

Abstract
Ribonucleotide analog inhibitors of the RNA-dependent RNA polymerase of hepatitis C virus (HCV) represent one of the most exciting recent developments in HCV antiviral therapy. Although it is well established that these molecules cause chain termination by competing at the triphosphate level with natural nucleotides for incorporation into elongating RNA, strategies to rationally optimize antiviral potency based on enzyme kinetics remain elusive. In this study, we used the isolated HCV polymerase elongation complex to determine the pre-steady-state kinetics of incorporation of 2′F-2′C-Me-UTP, the active metabolite of the anti-HCV drug sofosbuvir. 2′F-2′C-Me-UTP was efficiently incorporated by HCV polymerase with apparent Kd (equilibrium constant) and kpol (rate of nucleotide incorporation at saturating nucleotide concentration) values of 113 ± 28 μM and 0.67 ± 0.05 s−1, respectively, giving an overall substrate efficiency (kpol/Kd) of 0.0059 ± 0.0015 μM−1 s−1. We also measured the substrate efficiency of other UTP analogs and found that substitutions at the 2′ position on the ribose can greatly affect their level of incorporation, with a rank order of OH > F > NH2 > F-C-Me > C-Me > N3 > ara. However, the efficiency of chain termination following the incorporation of UMP analogs followed a different order, with only 2′F-2′C-Me-, 2′C-Me-, and 2′ara-UTP causing complete and immediate chain termination. The chain termination profile of the 2′-modified nucleotides explains the apparent lack of correlation observed across all molecules between substrate efficiency at the single-nucleotide level and their overall inhibition potency. To our knowledge, these results provide the first attempt to use pre-steady-state kinetics to uncover the mechanism of action of 2′-modified NTP analogs against HCV polymerase.
INTRODUCTION
Hepatitis C virus (HCV) is believed to have infected approximately 175 million individuals worldwide, with an estimated 2 to 4 million new infections each year (1). In the United States, approximately 3.2 million people are chronically infected with HCV (Centers for Disease Control and Prevention [http://www.cdc.gov/hepatitis/HCV/Index.htm]). The primary mode of transmission for HCV is via exposure to infected blood, including transfusions from infected donors, and through intravenous use of illicit drugs. Although a minority of all HCV infections will spontaneously resolve without any clinical outcome, an estimated 80% of cases will progress into chronic hepatitis, leading to a significant proportion of cirrhosis and cases of hepatocellular carcinoma (2). This makes HCV the leading cause of liver transplantation in the United States.
Hepatitis C virus is a member of the Flaviviridae family (3). It contains a single, positive-strand RNA genome of about 9.5 kb. The viral genome encodes only one open reading frame translated to a polyprotein of approximately 3,000 amino acids. The NS5B protein is composed of 591 amino acids that are cleaved at the C-terminal end of the polyprotein. NS5B acts as the RNA-dependent RNA polymerase (RdRp), with critical functions in RNA replication and transcription. Similar to other known RdRps, NS5B contains six conserved motifs, designated A through F. The amino acids involved in the catalytic activity of NS5B are located within motif A (aspartate at position 220) and in the catalytic triad GDD at positions 318 to 320 in motif C (4). The orientation of these residues in the active site of NS5B and their contribution to the catalytic activity was supported by the crystal structure of the soluble form of NS5B truncated from its C-terminal transmembrane region (delta-21) (4–6). Using the polymerase right-hand analogy model, the HCV NS5B delta-21 protein also features finger, palm, and thumb subdomains. Unlike the traditional open-hand conformation shared by many DNA polymerases, HCV NS5B has an encircled active site due to extensive interactions between the finger and thumb subdomains. These contacts are believed to restrict the flexibility of the subdomains and favor the first steps, or initiation, of RNA synthesis leading to the formation of the primer strand. Therefore, it is believed that primer extension by NS5B during the elongation step requires important structural changes involving an opening of the thumb and fingers (7, 8). The capacity to experimentally trap and characterize the elongation complex (EC) has been a significant milestone to help understand replication of RNA by HCV NS5B (9, 10). Under the elongation mode, the average rate of nucleotide incorporation by NS5B EC is about 5 to 20 bases per second, in agreement with previous estimated rates of 200 to 700 bases per minute for NS5B replicating longer RNA templates (11, 12).
