

Abstract
Herpes simplex virus (HSV) infections can cause considerable morbidity. Transmission of HSV-2 has become a major health concern, since it has been shown to promote transmission of other sexually transmitted diseases. Pritelivir (AIC316, BAY 57-1293) belongs to a new class of HSV antiviral compounds, the helicase-primase inhibitors, which have a mode of action that is distinct from that of antiviral nucleoside analogues currently in clinical use. Analysis of pharmacokinetic-pharmacodynamic parameters is a useful tool for the selection of appropriate doses in clinical trials, especially for compounds belonging to new classes for which no or only limited data on therapeutic profiles are available. For this purpose, the effective dose of pritelivir was determined in a comprehensive mouse model of HSV infection. Corresponding plasma concentrations were measured, and exposures were compared with efficacious concentrations derived from cell cultures. The administration of pritelivir at 10 mg/kg of body weight once daily for 4 days completely suppressed any signs of HSV infection in the animals. Associated plasma concentrations adjusted for protein binding stayed above the cell culture 90% effective concentration (EC90) for HSV-1 for almost the entire dosing interval. Interestingly, by increasing the dose 6-fold and prolonging the treatment duration to 8 days, it was possible to treat mice infected with an approximately 30-fold pritelivir-resistant but fully pathogenic HSV-1 virus. Corresponding plasma concentrations exceeded the EC90 of this mutant for <8 h, indicating that even suboptimal exposure to pritelivir is sufficient to achieve antiviral efficacy, possibly augmented by other factors such as the immune system.
INTRODUCTION
Infections by herpes simplex virus 1 (HSV-1) and HSV-2 lead to lifelong persistence of the virus, with frequent and sometimes painful recurrences. While HSV-1 persists predominantly in the trigeminal ganglia, causing oral lesions upon reactivation, HSV-2 manifests in the genital region after latent infection of the sacral ganglia and is mainly transmitted sexually. Infections in newborns or immunocompromised subjects can become life-threatening. Furthermore, genital herpes can be associated with severe psychological distress and may promote transmission of other sexually transmitted diseases, such as HIV (1).
Nucleoside analogues (acyclovir and penciclovir, as well as their orally bioavailable prodrugs valacyclovir and famciclovir, respectively) are widely used for treatment either as episodic therapy for a short period or as daily suppressive therapy for months or even years; however, latent virus is not eradicated. Recurrences still occur after cessation of episodic therapy and sometimes even during suppressive treatment (2, 3). Acute symptoms are significantly reduced only when treatment is initiated early in the course of the disease (4). In addition, HSV infections resistant to nucleoside analogues are recognized as a clinical problem among immunocompromised patients (5). The prevalence of resistance is reported to be about 5% among these patients but can reach up to 14 to 30% among patients with allogeneic bone marrow transplants (6). Therefore, there is a need for effective alternatives to nucleoside analogue inhibitors, to provide more-efficient therapy (even after delayed onset) and to counteract resistance.
Pritelivir (AIC316, BAY 57-1293) is a member of the group of helicase-primase inhibitors, which represent a novel class of anti-HSV compounds that may be promising candidates for such improved therapy (7). These molecules target the viral helicase-primase enzyme complex, which comprises three proteins, encoded by the UL5 (helicase), UL52 (primase), and UL8 (scaffold protein shown to promote primer synthesis) genes, and is crucial for viral DNA replication (8, 9). Pritelivir was shown to be more potent in cell culture than nucleoside analogues and provided superior efficacy in several animal models, even after delayed onset of treatment (mimicking the clinical situation) (10–13). Moreover, due to its different mode of action, pritelivir does not require activation by the viral thymidine kinase and is active against nucleoside analogue-resistant HSV strains (14). Initial clinical data showed that pritelivir treatment led to significant dose-dependent reductions in HSV shedding, genital lesions, and the amounts of virus shed in otherwise healthy persons with genital herpes (15).
All mutations mediating resistance to pritelivir identified so far are located in the UL5 helicase gene, close to or within functional motif IV except for a single amino acid exchange in the UL52 primase (16, 17). The growth rates and pathogenicity of pritelivir-resistant mutants vary and depend on the particular amino acid substitution mediating resistance (18).
As a useful tool for the selection of doses and dosing regimens for clinical trials, especially in the early stages in drug development, the pharmacokinetic-pharmacodynamic (PK-PD) correlation for a compound, i.e., the time course of the drug in the body versus the effective concentration, can be explored (19). By comparing exposures that show efficacy in cell culture or animal models with exposures derived from PK trials in humans, the appropriate doses and dosing regimens for efficacy trials can be deduced (20).
In order to establish a PK-PD correlation for pritelivir, a murine neck infection model was used. It was shown previously that once-daily oral therapy with pritelivir was effective for treatment of wild-type HSV-1 strains in this model and exhibited superior activity, compared with famciclovir (12). Pritelivir showed comparable activities against HSV-1 and HSV-2 in vitro and in vivo (11, 14). Therefore, results from this model should be transferable to HSV-2, which is currently the focus of clinical drug development. The purposes of this work were to determine the lowest effective dose of pritelivir sufficient to completely suppress viral replication and to correlate the corresponding drug exposures with efficacy data derived from cell culture studies. Furthermore, the question of whether a pritelivir-resistant but replication-competent and fully pathogenic HSV-1 strain can be efficiently treated with higher and longer-lasting pritelivir exposures was explored by adjusting the dose and dosing regimen. Results were used to support dose selection for clinical trials.
MATERIALS AND METHODS
Viruses.
The parental strain was HSV-1 SC16 that had been plaque purified three times (SC16 cl-2). The single resistance-mediating UL5 substitution K356Q was transferred to SC16 cl-2 to produce recombinant cl-2-r1-Rec (21). This was accomplished by PCR amplification of a 2.1-kb product containing the target mutation. Transfection of the DNA was carried out in 293T cells, which were subsequently infected with SC16 cl-2. Resistant plaques were selected using 3.0 μM pritelivir, at a frequency approximately 50-fold above background (10−6 PFU). Transfer of the required mutation was confirmed by sequencing.
Antiviral compound.
For the HSV-1 plaque assay, pritelivir (molecular weight, 402.5) was stored as a 50 mM stock solution in dimethyl sulfoxide (DMSO). For animal experiments, pritelivir was dissolved in distilled water containing 1% carboxymethyl cellulose (CMC), with sonication for 5 min. Stock solutions containing 1.0, 3.0, and 12 mg/ml were stored in aliquots at −20°C and were thawed immediately before use.
