differ, i.e., while reverse transcription in HIV-1 takes place immediately after the virus enters the cell, reverse transcription in FVs occurs predominantly in the virus-producing cell before the virus leaves the infected cell. Thus, FVs harbor DNA in the virus particle (39–41). These differences might have an impact on the development of resistant viruses and might make results difficult to compare and interpret.
Conclusions.
We tested the effect of five HIV-1 RHIs known to interact with different HIV-1 RT pockets on PFV RT in vitro: four selective RHIs (RDS1643, DHQ, β-thujaplicinol, and VU6) and one HIV-1 RDDP and RNase H dual inhibitor (NSC657589). Moreover, for comparative purposes, we included in the study the nonnucleoside inhibitor efavirenz. All RHIs inhibited the PFV RNase H function at low-micromolar or nanomolar concentrations. These results are comparable to the ones reported for HIV-1 RNase H, indicating similar binding in the two proteins (Table 1). Among the selective RHIs tested, RDS1643, DHQ, and VU6 inhibited the FV RT-associated RDDP activity, suggesting differences with HIV-1 RT in the structure and flexibility of the protein. NSC657589 was confirmed in its ability to inhibit both functions in PFV RT. Efavirenz was inactive on PFV PR-RT, indicating the lack of the nonnucleoside binding pocket, as reported for other monomeric RTs, confirming that NSC657589 does not bind to the nonnucleoside binding pocket.
Since the structure of PFV RNase H has been determined by NMR spectroscopy (7, 8), NMR analysis was used to characterize the interactions of RDS1643 with the protein. In the presence of magnesium ions, NMR titration experiments revealed several conformational changes of the residues next to the active-site amino acids, indicating an interaction between the keto groups of RDS1643 and the magnesium ions in the RNase H active site. Moreover, chemical shifts of noncatalytic residues in the PFV RNase H active-site region (T641, I647, Y672, and W703) might provide a hydrophobic pocket for the aromatic moiety of RDS1643 and thus improve its binding affinity. Due to the high structural similarity of PFV and HIV-1 RNase H, docking experiments and structural overlays enabled us to propose an inhibitor binding site in HIV-1 RNase H. This includes the corresponding HIV-1 RNase H residues T473, L479, Y501, and V518, some of which are conserved, since they are located in the RNase H primer grip area. Our results suggest that PFV RNase H can be used as a model enzyme to analyze the binding of HIV-1 RHIs, while it cannot be used to analyze HIV-1 NNRTI binding.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by RAS grant LR 7/2007 CRP-24915, the Deutsche Forschungsgemeinschaft (grant Wo630/7-3), and the University of Bayreuth, Germany.
We thank Ramona Heissmann for purifying the PFV PR-RT and PFV RNase H.
Footnotes
Published ahead of print 5 May 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00056-14.
REFERENCES
- 1.Rinke CS, Boyer PL, Sullivan MD, Hughes SH, Linial ML. 2002. Mutation of the catalytic domain of the foamy virus reverse transcriptase leads to loss of processivity and infectivity. J. Virol. 76:7560–7570. 10.1128/JVI.76.15.7560-7570.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boyer PL, Stenbak CR, Clark PK, Linial ML, Hughes SH. 2004. Characterization of the polymerase and RNase H activities of human foamy virus reverse transcriptase. J. Virol. 78:6112–6121. 10.1128/JVI.78.12.6112-6121.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hartl MJ, Mayr F, Rethwilm A, Wöhrl BM. 2010. Biophysical and enzymatic properties of the simian and prototype foamy virus reverse transcriptases. Retrovirology 7:5. 10.1186/1742-4690-7-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hartl MJ, Wöhrl BM, Rösch P, Schweimer K. 2008. The solution structure of the simian foamy virus protease reveals a monomeric protein. J. Mol. Biol. 381:141–149. 10.1016/j.jmb.2008.05.064 [DOI] [PubMed] [Google Scholar]