Several nucleotide analogs inhibiting HCV NS5B have been reported (13–22). These inhibitors must enter liver cells as nonphosphorylated nucleosides (or as monophosphate prodrugs) before being converted to the active triphosphates by cellular kinases. Although it is well established that nucleotide analogs compete with the natural substrate for incorporation into viral RNA and cause chain termination, strategies to rationally optimize antiviral potency based on enzyme kinetics have been elusive.
In this study, we isolated the stalled NS5B EC to measure the efficiency of incorporation of modified nucleotides with a 3′-OH group under single-turnover conditions. The active metabolite of the anti-HCV drug sofosbuvir, β-d-2′-deoxy-2′-α-fluoro-2′-β-C-methyluridine triphosphate (2′F-2′C-Me-UTP), was used as the benchmark because it is a clinically relevant molecule that is known to inhibit HCV polymerase without causing significant toxicity in vitro and in vivo (23). By comparing 2′F-2′C-Me-UTP to other 2′-substituted nucleotides in our polymerase assay, we found that substitutions at the 2′ position on the sugar moiety had a profound impact not only on the efficiency of incorporation of the nucleotide analog itself but also on the incorporation of the next correct nucleotide. These two kinetic parameters reconciled the differences in inhibition potencies (50% inhibitory concentrations [IC50s]) found among nucleotide analogs. To our knowledge, these results provide the first detailed mechanistic study of the role of 2′-position modifications on the efficiency of incorporation and chain termination of nucleotide analogs used against HCV polymerase.
MATERIALS AND METHODS
Chemicals, nucleic acids, and protein.
All ultrapure-grade nucleoside triphosphates (NTPs) were purchased from TriLink Biotechnologies (San Diego, CA) or synthesized at Alios BioPharma Inc. (South San Francisco, CA). Tritiated nucleotides were purchased from PerkinElmer (Waltham, MA). Heparin sodium salt (195.9 USP units/mg) was purchased from Sigma-Aldrich (St. Louis, MO). Solutions of MgCl2, EDTA, NaCl, and Tris-Cl buffers were purchased from Ambion (Austin, TX). Dithiothreitol (DTT) was purchased from Sigma-Aldrich (St. Louis, MO). Trichloroacetic acid was purchased from Fisher Scientific (Waltham, MA). The 20-mer RNA templates for single UMP incorporation (3′-CCUCUCUUCGACCUCUCUCC-5′), single GMP incorporation (3′-CCUAUAUUAGCAAUAUCUAA-5′), and 5′ monophosphorylated dinucleotide primer (pGG) were chemically synthesized by Dharmacon Inc. (Chicago, IL). The N-terminal hexa-His-tagged NS5BΔ21 (BK strain, GT1b) was cloned, expressed, and purified by Emerald Bio (Bainbridge Island, WA) in a manner similar to that previously described for a β-hairpin loop deletion NS5B construct (JFH-1 isolate, GT2a) (24).
Isolation of the NS5B EC.
The primer extension and pause reaction mixture containing 6.5 μM NS5B, 20 μM pGG, 20 μM RNA template, 25 μM ATP, and 12.5 μM GTP in the optimized reaction buffer (40 mM Tris-Cl, pH 7.0, 40 mM NaCl, 5 mM DTT, and 2 mM MgCl2) was run for 2 h at 30°C to generate a 9-mer, followed by the addition of 20 μM CTP containing 13 nM 33P-radiolabeled CTP, and incubated for 30 s at 30°C to generate a 10-mer. As previously discovered (9, 25), all of the active EC was pelleted during the reaction and isolated by centrifugation at 16,000 × g for 5 min at room temperature using a benchtop centrifuge (model 5415 D; Eppendorf). After centrifugation, the supernatant was removed and the remaining pellet was washed twice by additional resuspension in the wash buffer (40 mM Tris-Cl, pH 7.0, 20 mM NaCl, 5 mM DTT, and 2 mM MgCl2) and centrifugation to remove residual contaminants. The final pellet containing the EC was resuspended and solubilized in a physiologically relevant reaction buffer (40 mM Tris-Cl, pH 7.4, 5 mM DTT, 2 mM MgCl2, and 150 mM NaCl) to serve as the isolated EC for this study. The supernatant or the resuspended pellet was mixed with the quench buffer (90% formamide, 50 mM EDTA, 0.1% bromophenol blue, and 0.1% xylene cyanol) and analyzed by polyacrylamide gel electrophoresis (PAGE). To test the processivity of the NS5B-RNA complex, heparin (0.2 mg/ml) was added either at the start of the extension and pause reaction or after purification of the NS5B EC, followed by the addition of ATP, GTP, and UTP (40 μM each) and incubation for 30 s at 30°C.