HSV-1 plaque reduction assay.
The efficacy of pritelivir in vitro against both wild-type and resistant HSV-1 was determined by a plaque reduction assay (PRA) in Vero cells (12). Briefly, approximately 100 PFU of virus was inoculated into 12-well plates (Nunc, Denmark) containing approximately 2 × 105 Vero cells/well. After adsorption for 60 min at 37°C in a humidified atmosphere of 5% CO2, a Dulbecco's modified Eagle's medium (DMEM) overlay containing 1% newborn calf serum and high-density CMC (CMC-DMEM) and different concentrations of pritelivir were added to each well. The plaques were fixed, stained, and enumerated after 48 h of incubation. The mean number of plaques at each concentration (expressed as the percentage of the mean of the control wells, which contained no drug) was plotted against the log10 drug concentration and evaluated with GraphPad Prism 4.
Murine experiments.
Animal experiments were performed according to the Home Office (United Kingdom) guidelines or in accordance with an institutional proposal (proposal 84-02.05.20.12.045/2012A-01 or 84-02.05.20.12.046/2012A-02) approved by the Landesamt für Natur, Umwelt, und Verbraucherschutz Nordrhein-Westfalen (Recklinghausen, Germany). Animal care and use were performed in accordance with federal guidelines (German Animal Welfare Act §8). Mice that were in extremis or showing irreversible neurological signs or severe weight loss (>15%) were culled; the times of the deaths of these mice were recorded as the time from inoculation plus 1 day.
Mice were female BALB/c mice obtained from Harlan UK at weights of approximately 16 g. The mice were acclimatized for 1 week before use. The neck-ear skin infection model was used as described previously (12, 18, 21). Briefly, the skin on the right neck was shaved, and 3 days later a dose of 5 × 106 PFU/ml in 10 μl DMEM was applied to the skin, which was then scarified in a crosshatch pattern. Mice were inoculated with wild-type or mutant virus, and one control group was mock infected. Therapy was by oral gavage (100 μl) once per day, starting 1 day postinfection (p.i.). Controls were given the vehicle only (1% CMC in distilled water). General appearance and specific clinical signs (e.g., lesion score, body weight, and mortality) were noted at the same time each day. The development of lesions on the neck (primary site) and pinna (zosteriform spread) was assessed subjectively, using a magnifying glass. Lesions were scored first at the primary inoculation site and then at the secondary site, according to the following arbitrary scale: 0, no clinical signs; 1, one vesicle and swelling; 2, more than one vesicle; 3, local erosion; 4, ulceration of the local lesion; 5, primary lesions plus isolated zosteriform lesion; 6, mild ulceration of confluent zosteriform lesions; 7, moderate ulceration of confluent zosteriform lesions; 8, severe ulceration of confluent zosteriform lesions. This scoring system was adapted from one proposed by Nagafuchi et al. (22). On days 1, 3, 5, and 8 p.i., groups of three mice were killed and their tissues were sampled. The skin local to the inoculation site, the ipsilateral ear pinna, and brainstem tissues was homogenized and tested for infectious virus, using methods described previously (18, 21).
For the first experiment, five infected groups (each containing 20 mice) were treated with pritelivir at 0.5, 1.0, 5.0, 10, or 15 mg/kg of body weight once per day for 4 consecutive days by oral gavage, starting at day 1 p.i. Controls (two groups, one containing uninfected mice and the other containing infected but untreated mice) were given placebo. In the next experiment, mice were treated with pritelivir at 5 or 15 mg/kg/day for 4 days. In the following experiment, mice were treated with 15 mg/kg/day for 4 or 8 days. Finally, another group of infected mice was treated with 60 mg/kg/day for 4 or 8 days. To minimize the use of animals, in the second experiment the tissues of wild-type virus-infected mice were assessed for infectious virus only on days 3 and 5 p.i., to confirm the effectiveness of the compound.
Mice in a satellite group for PK analyses were treated once orally with 0.2 ml pritelivir in 1% CMC. Thirty-six mice in each dose group (10 and 60 mg/kg) were dosed at a time of 0 min. At defined time points (10 min, 30 min, 1 h, 2 h, 3 h, 4 h, 6 h, 8 h, 12 h, 24 h, 36 h, and 48 h after dosing), 3 mice per time point and dose group were sacrificed following CO2 anesthesia, and blood was collected by heart puncture, into lithium-heparin tubes. In addition, two mice without treatment were sacrificed to serve as controls. Blood was stored on ice until centrifugation (20,800 × g for 10 s at 4°C), which separated blood cells from plasma. From each centrifuged sample, approximately 200 μl of plasma was transferred to a new vial. Samples were mixed briefly (Vortex mixer) and stored immediately at −20°C until further use.
Analyses of plasma samples.
Samples were prepared using protein precipitation with an organic solvent. Briefly, after the samples were thawed, 100 μl of plasma was transferred to a deep-well plate and 300 μl of precipitation solution (acetonitrile containing 1% acetic acid) containing the internal standard ([13C2H315N]pritelivir) was added. After centrifugation at 1,360 × g for 10 min, 100 μl of the supernatant was mixed with 60 μl water and subjected to high-performance liquid chromatography (HPLC)-mass spectrometry (MS) (Applied Biosystems HPLC-MS/MS 3200 QTrap, Agilent 1200 RR system, and PAL HTC autosampler). Samples were separated with an Ascentis RP-amide column (Supelco), using an injection volume of 20 μl; water containing 1% acetic acid (mobile phase A) and methanol-acetonitrile-acetic acid at 50:50:1 (vol/vol/vol; mobile phase B) was used in a gradient that was held at 0% mobile phase B for 0.5 min, linearly increased to 70% mobile phase B in 2.5 min, and then held at 70% mobile phase B for 1.5 min, at a flow of 0.25 ml/min. The column was then flushed with 95% mobile phase B for 1 min and reequilibrated for 4.5 min. Detection was performed in the API 3200 QTrap system with a TurboIonSpray ion source operating in the positive mode, with an ionization voltage of 5,000 V and a declustering potential of 31 V. The mass transitions for the multiple-reaction monitoring were m/z 403.2 to 196.1 and 408.2 to 196.1 for pritelivir and its internal standard, respectively, at a collision energy of 27 eV. The retention time was 4.46 min for both pritelivir and its stable-isotope-labeled internal standard. The calibration range was from 10 ng/ml to 9,993 ng/ml, with interday accuracies of the mean ranging from −5.75% to 6.25% and precision values of ≤10.9%. The mean accuracies of in-study validation samples were between −5.01% and −1.39%, with interday precision values of ≤3.49%. The prestudy validation samples revealed intraday accuracies of the mean between −8.66% and 7.26% and precision values of ≤8.61%. The interday accuracies of the mean ranged from −5.82% to 4.40%, with precision values of ≤6.34%. All results obtained were within the acceptance criteria for bioanalytical method validation, as published by the FDA and the European Medicines Agency (23, 24).