- 5.Hartl MJ, Bodem J, Jochheim F, Rethwilm A, Rösch P, Wöhrl BM. 2011. Regulation of foamy virus protease activity by viral RNA: a novel and unique mechanism among retroviruses. J. Virol. 85:4462–4469. 10.1128/JVI.02211-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goff SP. 2007. Retroviridae: The retroviruses and their replication, p 1999–2069 In Knipe DM, Howley PM. (ed), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 7.Leo B, Hartl MJ, Schweimer K, Mayr F, Wöhrl BM. 2012. Insights into the structure and activity of prototype foamy virus RNase H. Retrovirology 9:14. 10.1186/1742-4690-9-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leo B, Schweimer K, Rösch P, Hartl MJ, Wöhrl BM. 2012. The solution structure of the prototype foamy virus RNase H domain indicates an important role of the basic loop in substrate binding. Retrovirology 9:73. 10.1186/1742-4690-9-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Evans DB, Brawn K, Deibel MR, Jr, Tarpley WG, Sharma SK. 1991. A recombinant ribonuclease H domain of HIV-1 reverse transcriptase that is enzymatically active. J. Biol. Chem. 266:20583–20585 [PubMed] [Google Scholar]
- 10.Hostomsky Z, Hostomska Z, Hudson GO, Moomaw EW, Nodes BR. 1991. Reconstitution in vitro of RNase H activity by using purified N-terminal and C-terminal domains of human immunodeficiency virus type 1 reverse transcriptase. Proc. Natl. Acad. Sci. U. S. A. 88:1148–1152. 10.1073/pnas.88.4.1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith JS, Gritsman K, Roth MJ. 1994. Contributions of DNA polymerase subdomains to the RNase H activity of human immunodeficiency virus type 1 reverse transcriptase. J. Virol. 68:5721–5729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Esposito F, Corona A, Tramontano E. 2012. HIV-1 reverse transcriptase still remains a new drug target: structure, function, classical inhibitors, and new inhibitors with innovative mechanisms of actions. Mol. Biol. Int. 2012:586401. 10.1155/2012/586401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tramontano E. 2006. HIV-1 RNase H: recent progress in an exciting, yet little explored, drug target. Mini Rev. Med. Chem. 6:727–737. 10.2174/138955706777435733 [DOI] [PubMed] [Google Scholar]
- 14.Tramontano E, Di Santo R. 2010. HIV-1 RT-associated RNase H function inhibitors: recent advances in drug development. Curr. Med. Chem. 17:2837–2853. 10.2174/092986710792065045 [DOI] [PubMed] [Google Scholar]
- 15.Distinto S, Maccioni E, Meleddu R, Corona A, Alcaro S, Tramontano E. 2013. Molecular aspects of the RT/drug interactions. Perspective of dual inhibitors. Curr. Pharm. Des. 19:1850–1859. 10.2174/1381612811319100009 [DOI] [PubMed] [Google Scholar]
- 16.Rausch JW. 2013. Ribonuclease inhibitors: structural and molecular biology, p 143–172 In Le Grice SFJ, Gotte M. (ed), Human immnodeficiency virus reverse transcriptase. A bench-to-bedside success. Springer, London, United Kingdom [Google Scholar]