Measurement of K1/2 (concentration of NTP at which half of the total percentage of the product was formed) and individual Kd (equilibrium constant) and kpol (rate of nucleotide incorporation at saturating nucleotide concentration) values using single-nucleotide incorporation.
For single UMP incorporation, UTP was added at up to 10 μM to the isolated EC for 20, 40, 60, or 80 s at 30°C. For single UMP analog incorporation, each UTP analog was added at up to 100 μM to the EC for 5 min. For single GMP incorporation, GTP was added to the EC (with CMP on the template) at up to 10 μM for 10, 20, 30, or 60 s at 30°C. The reactions were stopped by mixing with quench buffer. The time courses of single 2′F-2′C-Me-UMP incorporation at various 2′F-2′C-Me-UTP concentrations were performed to measure the pre-steady-state kinetic parameters of 2′F-2′C-Me-UMP incorporation. A solution containing 2′F-2′C-Me-UTP at 0.92, 1.7, 8.2, 24, 74, 222, 667, or 2,000 μM was mixed with isolated EC to start the incorporation reactions at 30°C. At 5, 10, 20, 40, 80, 160, and 320 s, the reactions were stopped by mixing with quench buffer. Calculation of rate constants was performed as detailed in “Data analysis,” below.
Inhibition potency assay under steady-state kinetics.
The HCV polymerase activity was measured as the incorporation of radiolabeled nucleotide monophosphates into acid-insoluble RNA products using HCV NS5B and complementary internal ribosome entry site (IRES)-derived RNA templates as described previously (14), with the following modifications: HCV polymerase reaction mixtures contained 50 nM 5′-untranslated region (UTR) RNA template, 1 μM tritiated CTP (18.8 Ci/mmol), 1 μM ATP, 1 μM GTP, 0.5 μM UTP, 40 mM Tris-HCl (pH 8.0), 20 mM NaCl, 3 mM DTT, 4 mM MgCl2, serial diluted inhibitor, and 100 nM NS5B enzyme in 96-well MultiScreen plates (EMD Millipore, Billerica, MA). Reaction mixtures were incubated for 2 h at 30°C and stopped by the addition of equal volumes of 20% (vol/vol) trichloroacetic acid. Microscint-20 (PerkinElmer, Waltham, MA) was added to the acid-insoluble RNA products and read on a MicroBeta Trilux (PerkinElmer, Waltham, MA).
Chain termination in the presence of the next correct nucleotides.
After the NS5B EC with the 9-mer RNA was assembled in the extension and pause reaction, 20 μM CTP containing 33 nM 33P-radiolabeled CTP was added to the reaction and reacted for 30 s at 30°C to generate the EC containing a 10-mer RNA with [α-33P]CMP at the 3′ end. The 10-mer EC then was incubated with either 100 μM UTP for 40 s or 100 μM UTP analog for 5 min to generate an 11-mer RNA with the U analog at the 3′ end. The complex was isolated by centrifugation as described above. To test for chain termination, the solution containing the 11-mer EC was mixed with ATP and GTP, each at 100 μM for 20 s, and then stopped by the addition of quench buffer.
Efficiency of GTP incorporation after U analogs.