PK calculations.
PK analysis was performed on the mean values obtained by using WinNonLin software (version 5.2; Pharsight) with a noncompartmental model and extravascular input. Chromatograms were recorded and integrated with the use of Analyst (version 1.4.2; Applied Biosystems). Accuracy (bias) was calculated as the percent mean deviation of the nominal concentration. Precision was calculated as the coefficient of variation.
Statistics.
A two-way analysis of variance (ANOVA) with repeated measures of each clinical parameter (lesion score, body weight, or ear thickness) was performed to determine statistically significant overall differences among the groups of mice (P < 0.05). When significant differences were detected, the Tukey post hoc test was performed to confirm which groups contributed to such differences. On a given day, the statistically significant difference among the groups was determined by one-way ANOVA.
For the infectious virus titers for the neck skin, ear pinna, and brainstem, data points were geometric means (and standard deviations) from three mice sampled at each time point. The limit of detection was 0.7 log10 PFU/sample for all tissues; differences between titers for the wild-type and mutant viruses at particular time points were compared by one-way ANOVA. Virus titers below the level of detection of 0.7 log10 PFU/tissue were taken to be 0.6 log10 PFU/tissue for statistical calculations.
RESULTS
Susceptibility of HSV-1 strain SC16 cl-2 and derivative mutant cl-2-r1-Rec to pritelivir.
To confirm previously published results in our laboratory, the 50% effective concentration (EC50) and 90% effective concentration (EC90) of pritelivir for both the parental HSV-1 strain SC16 cl-2 (12) and the mutant strain cl-2-r1-Rec, carrying a lysine-to-glutamine substitution in the helicase protein at amino acid position 356 (K356Q) mediating resistance to pritelivir in vitro (21), were determined with a plaque reduction assay (PRA) (Fig. 1). The mean EC50 and EC90 values of pritelivir for the SC16 cl-2 strain were 0.01 μM and 0.03 μM, respectively (means derived from 3 independent experiments), and those for the cl-2-r1-Rec strain were 0.39 μM and 0.81 μM, respectively (means derived from 3 independent experiments). Hence, the mutant strain cl-2-r1-Rec showed a 27-fold decrease in sensitivity with regard to EC90, confirming the previously described resistance phenotype (21).
FIG 1.
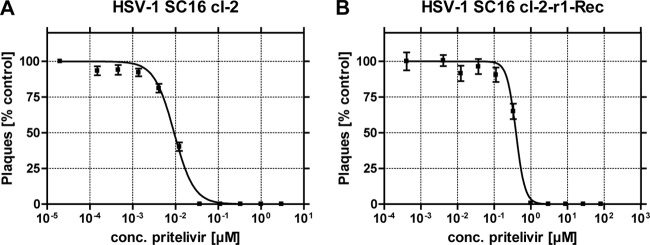
Sensitivity of the parental virus HSV-1 SC16 cl-2 (A) and the pritelivir-resistant mutant HSV-1 cl-2-r1-Rec (B) in a plaque reduction assay (PRA) (representative experiment). The 50% effective concentration (EC50) and 90% effective concentration (EC90) doses were measured by means of the PRA. Plaques were counted after 48 h of incubation. Data points represent the means of six replicates, with standard deviations.
Determination of the lowest effective dose and corresponding exposure in the murine skin infection model.
In order to determine the lowest effective dose of pritelivir sufficient to completely suppress viral replication, the murine neck-ear zosteriform HSV-1 infection model was used, in which pritelivir has already been shown to be highly efficacious in comparison with famciclovir (12). An experiment with immunocompetent mice was carried out with doses between 0.5 and 15 mg/kg/day for once-daily oral dosing for 4 days starting on day 1 p.i., with 4 days of follow-up. Mice were infected in the skin of the neck using the laboratory HSV-1 strain SC16 cl-2. An inoculum of 5.0 × 103 PFU was employed, as determined by back-titration. The animals were examined daily, including measurements of body weight and thickness of the right ear, and HSV lesions at the infection site were classified according to a well-established scoring system (see Materials and Methods) (25). On days 1, 3, 5, and 8 p.i., mice were killed and tissue samples were taken approximately 3 h after dosing. Skin from the inoculation site, right ear pinna, and brainstem was collected, and titers of infectious virus were determined by the PRA in Vero cells.
All placebo-treated mice developed clinical signs of infection (Fig. 2A), and approximately 40% of mice died between days 6 and 8 p.i. Without active treatment, levels of infectious virus in the skin at the inoculation site reached a peak at day 5 p.i., with titers of approximately 104 PFU/sample already being detectable at day 1 p.i. (Fig. 2B). High titers remained in skin, ear pinna, and brain tissues at day 8 p.i. (Fig. 2B to D).
FIG 2.

Effects of oral therapy in mice treated with various doses of pritelivir or placebo once per day from day 1 to day 4 after infection with the parental laboratory strain HSV-1 SC16 cl-2 in the neck skin infection model. Mice were inoculated with HSV-1 SC16 cl-2 by application of a dose of 5 × 105 PFU/ml to the skin on the right neck, which was then scarified in a crosshatch pattern. The indicated treatment was started 1 day after infection. The treatment period is indicated by the horizontal black bars. (A) Lesion scores. Data points are mean values of the lesion scores obtained from groups of 5 mice. (B to D) Infectious virus titers in tissue samples, i.e., skin from the inoculation site (B), ear pinnae (C), or brainstem (D), taken from three infected mice from each treatment group on days 1, 3, 5, and 8 p.i. Data points are geometric mean titers per tissue sample for three mice tested at each time point, with standard deviations. *, virus titers were significantly lower (P < 0.05) than those in placebo-treated control mice. The limit of detection was 5 PFU/sample.