- 17.Himmel DM, Maegley KA, Pauly TA, Bauman JD, Das K, Dharia C, Clark AD, Jr, Ryan K, Hickey MJ, Love RA, Hughes SH, Bergqvist S, Arnold E. 2009. Structure of HIV-1 reverse transcriptase with the inhibitor beta-thujaplicinol bound at the RNase H active site. Structure 17:1625–1635. 10.1016/j.str.2009.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Himmel DM, Sarafianos SG, Dharmasena S, Hossain MM, McCoy-Simandle K, Ilina T, Clark AD, Jr, Knight JL, Julias JG, Clark PK, Krogh-Jespersen K, Levy RM, Hughes SH, Parniak MA, Arnold E. 2006. HIV-1 reverse transcriptase structure with RNase H inhibitor dihydroxy benzoyl naphthyl hydrazone bound at a novel site. ACS Chem. Biol. 1:702–712. 10.1021/cb600303y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kirschberg TA, Balakrishnan M, Squires NH, Barnes T, Brendza KM, Chen X, Eisenberg EJ, Jin W, Kutty N, Leavitt S, Liclican A, Liu Q, Liu X, Mak J, Perry JK, Wang M, Watkins WJ, Lansdon EB. 2009. RNase H active site inhibitors of human immunodeficiency virus type 1 reverse transcriptase: design, biochemical activity, and structural information. J. Med. Chem. 52:5781–5784. 10.1021/jm900597q [DOI] [PubMed] [Google Scholar]
- 20.Lansdon EB, Liu Q, Leavitt SA, Balakrishnan M, Perry JK, Lancaster-Moyer C, Kutty N, Liu X, Squires NH, Watkins WJ, Kirschberg TA. 2011. Structural and binding analysis of pyrimidinol carboxylic acid and N-hydroxy quinazolinedione HIV-1 RNase H inhibitors. Antimicrob. Agents Chemother. 55:2905–2915. 10.1128/AAC.01594-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Su HP, Yan Y, Prasad GS, Smith RF, Daniels CL, Abeywickrema PD, Reid JC, Loughran HM, Kornienko M, Sharma S, Grobler JA, Xu B, Sardana V, Allison TJ, Williams PD, Darke PL, Hazuda DJ, Munshi S. 2010. Structural basis for the inhibition of RNase H activity of HIV-1 reverse transcriptase by RNase H active site-directed inhibitors. J. Virol. 84:7625–7633. 10.1128/JVI.00353-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pari K, Mueller GA, DeRose EF, Kirby TW, London RE. 2003. Solution structure of the RNase H domain of the HIV-1 reverse transcriptase in the presence of magnesium. Biochemistry 42:639–650. 10.1021/bi0204894 [DOI] [PubMed] [Google Scholar]
- 23.Christen MT, Menon L, Myshakina NS, Ahn J, Parniak MA, Ishima R. 2012. Structural basis of the allosteric inhibitor interaction on the HIV-1 reverse transcriptase RNase H domain. Chem. Biol. Drug Des. 80:706–716. 10.1111/cbdd.12010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gong Q, Menon L, Ilina T, Miller LG, Ahn J, Parniak MA, Ishima R. 2011. Interaction of HIV-1 reverse transcriptase ribonuclease H with an acylhydrazone inhibitor. Chem. Biol. Drug Des. 77:39–47. 10.1111/j.1747-0285.2010.01052.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stahl SJ, Kaufman JD, Vikić-Topić S, Crouch RJ, Wingfield PT. 1994. Construction of an enzymatically active ribonuclease H domain of human immunodeficiency virus type 1 reverse transcriptase. Protein Eng. 7:1103–1108. 10.1093/protein/7.9.1103 [DOI] [PubMed] [Google Scholar]
- 26.Shaw-Reid CA, Munshi V, Graham P, Wolfe A, Witmer M, Danzeisen R, Olsen DB, Carroll SS, Embrey M, Wai JS, Miller MD, Cole JL, Hazuda DJ. 2003. Inhibition of HIV-1 ribonuclease H by a novel diketo acid, 4-[5-(benzoylamino)thien-2-yl]-2,4-dioxobutanoic acid. J. Biol. Chem. 278:2777–2780. 10.1074/jbc.C200621200 [DOI] [PubMed] [Google Scholar]
- 27.Davies JF, Jr, Hostomska Z, Hostomsky Z, Jordan SR, Matthews DA. 1991. Crystal structure of the ribonuclease H domain of HIV-1 reverse transcriptase Science 252:88–95. 10.1126/science.1707186 [DOI] [PubMed] [Google Scholar]
- 28.Tramontano E, Esposito F, Badas R, Di Santo R, Costi R, La Colla P. 2005. 6-[1-(4-Fluorophenyl)methyl-1H-pyrrol-2-yl)]-2,4-dioxo-5-hexenoic acid ethyl ester a novel diketo acid derivative which selectively inhibits the HIV-1 viral replication in cell culture and the ribonuclease H activity in vitro. Antiviral Res. 65:117–124. 10.1016/j.antiviral.2004.11.002 [DOI] [PubMed] [Google Scholar]