After the NS5B EC with the 9-mer RNA was assembled in the extension and pause reaction, 33 nM [α-33P]CTP was added to the reaction and reacted for 30 s at 30°C to generate the EC containing a 10-mer RNA with [α-33P]CMP at its 3′ end. The complex was isolated by centrifugation as described above. The isolated EC was reacted with each UTP analog at 100 μM for 5 min to generate the 11-mer RNA EC with the U analog at its 3′ end. The solution containing this EC was mixed with GTP at various concentrations to start the incorporation reactions. After incubation for a fixed time period, the reactions were stopped by the addition of quench buffer.
Product analysis for gel-based assays.
The quenched reactions were denatured for 3 min at 95°C before electrophoresis. The samples were loaded onto a 22% denaturing polyacrylamide gel with 7 M urea (National Diagnostics, Atlanta, GA), and electrophoresis was performed at 80 W using a Sequi-Gen GT system from Bio-Rad (Hercules, CA). Gels were dried at 80°C for 1 h with a model 583 gel drier (Bio-Rad, Hercules, CA). Dried gels were exposed to storage phosphor screens and visualized by a Typhoon 9400 scanner (GE Healthcare, Piscataway, NJ). Intensities of the substrate and product bands on the gel were quantified using ImageQuant software (version 5.2; GE Healthcare, Piscataway, NJ), and the percentage of RNA products was calculated by dividing the intensity of the product by the sum of the intensity of the substrate and the product band for each lane.
Simulation of the single-turnover, single-nucleotide incorporation reaction.
The progress curves of the single-turnover, single-nucleotide incorporation were obtained by simulation using KinTek Explorer software (KinTek Inc., Austin, TX) (26). The two-step nucleotide incorporation mechanism (Fig. 1A) was used for the simulation.
FIG 1.
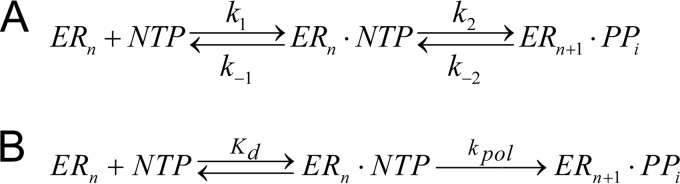
Two-step nucleotide incorporation mechanism. (A) ERn is defined as the enzyme/primer/template complex, NTP is the nucleotide triphosphate substrate, and ERn + 1 is the product of the reaction that contains the primer extended by one nucleotide. For the simulation, k1 = 100 μM−1 s−1, k−1 = 10,000 s−1, k2 = 1 s−1, and k−2 = 0 were applied. The initial concentration for ERn was set at 0.1 μM, and the starting concentrations for NTP were set at various points (800, 400, 200, 100, 50, 25, 12.5, 6.25, 3.125, 1.56, and 0.78 μM). The output for the simulation is the percentage of ERn + 1 in the initial concentration of ERn. The simulated progress curves of the percentage of ERn + 1 increase then were generated at up to 60 s and are shown in Fig. 5A. (B) Classic two-step reaction mechanism for a polymerase reaction.
Data analysis.
Single-turnover nucleotide incorporation experiments were repeated at least two times, and the data were analyzed by nonlinear regression using the program Prism 5 (GraphPad Software, La Jolla, CA). Nucleotide incorporation data at various nucleotide concentrations at selected time points were fitted to the Hill equation:
| (1) |
where Y is the percentage of RNA product, S is the concentration of the testing nucleotide or nucleotide analog, %Min is the offset for data fitting that accounts for background and misincorporated products, %Max − %Min is the total percentage of the product formed during the reaction, and K1/2 is the concentration of NTP at which half of the total percentage of the product was formed. The substrate efficiency or specificity parameter, kpol/Kd, was calculated from K1/2 by the following equation (see the supplemental material for equation derivation):
| (2) |
where Δt is the time of the reaction.