Pritelivir was administered orally to groups of mice once daily for 4 days starting on day 1 p.i., using doses of 0.5, 1.0, 5, 10, and 15 mg/kg. None of the infected mice treated at any dose died. The higher treatment doses (10 and 15 mg/kg) completely prevented the development of visible lesions (Fig. 2A). The lower doses (0.5 and 1 mg/kg) showed no reduction in lesion development, compared to the control (mean score of ≥4), whereas the middle dose (5 mg/kg) decreased the mean lesion score below 1.5. Treatment with pritelivir also reduced virus titers in the skin on day 3 p.i. for all except the lowest dose (0.5 mg/kg) (Fig. 2B). When 1.0 mg/kg was employed, infectious virus levels were reduced below the level of detection on day 3 p.i. but infectious virus was detected on day 5 p.i. When mice were treated with the lower doses (0.5 or 1.0 mg/kg/day for 4 days), infectious virus was also detected in the ear pinna and brainstem on both days 5 and 8 p.i. (Fig. 2C and D). However, it was noted that the higher doses of 5.0, 10, and 15 mg/kg produced clearance of infectious virus significantly below the level of detection in all tissues tested at 5 or 8 days p.i. (P < 0.03).
Pritelivir plasma concentrations after a single dose of 10 mg/kg.
Taking into account both clinical signs and virus titers in the tissues, the 50% effective dose (ED50) was estimated to be between 1 and 5 mg/kg/day, whereas 10 mg/kg/day was able to completely suppress HSV-1 replication in infected animals without any visible lesions. In order to determine the PK profile for this dose, mice were treated with a single dose of 10 mg/kg. Plasma samples from 3 animals at each time point were obtained 10 min, 30 min, 1 h, 2 h, 3 h, 4 h, 6 h, 8 h, 12 h, 24 h, 36 h, and 48 h after dosing, and pritelivir concentrations in plasma were determined by HPLC-MS/MS. Basic PK parameters were calculated using standard software (Table 1).
TABLE 1.
Pharmacokinetic parameters derived from total mean concentrations in female mice (n = 3 per time point) after single oral applications of 10 or 60 mg/kg pritelivir
| Parametera | Data for mice given pritelivir dose of: |
|
|---|---|---|
| 10 mg/kg | 60 mg/kg | |
| AUC0–48 (h·μg/ml) | 233 | 951 |
| AUC0–24 (h·μg/ml) | 219 | 852 |
| AUC0–∞ (h·μg/ml) | 233 | 956 |
| AUC0–∞/D (h·kg·μg/ml/mg) | 23.3 | 15.9 |
| AUC∞ex (%) | 0.188 | 0.535 |
| Cmax (μg/ml) | 17.7 | 68.7 |
| Cmax/D (kg·μg/ml/mg) | 1.77 | 1.14 |
| C24h (μg/ml) | 1.09 | 6.73 |
| C24h/D (kg·μg/ml/mg) | 0.109 | 0.112 |
| CL/F (ml/h/kg) | 42.8 | 62.7 |
| t1/2z (h) | 5.38 | 6.64 |
| Tmax (h) | 4.00 | 3.00 |
AUC0–48, area under the curve for 0 to 48 h; AUC0–24, area under the curve for 0 to 24 h; AUC0–∞, area under the curve for 0 h to infinity; D, dose; AUC∞ex, extrapolated proportion of AUC0–∞; Cmax, maximum plasma concentration; C24h, plasma concentration 24 h after dosing; CL/F, oral clearance; t1/2z, terminal half-life; Tmax, time to reach Cmax.
A single dose of 10 mg/kg resulted in a maximum plasma concentration (Cmax) of 17.6 μg/ml and an area under the curve for the dosing interval of 0 to 24 h (AUC0-24h) of 219 μg·h/ml, with a terminal half-life (t1/2) of about 5 h. The plasma concentration 24 h after dosing (C24h) decreased to 1.09 μg/ml.
Dose regimen adjustment for the treatment of a resistant mutant.
To determine whether it was possible to treat a fully pathogenic and replication-competent resistant virus by adjusting the dose and dose regimen, mice were infected with the pritelivir-resistant HSV-1 mutant cl-2-r1-Rec. This mutant showed growth to higher titers, in comparison with wild-type virus, in Vero cell culture. This mutant was also pathogenic in mice, in which its virulence was equal to or greater than that of the wild-type virus strain SC16 cl-2 (18, 21).
In a first experiment, mice were infected in the skin of the neck with SC16 cl-2 or the pritelivir-resistant mutant cl-2-r1-Rec with a target inoculum at 5.0 × 104 PFU/mouse, which was confirmed by back-titration after inoculation. The animals were allocated to three treatment groups and were treated with either placebo or pritelivir at 5 or 15 mg/kg once daily for 4 days, starting 1 day after infection.
Without treatment, both viruses produced vigorous disease responses, with clinical signs apparent from day 3 or 4 p.i., and the lesion scores reached maximum values by day 6 or 7 p.i. (Fig. 3A). Since data for wild-type SC16 cl-2 confirmed the results with 5 and 15 mg/kg shown in Fig. 2, only data derived from the mutant cl-2-r1-Rec are shown.
FIG 3.

Effects of oral therapy in mice treated with 5 or 15 mg/kg of pritelivir or placebo once per day from day 1 to day 4 after infection with the pritelivir-resistant strain HSV-1 cl-2-r1-Rec in the neck skin infection model. Mice were inoculated with HSV-1 cl-2-r1-Rec by application of a dose of 5 × 106 PFU/ml to the skin on the right neck, which was then scarified in a crosshatch pattern. The treatment period is indicated by the horizontal black bars. (A) Lesion scores. Data points are mean values of the lesion scores obtained from groups of 5 mice. (B to D) Infectious virus titers in tissue samples, i.e., skin from the inoculation site (B), ear pinnae (C), or brainstem (D), taken from three infected mice from each treatment group on days 1, 3, 5, and 8 p.i. Data points are geometric mean titers per tissue sample for three mice tested at each time point, with standard deviations. Virus titers were not significantly different from those for the placebo-treated control mice. The limit of detection was 5 PFU/sample.
The infectious virus titers observed in the target tissues (skin of inoculation site, ear pinna, and brainstem) were consistent with the clinical signs (Fig. 3A). The infectious virus titers at the infection site and in the tissues were similar for wild-type virus and the resistant mutant (Fig. 3B to D). These results confirmed that the pritelivir-resistant strain cl-2-r1-Rec was fully pathogenic.