- 29.Trott O, Olson AJ. 2010. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31:455–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suchaud V, Bailly F, Lion C, Tramontano E, Esposito F, Corona A, Christ F, Debyser Z, Cotelle P. 2012. Development of a series of 3-hydroxyquinolin-2(1H)-ones as selective inhibitors of HIV-1 reverse transcriptase associated RNase H activity. Bioorg. Med. Chem. Lett. 22:3988–3992. 10.1016/j.bmcl.2012.04.096.; 10.1016/j.bmcl.2012.04.096 [DOI] [PubMed] [Google Scholar]
- 31.Chung S, Himmel DM, Jiang JK, Wojtak K, Bauman JD, Rausch JW, Wilson JA, Beutler JA, Thomas CJ, Arnold E, Le Grice SF. 2011. Synthesis, activity, and structural analysis of novel α-hydroxytropolone inhibitors of human immunodeficiency virus reverse transcriptase-associated ribonuclease H. J. Med. Chem. 54:4462–4473. 10.1021/jm2000757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chung S, Wendeler M, Rausch JW, Beilhartz G, Gotte M, O'Keefe BR, Bermingham A, Beutler JA, Liu S, Zhuang X, Le Grice SF. 2010. Structure-activity analysis of vinylogous urea inhibitors of human immunodeficiency virus-encoded ribonuclease H. Antimicrob. Agents Chemother. 54:3913–3921. 10.1128/AAC.00434-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Distinto S, Esposito F, Kirchmair J, Cardia MC, Gaspari M, Maccioni E, Alcaro S, Markt P, Wolber G, Zinzula L, Tramontano E. 2012. Identification of HIV-1 reverse transcriptase dual inhibitors by a combined shape-, 2D-fingerprint- and pharmacophore-based virtual screening approach. Eur. J. Med. Chem. 50:216–229. 10.1016/j.ejmech.2012.01.056 [DOI] [PubMed] [Google Scholar]
- 34.Esposito F, Sanna C, Del Vecchio C, Cannas V, Venditti A, Corona A, Bianco A, Serrilli AM, Guarcini L, Parolin C, Ballero M, Tramontano E. 2013. Hypericum hircinum L. components as new single-molecule inhibitors of both HIV-1 reverse transcriptase-associated DNA polymerase and ribonuclease H activities. Pathog. Dis. 68:116–124. 10.1111/2049-632X.12051 [DOI] [PubMed] [Google Scholar]
- 35.Esposito F, Corona A, Zinzula L, Kharlamova T, Tramontano E. 2012. New anthraquinone derivatives as inhibitors of the HIV-1 reverse transcriptase-associated ribonuclease H function. Chemotherapy 58:299–307. 10.1159/000343101 [DOI] [PubMed] [Google Scholar]
- 36.Esposito F, Kharlamova T, Distinto S, Zinzula L, Cheng YC, Dutschman G, Floris G, Markt P, Corona A, Tramontano E. 2011. Alizarine derivatives as new dual inhibitors of the HIV-1 reverse transcriptase-associated DNA polymerase and RNase H activities effective also on the RNase H activity of non-nucleoside resistant reverse transcriptases. FEBS J. 278:1444–1457. 10.1111/j.1742-4658.2011.08057.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chung S, Miller JT, Johnson BC, Hughes SH, Le Grice SF. 2012. Mutagenesis of human immunodeficiency virus reverse transcriptase p51 subunit defines residues contributing to vinylogous urea inhibition of ribonuclease H activity. J. Biol. Chem. 287:4066–4075. 10.1074/jbc.M111.314781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ren J, Stammers DK. 2008. Structural basis for drug resistance mechanisms for non-nucleoside inhibitors of HIV reverse transcriptase. Virus Res. 134:157–170. 10.1016/j.virusres.2007.12.018 [DOI] [PubMed] [Google Scholar]
- 39.Yu SF, Baldwin DN, Gwynn SR, Yendapalli S, Linial ML. 1996. Human foamy virus replication: a pathway distinct from that of retroviruses and hepadnaviruses. Science 271:1579–1582. 10.1126/science.271.5255.1579 [DOI] [PubMed] [Google Scholar]
- 40.Yu SF, Sullivan MD, Linial ML. 1999. Evidence that the human foamy virus genome is DNA. J. Virol. 73:1565–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rethwilm A. 2010. Molecular biology of foamy viruses. Med. Microbiol. Immunol. 199:197–207. 10.1007/s00430-010-0158-x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.