Each time course of 2′F-2′C-Me-UMP incorporation was fit to a single exponential equation to derive the apparent rate of incorporation, k (equation 3). The apparent rate of incorporation was then plotted against nucleotide concentration, and the data were fit to a hyperbola equation (equation 4) to derive the values of kpol and Kd.
| (3) |
| (4) |
The level of discrimination of NTP analogs compared to that of natural NTPs by the polymerase was calculated as (kpol/Kd,NTP)/(kpol/Kd,NTP analog).
RESULTS
Isolation of HCV polymerase in the elongation mode.
The RdRp reaction leading to a 10-mer product was initiated by incubating the HCV polymerase NS5B with a dinucleotide GG primer (Fig. 2A). The incorporation of a single [33P]CMP molecule opposite template G at position 10 allows the radiolabeling of the 10-mer product; thus, the labeled RNA molecules can be analyzed by phosphorimaging. The initiation of primer synthesis by RNA polymerases is slow and inefficient. The lack of processivity during the initiation step was supported by the fact that addition of heparin at the beginning of the reaction completely inhibited product formation (Fig. 2B). However, formation of the 20-mer full-length RNA product from the 10-mer complex was resistant to heparin inhibition, which is indirect evidence of NS5B reaching the fast and processive elongation mode (Fig. 2A and B). As previously reported (9), we could separate the stalled enzyme-RNA elongation complex (EC) from free protein and unreacted substrates by sedimentation (Fig. 2C). Isolation and purification of the EC formed by NS5B in complex with a primer-template provided the basis for single-nucleotide incorporation experiments under pre-steady-state kinetics.
FIG 2.

Isolating the NS5B elongation complex (EC). (A) Principle of the initiation, labeling, and elongation reactions. Initiation of RNA synthesis by NS5B was performed in the presence of a GG-dinucleotide primer together with ATP and GTP for 2 h. Because of the template sequence, the NS5B EC was stalled at the 9-mer position. To track the RNA in the EC by phosphorimaging, 3′-end isotope labeling was performed by incorporation of [33P]CMP at the 10-mer position. The fast elongation of RNA synthesis could resume by adding UTP (1 min or less). The boldfaced “C” represents the radiolabeled nucleoside. (B) Processivity of NS5B complex during initiation and elongation. The enzyme processivity during 10-mer and 20-mer formation was monitored by adding heparin either at the beginning of the initiation reaction or together with UTP after the 10-mer EC was already formed. The error bars represent the standard deviations from two independent experiments. (C) High-resolution Tris-borate-EDTA–urea gel electrophoresis showing the formation of the radiolabeled 10-mer RNA product of NS5B activity in the presence of the [33P]CTP tracer (lane 1). After centrifugation of the reaction product for 5 min at 13,000 × g, the supernatant did not contain any 10-mer product (lane 2). The EC containing the 10-mer product formed a precipitate that could be washed two times with low-salt buffer and then redissolved in high-salt buffer (lane 3).
Pre-steady-state kinetics of 2′F-2′C-Me-UMP incorporation.
The minimum kinetic pathway for nucleotide incorporation catalyzed by HCV NS5B polymerase EC follows the classic two-step reaction mechanism for a polymerase reaction (9) (Fig. 1B), where Kd is the equilibrium dissociation constant for nucleotide binding, kpol is the rate of nucleotide incorporation at saturating nucleotide concentration, and kpol/Kd is the parameter that defines the overall specificity or catalytic efficiency of nucleotide incorporation, which is the key parameter we used for the rest of this study.
For the study of nucleotide incorporation under single-turnover conditions, the preformed HCV polymerase EC stalled at the 10-mer position was rapidly mixed with a reaction buffer containing β-d-2′-deoxy-2′-α-fluoro-2′-β-C-methyluridine triphosphate (2′F-2′C-Me-UTP) at various concentrations (Fig. 3A). The time courses of 11-mer formation were analyzed by data fitting to a single exponential equation to derive the apparent rates of incorporation at various concentrations of 2′F-2′C-Me-UTP (Fig. 3B). The rates were plotted against the concentrations, and the data were fit to a hyperbola equation to derive Kd and kpol. The Kd and kpol values for 2′F-2′C-Me-UTP were 113 ± 28 μM and 0.67 ± 0.05 s−1, respectively, giving an overall substrate efficiency, kpol/Kd, of 0.0059 ± 0.0015 μM−1 s−1 (Fig. 3C).