When wild-type virus-infected mice were treated with pritelivir at 5 or 15 mg/kg orally once daily on days 1 to 4 p.i. (inclusive), results were comparable to those shown in Fig. 2 for the same doses (data not shown). Therapy using 5 mg/kg, exceeding the ED50 for the wild-type virus, had no significant impact on clinical signs or virus titers for the pritelivir-resistant mutant cl-2-r1-Rec (Fig. 3). Therapy using 15 mg/kg for 4 days delayed the progression of zosteriform lesions (Fig. 3A) but had no significant influence on virus titers in the tissues analyzed (Fig. 3B to D).
Given these results, the aim was to produce a potential beneficial effect of pritelivir treatment by increasing the treatment duration and/or the treatment dose. However, prolongation of treatment to 8 days with a dose of 15 mg/kg showed no improvement over treatment for 4 days (data not shown). Therefore, the next step was to increase the dose to 60 mg/kg and to treat the infected animals for 4 and 8 days.
When the higher pritelivir dose of 60 mg/kg was used but the treatment duration was kept as 4 days, lesion development was delayed until day 5 p.i. (Fig. 4A), with complete prevention of weight loss and mortality, whereas an increase in ear thickness could not be prevented (Fig. 4B to D). However, treatment using 60 mg/kg for mice infected with the resistant mutant resulted in significant decreases in infectious virus titers in skin and ear tissues (Fig. 4E and F). By day 8 p.i., the levels of infectious virus in the skin at the inoculation site were below the level of detection for all 6 mice sampled from the 60-mg/kg treatment groups. In contrast, the skin tissues from the placebo-treated group (one of three sampled mice) still formed visible virus plaques in cell culture.
FIG 4.

Effects of oral therapy in mice treated with 60 mg/kg of pritelivir or placebo once per day on either days 1 to 4 or days 1 to 8 after infection with the pritelivir-resistant strain HSV-1 cl-2-r1-Rec in the neck skin infection model. Mice were inoculated with HSV-1 1 cl-2-r1-Rec by application of a dose of 5 × 106 PFU/ml to the skin on the right neck, which was then scarified in a crosshatch pattern. The treatment period is indicated by the horizontal black bars (4 days) or by both black and gray bars (8 days). (A) Lesion scores. Data points are mean values of the lesion scores obtained from groups of 5 mice. (B) Survival. (C) Body weights relative to baseline (day 0). (D) Ear thickness values relative to baseline (day 0). (E to G) Infectious virus titers in tissue samples, i.e., skin from the inoculation site (E), ear pinnae (F), or brainstem (G), taken from three infected mice from each treatment group on days 1, 3, 5, and 8 p.i. Data points are geometric mean titers per tissue sample for three mice tested at each time point, with standard deviations. *, virus titers were significantly lower (P < 0.05) than those in placebo-treated control mice. The limit of detection was 5 PFU/sample.
Prolongation of the treatment duration to 8 days, starting on day 1 p.i., also prevented mortality and weight loss (Fig. 4B and C). Clinical signs were still present starting at day 5 p.i. but lesion scores were significantly reduced (P = 0.004 to 0.03), by almost one-half, in comparison with the 4-day treatment group on days 7 to 10 p.i. (Fig. 4A). Furthermore, the mean ear thickness values for the mice treated longer were lower than those for mice treated for 4 days, and this difference was significant on day 6 p.i. (P = 0.03) and day 7 p.i. (P = 0.04) (Fig. 4D). Expanding the treatment duration to 8 days did not completely prevent the spread of the resistant virus to the ear pinna on day 3 p.i. However, virus titers in the ear tissues were significantly lower (P = 0.02) than values for the untreated mutant-infected control group from day 8 p.i., and values were lower by up to 3 log10 PFU in mice that received 60 mg/kg for 8 days, compared with untreated mice and mice treated for 4 days only (Fig. 4F). For both 4- and 8-day therapy, infectious virus levels in the brainstem remained below detection levels in samples from day 8 p.i. onward (Fig. 4G).
Pritelivir plasma concentrations after a single dose of 60 mg/kg.
Determination of the PK profile in mice with a single dose of 60 mg/kg, keeping all other settings and conditions constant as for the 10-mg/kg dose, resulted in a Cmax of 68.7 μg/ml and an AUC0-24h of 852 μg·h/ml, with a t1/2z of almost 7 h (Table 1). The plasma concentration dropped to 6.73 μg/ml after 24 h.
PK-PD correlation in mice.
It is generally agreed that, in order to be efficacious, plasma concentrations of antiviral compounds should be maintained at levels that are able to completely suppress viral replication over the entire dosing interval (19). Furthermore, only the free fraction, and not the protein-bound fraction, of the drug in plasma is usually taken into account. In order to determine whether the exposures that were reached with 10 and 60 mg/kg exceeded the concentrations required for suppression of viral replication, the plasma concentrations of pritelivir achieved in the murine infection model were correlated with the EC90 values in cell culture (Fig. 5).
FIG 5.

Plasma concentration-time curves for pritelivir after oral administration of a single dose of 10 or 60 mg/kg to uninfected mice. Uninfected mice were treated with a single dose of pritelivir as indicated. At defined time points, 3 mice were sacrificed and blood was collected. In addition, two mice without treatment were sacrificed to serve as controls. The concentrations of pritelivir in plasma were determined by HPLC, and mean values were plotted against sampling times after correction for protein binding in murine plasma. Data points denote the arithmetic mean pritelivir concentration at each time point, with standard deviations (n = 3). Lines parallel to the x axis, cell culture EC50 and EC90 values for SC16 cl-2 (wild-type) and cl-2-r1-Rec (pritelivir-resistant mutant), as indicated.
In the PRA, pritelivir exhibited an EC50 of 0.01 μM (equal to 4 ng/ml) and an EC90 of 0.03 μM (12 ng/ml) for HSV-1 SC16 cl-2. The unbound fraction (fu) of pritelivir in the cell culture medium used in this assay is approximately 97%, as determined by equilibrium analysis with 14C-labeled compound (data not shown); therefore, protein binding can be neglected. Taking into account the fraction of pritelivir in murine plasma that is not bound to plasma proteins (0.8%, as obtained from equilibrium dialysis experiments; data not shown), the maximum plasma concentration (Cmax) and the plasma concentration 24 h after dosing (C24h) correspond to unbound concentrations of 141 ng/ml and 9 ng/ml, respectively. Therefore, pritelivir plasma concentrations were maintained above the unbound EC90 of 12 ng/ml for almost the entire dosing interval.