FIG 3.

Pre-steady-state kinetics of incorporation of 2′F-2′C-Me-UMP. (A) Principle of the reaction. Once the stalled 10-mer EC was isolated, increasing concentrations of 2′F-2′C-Me-UTP at 0.46 (●), 1.4 (■), 4.1 (▲), 12.4 (▼), 37 (◆), 111 (○), 333 (□), and 1,000 μM (◇) were added to start the 10- to 11-mer extension reactions. The reactions were quenched by adding formamide with 50 mM EDTA at various time points. The boldfaced and underlined “C” represents the radiolabeled nucleoside. The box around “A” represents the opposing nucleoside to the incoming nucleotide. (B) Time courses of 11-mer formation at various 2′F-2′C-Me-UTP concentrations. Each time course was fit to a single exponential equation to obtain the rate of analog incorporation at each concentration. (C) The rates of incorporation were plotted against analog concentrations, and the data were fit to a hyperbola equation to derive the maximum rate of 2′F-2′C-Me-UMP incorporation, with a kpol of 0.67 ± 0.05 s−1 and a dissociation equilibrium constant (Kd) of 113 ± 28 μM. The calculated overall catalytic efficiency (kpol/Kd) for 2′F-2′C-Me-UMP incorporation is 0.0059 ± 0.0015 μM−1 s−1. The error bars represent the standard deviations from two independent experiments.
Chain termination caused by incorporated 2′F-2′C-Me-UMP.
Chain termination can be defined as the incapacity of a polymerase, following incorporation of a nucleotide analog, to resume nucleic acid synthesis in the presence of the next correct nucleotide. In our assay, the NS5B EC was first stalled to the 11-mer in the presence of either natural UTP or of 2′F-2′C-Me-UTP before the addition of the next correct nucleotide (Fig. 4A). In the case of natural UTP, primer extension was fully resumed by adding GTP and ATP for 20 s, as judged by the formation of the full-length 20-mer product (Fig. 4B). In comparison, the incorporation of 2′F-2′C-Me-UMP fully prevented the extension of the stalled 11-mer product in the presence of the next correct nucleotide for 20 s.
FIG 4.
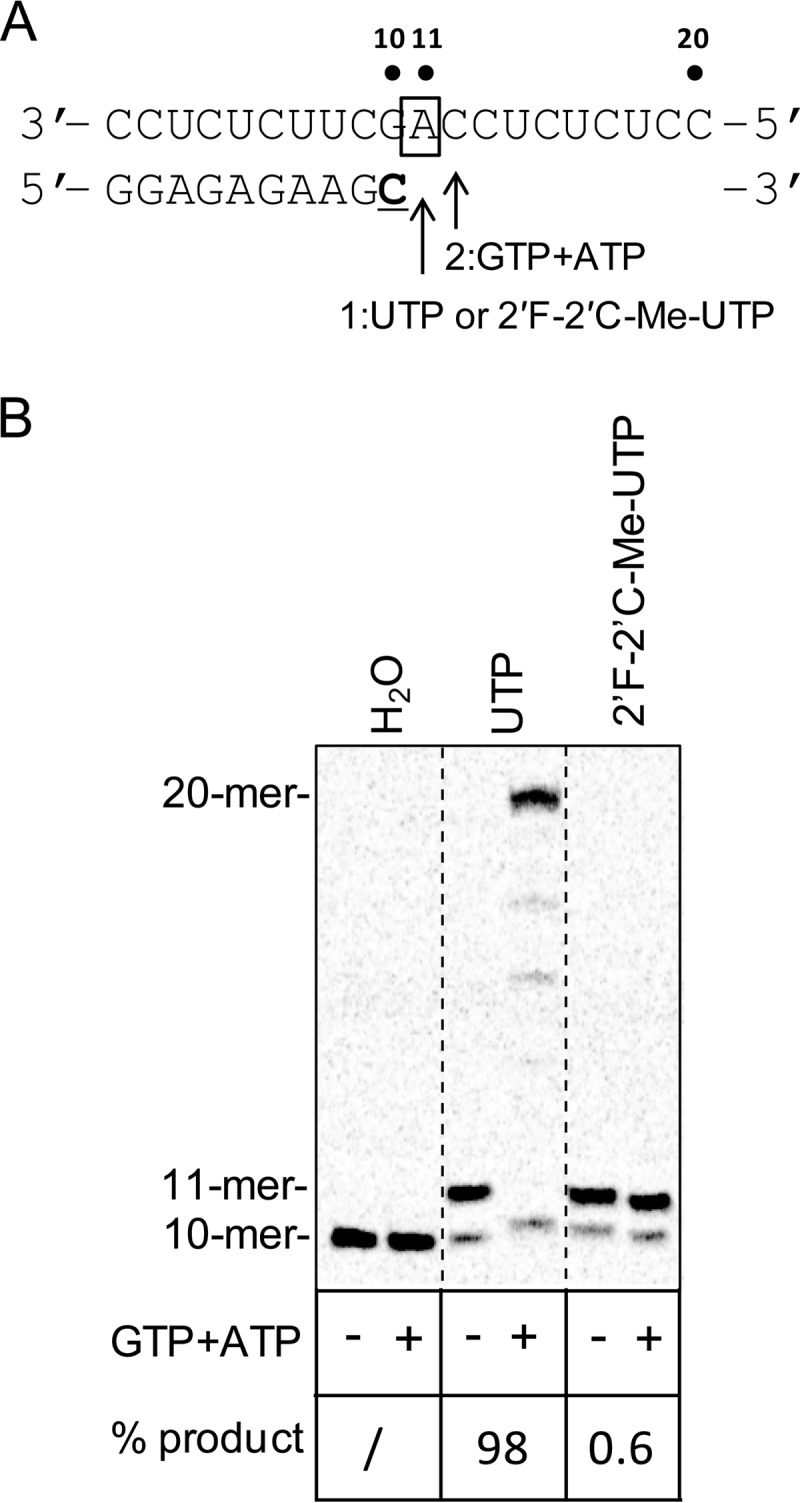
Chain termination with 2′F-2′C-Me-UMP. (A) Principle of the reaction. Once the stalled 10-mer EC was isolated, adding either UTP (40 s) or 2′F-2′C-Me-UTP (5 min) enabled the formation of the 11-mer product. After this step, GTP and ATP were added for 20 s as the next correct nucleotides to monitor 20-mer formation. The boldfaced and underlined “C” represents the radiolabeled nucleoside. The box around “A” represents the opposing nucleoside to the incoming nucleotide. (B) High-resolution Tris-borate-EDTA–urea gel electrophoresis showing the extension of 10- to 11-mer RNA products with 2′F-2′C-Me-UTP, followed by the addition of the next correct nucleotides (+). The formation of RNA products longer than the 11-mer was expressed as the percentage of the initial amount of 11-mer formed for either UTP or 2′F-2′C-Me-UTP in the absence of GTP and ATP (−). “/” indicates that no product was detected.
A simple method for single-turnover kinetics of nucleotide incorporation.
As we have shown with 2′F-2′C-Me-UTP, the determination of kpol and Kd under single-turnover enzyme reaction conditions can be obtained by measuring the time courses of product formation at various substrate concentrations. However, this method is not practical to characterize NTP analogs in a high-throughput manner. In this study, we demonstrated that kpol/Kd values can also be calculated from a single-time-point reaction without the need to determine kpol and Kd individually. By simulating the kinetics of the reaction under single-turnover conditions, we observed that at any time point (t1 to t5) the NTP concentration at which 50% products are formed, defined as K1/2, shifts to a lower concentration for longer time periods (Fig. 5A). At any given time, the K1/2 value can be obtained by fitting the data of product formation as a function of NTP concentration using the Hill equation (see Fig. S1 in the supplemental material). The relationship between K1/2 and kpol/Kd follows a simple equation that is dependent on the time of the reaction (see equation 2). As demonstrated in the simulation, the calculated value of kpol/Kd from each time point was similar to the initial value of kpol/Kd set for the reaction (see Fig. S1). As a practical example, single-nucleotide incorporation with the purified NS5B EC was performed using increasing concentrations of natural UTP (Fig. 5B). For each reaction time, ranging from 20 to 80 s, the K1/2 value was used to calculate the catalytic efficiency of UMP incorporation, (kpol/Kd)UTP, as 0.16 ± 0.02 μM−1 s−1 (Fig. 5C and Table 1). While K1/2 values decreased proportionally with the increase of reaction time, the overall catalytic efficiency was independent of the time of reaction. Similar results were obtained with single GMP incorporation opposite template C, with (kpol/Kd)GTP of 0.20 ± 0.04 μM−1 s−1 (Table 2).