After correction for fu, the single pritelivir dose of 60 mg/kg in mice yielded a Cmax of 549 ng/ml and a C24h of 54 ng/ml. The EC50 for the mutant cl-2r1-Rec was 0.39 μM (156 ng/ml) and the EC90 was 0.81 μM (325 ng/ml). As shown in Fig. 5, the plasma concentration in mice after a single dose of 60 mg/kg exceeded the cell culture EC50 for HSV-1 mutant cl-2-r1-Rec for approximately 20 h, but the EC90 was exceeded for only approximately 10 h.
DISCUSSION
The aim of this work was to investigate the nonclinical PK-PD profile of the novel anti-HSV helicase-primase inhibitor pritelivir in mice. For doses of 0.5 to 15 mg/kg administered once daily for 4 days, it was shown that, starting with pritelivir at 5 mg/kg, replication of wild-type virus was completely suppressed in our murine skin infection model. A 50% effective dose (ED50) between 1 and 5 mg/kg/day was established. This result is in good agreement with data from previously reported animal models, e.g., the murine lethal-challenge model, in which the ED50 was determined to be 1.5 mg/kg/day after intranasal infection of the animals (11). The amount of virus in the inoculum used to determine the effective dose of pritelivir described here was only 5 × 103 PFU, which was 10-fold below the target inoculum of 5 × 104 PFU. This may explain the reduced mortality rate and the delay in the occurrence of clinical signs, in comparison with other experiments (12). However, in the subsequent experiments reported here, the effectiveness of pritelivir administered at 5 mg/kg/day for 4 days to prevent viral replication was confirmed with a target inoculum of 5 × 104 PFU.
In contrast to previously published experiments with a different mutant strain (25), this is the first report of efficacy of a helicase-primase inhibitor against a drug-resistant HSV mutant in an animal infection model. Given the approximately 30-fold decrease in the sensitivity to pritelivir of the variant carrying the K356Q substitution in cell culture, with regard to the EC90, it was expected that the virus would show a reduced response to therapy. This hypothesis was confirmed since there was little or no effect of treatment using pritelivir at 5 mg/kg, a dose which was effective for the treatment of wild-type virus in the same experiment. Even treatment with 15 mg/kg for 4 days had no significant effect on the replication of the pritelivir-resistant mutant in the target tissues, and the efficacy of treatment was not improved when therapy was extended to 8 days (data not shown). However, it was shown that increasing the daily dose to 60 mg/kg for 4 days can prevent mortality and body weight reduction and had an influence on clinical signs and virus titers of the mutant in tissues. Although therapy with 60 mg/kg delayed lesion development for 1 day up to day 4 p.i. (last day of therapy), lesions became visible from day 5 p.i. and showed progressive development. Without continuing doses beyond day 4 p.i., the mean lesion score almost reached the value for placebo treatment by day 7 p.i. However, extension of therapy to day 8 p.i. showed a clear benefit over therapy for 4 days. Although some lesions started to appear at day 5 p.i., the severity was significantly less than that in the untreated controls. Furthermore, none of the treated mice reached wild-type lesion scores during the course of the disease. With respect to virus titers in tissues, no significant difference was seen at the infection site with treatment at 60 mg/kg for 4 days versus 8 days. With increasing treatment duration, the virus titer in the ear pinna was significantly reduced by day 8 p.i. (P = 0.02); however, even treatment with 60 mg/kg for only 4 days was able to eventually decrease the virus titer below the detection limit by day 12.
Single daily oral doses of pritelivir at 10 mg/kg for 4 days were able to completely suppress mortality, clinical signs, and replication, as well as the spread of wild-type virus, in the animal model. The comparison of the time-plasma concentration profile at 10 mg/kg with the EC90 in cell culture revealed a clear correlation, confirming the efficacy of this dose; plasma concentrations remained well above the EC90 corrected for protein binding for almost the entire dosing interval of 24 h. This is in good agreement with the observation that a daily dose of 5 mg/kg was sufficient to prevent mortality and viral spread and was able to significantly reduce clinical signs in the infection model. By increasing the dose by only 6-fold to 60 mg/kg/day and prolonging the treatment duration to 8 days, it was possible to treat infections with the mutant HSV-1 strain cl-2-r1-Rec, which was 27-fold less sensitive to pritelivir in cell culture than was the wild-type virus. Corresponding plasma concentrations exceeded the estimated EC90 of the mutant, corrected for protein binding, for only about 10 h, and the EC50 of cl-2-r1-Rec was covered only for approximately 20 h. Nevertheless, this dose led to significant reductions of the viral titers in the analyzed tissues and was sufficient to prevent mortality and body weight reductions in infected animals.
Acute HSV infection in an immunocompetent host is generally a self-limiting disease. Since pritelivir plasma concentrations stayed above the EC50 for the variant carrying the K356Q substitution for more than two-thirds of the dosing interval, it can be assumed that viral replication was impaired, if not fully blocked, by treatment with 60 mg/kg. Since the mice used in this study were fully immunocompetent, pritelivir treatment might have supported the immune system to better control the infection in treated animals, compared with animals that did not receive treatment and therefore had to deal with actively replicating virus. This explanation would be in good agreement with our observation that prolongation of the treatment duration to 8 days was required to gain full beneficial effects, because longer treatment with pritelivir counteracted viral replication more effectively and possibly facilitated faster immune clearance of the infection.
PK factors may also contribute to the clearance of infection in pritelivir-treated animals inoculated with the mutant strain. Plasma samples used to determine the exposure of infected mice treated with 60 mg/kg for 4 or 8 days were derived from single-dose administration in uninfected animals. It is unlikely that HSV-1 infection itself has any potential influence on the PK of pritelivir (there is no evidence, to date, that HSV-1 infection has any effect on drug metabolism in mice). Moreover, no pritelivir metabolites with relevant antiviral activity have been identified to date. Thus, it can be concluded that the pharmacological effects observed are driven by the parent compound. There may be an accumulation of pritelivir in the animals after multiple doses. However, due to the half-life of about 5 to 7 h in mice, the compound is not expected to show relevant accumulation with a dosing interval of 24 h (26). Furthermore, our considerations focused on the concentrations of the unbound fraction of the compound in plasma, which may be misleading because the sites of activity are located in the skin and neuronal tissue. Whole-body autoradiography with radiolabeled compound showed comparable pritelivir concentrations in most body tissues and plasma, as well as penetration of the blood-brain barrier (data not shown). However, the actual concentrations of free, not protein-bound, compound in the respective tissues are unknown. Therefore, we may even underestimate the amount of available drug at the viral replication sites in the skin and central nervous system. It should also be noted that the mutant strain carrying the K356Q substitution used here showed only 27-fold resistance. Further experiments are needed to investigate the effects of increased doses and treatment durations on mutants with even lower sensitivity to pritelivir (25).
Correlation of PK-PD parameters is a very useful tool to select doses and dosing regimen for clinical trials, especially those addressing efficacy (19). As discussed above, daily doses of 10 mg/kg for 4 days were able to prevent any signs of HSV-1 infection in treated animals. Corresponding pritelivir plasma concentrations in mice after a single oral dose of 10 mg/kg remained above the EC90 derived from cell cultures for almost the entire dosing interval of 24 h, pointing to a good PK-PD correlation. In a phase 1 clinical trial in healthy volunteers, once-daily dosing with 25 mg pritelivir led to a mean Cmax of 1.36 μg/ml at steady state and a mean predose trough concentration (Ctrough) of 0.833 μg/ml, with a mean terminal half-life of 69 h (n = 12) (A. Birkmann, D. Kropeit, D. McCormick, H. Zimmermann, and H. Ruebsamen-Schaeff, presented at the 24th International Conference on Antiviral Research, 5 to 8 May 2011). Correction for protein binding in human plasma (fu = 2.8%, determined by equilibrium analysis) resulted in an unbound mean Cmax of 38.0 ng/ml and an unbound Ctrough of 23.3 ng/ml, exceeding the EC90 for HSV-1 of 12 ng/ml derived from cell cultures.
Based on the good nonclinical PK-PD correlation, a dose of 75 mg per day was selected as the highest daily dose in a phase 2 proof-of-concept and dose-finding trial in HSV-2-positive persons with genital herpes (15). Besides the PK-PD data, this dose selection took into account variability in plasma exposures among individuals, as well as variability in susceptibility among clinical isolates and between HSV 1 and 2 (14). In this trial, dose-dependent reductions in viral shedding, the amounts of virus shed, and clinical lesions were shown starting with 25 mg administered once daily. The greatest efficacy was seen in the group treated once daily with 75 mg (15).
In summary, a good correlation between PK and PD parameters was shown for pritelivir in nonclinical studies. By increasing the dose only 6-fold and doubling the treatment duration, animals infected with an approximately 30-fold resistant HSV-1 mutant can be efficiently treated with pritelivir, indicating that even suboptimal exposure to pritelivir appears to be sufficient for antiviral efficacy. Nonclinical PK-PD correlations were predictive for dose selection in clinical efficacy trials.
ACKNOWLEDGMENTS
S.B. received support from the Cambridge Commonwealth Trust by means of a Cambridge Nehru Scholarship and the Isaac Newton Trust, Trinity College (Cambridge, United Kingdom). This work was supported by AiCuris GmbH & Co. KG.
Footnotes
Published ahead of print 21 April 2014
REFERENCES
- 1.Freeman EE, Weiss HA, Glynn JR, Cross PL, Whitworth JA, Hayes RJ. 2006. Herpes simplex virus 2 infection increases HIV acquisition in men and women: systematic review and meta-analysis of longitudinal studies. AIDS 20:73–83. 10.1097/01.aids.0000198081.09337.a7 [DOI] [PubMed] [Google Scholar]
- 2.Douglas JM, Critchlow C, Benedetti J, Mertz GJ, Connor JD, Hintz MA, Fahnlander A, Remington M, Winter C, Corey L. 1984. A double-blind study of oral acyclovir for suppression of recurrences of genital herpes simplex virus infection. N. Engl. J. Med. 310:1551–1556. 10.1056/NEJM198406143102402 [DOI] [PubMed] [Google Scholar]
- 3.Tyring SK, Douglas JM, Jr, Corey L, Spruance SL, Esmann J. 1998. A randomized, placebo-controlled comparison of oral valacyclovir and acyclovir in immunocompetent patients with recurrent genital herpes infections. Arch. Dermatol. 134:185–191 [DOI] [PubMed] [Google Scholar]
- 4.Reichman RC, Badger GJ, Mertz GJ, Corey L, Richman DD, Connor JD, Redfield D, Savoia MC, Oxman MN, Bryson Y, Tyrrell DL, Portnoy J, Creigh-Kirk T, Keeney RE, Ashikaga T, Dolin R. 1984. Treatment of recurrent genital herpes simplex infections with oral acyclovir: a controlled trial. JAMA 251:2103–2107 [PubMed] [Google Scholar]
- 5.Bacon TH, Levin MJ, Leary JJ, Sarisky RT, Sutton D. 2003. Herpes simplex virus resistance to acyclovir and penciclovir after two decades of antiviral therapy. Clin. Microbiol. Rev. 16:114–128. 10.1128/CMR.16.1.114-128.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frobert E, Cortay JC, Ooka T, Najioullah F, Thouvenot D, Lina B, Morfin F. 2008. Genotypic detection of acyclovir-resistant HSV-1: characterization of 67 ACV-sensitive and 14 ACV-resistant viruses. Antiviral Res. 79:28–36. 10.1016/j.antiviral.2008.01.153 [DOI] [PubMed] [Google Scholar]
- 7.Birkmann A, Hewlett G, Ruebsamen-Schaeff H, Zimmermann H. 2011. Helicase-primase inhibitors as the potential next generation of highly active drugs against herpes simplex viruses. Future Virol. 6:1199–1209. 10.2217/fvl.11.28 [DOI] [Google Scholar]
- 8.Crute JJ, Tsurumi T, Zhu LA, Weller SK, Olivo PD, Challberg MD, Mocarski ES, Lehman IR. 1989. Herpes simplex virus 1 helicase-primase: a complex of three herpes-encoded gene products. Proc. Natl. Acad. Sci. U. S. A. 86:2186–2189. 10.1073/pnas.86.7.2186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cavanaugh NA, Ramirez-Aguilar KA, Urban M, Kuchta RD. 2009. Herpes simplex virus-1 helicase-primase: roles of each subunit in DNA binding and phosphodiester bond formation. Biochemistry 48:10199–10207. 10.1021/bi9010144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baumeister J, Fischer R, Eckenberg P, Henninger K, Ruebsamen-Waigmann H, Kleymann G. 2007. Superior efficacy of helicase-primase inhibitor BAY 57-1293 for herpes infection and latency in the guinea pig model of human genital herpes disease. Antivir. Chem. Chemother. 18:35–48 [DOI] [PubMed] [Google Scholar]
- 11.Betz UA, Fischer R, Kleymann G, Hendrix M, Rubsamen-Waigmann H. 2002. Potent in vivo antiviral activity of the herpes simplex virus primase-helicase inhibitor BAY 57-1293. Antimicrob. Agents Chemother. 46:1766–1772. 10.1128/AAC.46.6.1766-1772.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Biswas S, Jennens L, Field HJ. 2007. The helicase primase inhibitor, BAY 57-1293 shows potent therapeutic antiviral activity superior to famciclovir in BALB/c mice infected with herpes simplex virus type 1. Antiviral Res. 75:30–35. 10.1016/j.antiviral.2006.11.006 [DOI] [PubMed] [Google Scholar]
- 13.Kleymann G, Fischer R, Betz UA, Hendrix M, Bender W, Schneider U, Handke G, Eckenberg P, Hewlett G, Pevzner V, Baumeister J, Weber O, Henninger K, Keldenich J, Jensen A, Kolb J, Bach U, Popp A, Maben J, Frappa I, Haebich D, Lockhoff O, Rubsamen-Waigmann H. 2002. New helicase-primase inhibitors as drug candidates for the treatment of herpes simplex disease. Nat. Med. 8:392–398. 10.1038/nm0402-392 [DOI] [PubMed] [Google Scholar]
- 14.Field HJ, Huang ML, Lay EM, Mickleburgh I, Zimmermann H, Birkmann A. 2013. Baseline sensitivity of HSV-1 and HSV-2 clinical isolates and defined acyclovir-resistant strains to the helicase-primase inhibitor pritelivir. Antiviral Res. 100:297–299. 10.1016/j.antiviral.2013.08.024 [DOI] [PubMed] [Google Scholar]
- 15.Wald A, Corey L, Timmler B, Magaret A, Warren T, Tyring S, Johnston C, Kriesel J, Fife K, Galitz L, Stoelben S, Huang ML, Selke S, Stobernack HP, Ruebsamen-Schaeff H, Birkmann A. 2014. Helicase-primase inhibitor pritelivir for HSV-2 infection. N. Engl. J. Med. 370:201–210. 10.1056/NEJMoa1301150 [DOI] [PubMed] [Google Scholar]
- 16.Biswas S, Kleymann G, Swift M, Tiley LS, Lyall J, Guirre-Hernandez J, Field HJ. 2008. A single drug-resistance mutation in HSV-1 UL52 primase points to a difference between two helicase-primase inhibitors in their mode of interaction with the antiviral target. J. Antimicrob. Chemother. 61:1044–1047. 10.1093/jac/dkn057 [DOI] [PubMed] [Google Scholar]
- 17.Biswas S, Miguel RN, Sukla S, Field HJ. 2009. A mutation in helicase motif IV of herpes simplex virus type 1 UL5 that results in reduced growth in vitro and lower virulence in a murine infection model is related to the predicted helicase structure. J. Gen. Virol. 90:1937–1942. 10.1099/vir.0.011221-0 [DOI] [PubMed] [Google Scholar]
- 18.Biswas S, Tiley LS, Zimmermann H, Birkmann A, Field HJ. 2008. Mutations close to functional motif IV in HSV-1 UL5 helicase that confer resistance to HSV helicase-primase inhibitors, variously affect virus growth rate and pathogenicity. Antiviral Res. 80:81–85. 10.1016/j.antiviral.2008.04.005 [DOI] [PubMed] [Google Scholar]
- 19.Schmidt S, Barbour A, Sahre M, Rand KH, Derendorf H. 2008. PK/PD: new insights for antibacterial and antiviral applications. Curr. Opin. Pharmacol. 8:549–556. 10.1016/j.coph.2008.06.010 [DOI] [PubMed] [Google Scholar]
- 20.Schuck EL, Derendorf H. 2005. Pharmacokinetic/pharmacodynamic evaluation of anti-infective agents. Expert Rev. Anti Infect. Ther. 3:361–373. 10.1586/14787210.3.3.361 [DOI] [PubMed] [Google Scholar]
- 21.Biswas S, Jennens L, Field HJ. 2007. Single amino acid substitutions in the HSV-1 helicase protein that confer resistance to the helicase-primase inhibitor BAY 57-1293 are associated with increased or decreased virus growth characteristics in tissue culture. Arch. Virol. 152:1489–1500. 10.1007/s00705-007-0964-7 [DOI] [PubMed] [Google Scholar]
- 22.Nagafuchi S, Oda H, Mori R, Taniguchi T. 1979. Mechanism of acquired resistance to herpes simplex virus infection as studied in nude mice. J. Gen. Virol. 44:715–723. 10.1099/0022-1317-44-3-715 [DOI] [PubMed] [Google Scholar]
- 23.Center for Drug Evaluation and Research, Center for Veterinary Medicine. 2001. Bioanalytical method validation. Food and Drug Administration, Rockville, MD [Google Scholar]
- 24.Committee for Medicinal Products for Human Use. 2011. Guideline on bioanalytical method validation. EMEA/CHMP/EWP/192217/2009. European Medicines Agency, London, United Kingdom [Google Scholar]
- 25.Sukla S, Biswas S, Birkmann A, Lischka P, Ruebsamen-Schaeff H, Zimmermann H, Field HJ. 2010. Effects of therapy using a helicase-primase inhibitor (HPI) in mice infected with deliberate mixtures of wild-type HSV-1 and an HPI-resistant UL5 mutant. Antiviral Res. 87:67–73. 10.1016/j.antiviral.2010.04.008 [DOI] [PubMed] [Google Scholar]
- 26.Brett M, Weimann HJ, Cawello W, Zimmermann H, Pabst G, Sierakowski R, Gieschke R, Baumann A. 1999. Half-life, p 39–63 In Cawello W. (ed), Parameters for compartment free pharmacokinetics: standardisation of study design, data analysis and reporting. Shaker Verlag, Aachen, Germany [Google Scholar]