FIG 5.

Single-turnover kinetics of nucleotide incorporation. (A) Simulated progress curves of product formation at various nucleotide concentrations. Single-nucleotide incorporation was simulated using arbitrary values of Kd of 100 μM and kpol of 1 s−1, resulting in a theoretical specificity or substrate efficiency, kpol/Kd, of 0.01 s−1 μM−1. (B) Single incorporation of UMP opposite template A. High-resolution Tris-borate-EDTA–urea gel electrophoresis shows the extension of 10- to 11-mer RNA products. Increasing concentrations of UTP ranging from 0.0015 to 10 μM were added to start the extension reaction. Four different time points were measured to evaluate the effect of time on K1/2 and on kpol/Kd. The boldfaced “C” represents the radiolabeled nucleoside. The box around “A” represents the opposing nucleoside to the incoming nucleotide. (C) Quantitative analysis of product formation. For each time point, percent UMP incorporation was calculated and plotted as a function of UTP concentration. The data were fit to a sigmoidal dose-response equation (equation 1) to obtain K1/2 values, as reported in Table 1.
TABLE 1.
Enzymatic efficiency of single UMP incorporation opposite adenosine (U·A)a
| Time point (s) | K1/2 (μM) | kpol/Kd (μM−1 s−1) | Mean kpol/Kd (μM−1 s−1) |
|---|---|---|---|
| 20 | 0.2 | 0.18 | |
| 40 | 0.12 | 0.15 | 0.16 ± 0.02 |
| 60 | 0.08 | 0.14 | |
| 80 | 0.05 | 0.16 |
The RNA template was 3′-CCUCUCUUCGACCUCUCUCC-5′.
TABLE 2.
Enzymatic efficiency of single GMP incorporation opposite cytidine (G·C)a
| Time point (s) | K1/2 (μM) | kpol/Kd (μM−1 s−1) | Mean kpol/Kd (μM−1 s−1) |
|---|---|---|---|
| 10 | 0.44 | 0.16 | |
| 20 | 0.14 | 0.24 | 0.20 ± 0.04 |
| 30 | 0.097 | 0.24 | |
| 60 | 0.066 | 0.17 |
The RNA template was 3′-CCUAUAUUAGCAAUAUCUAA-5′.
Relationship between substrate efficiency (kpol/Kd) and inhibition potency (IC50) of 2′-modified nucleotide analogs.
We investigated whether substrate incorporation efficiency within a chemical series plays a role in defining the inhibition potency of nucleotide analogs. Five 2′-modified UTP analogs were selected for single-nucleotide incorporation experiments (Fig. 6A). In addition, 3′-dUTP was used as a control because it is an obligate chain terminator. All of the other nucleotides have a hydroxyl moiety at the 3′ position (3′-OH), which potentially enables further primer extension once the nucleotide analog is incorporated into the RNA. The incorporation of each UTP analog was measured at a single time point with increasing concentrations of the analog, and the corresponding K1/2 measurement was used to derive kpol/Kd values (Table 3). Compared to those for natural UTP, we found a wide range of catalytic efficiencies, from 0.029 ± 0.005 μM−1 s−1 for 2′F-UTP (6-fold discrimination) to 0.000076 ± 0.000010 μM−1 s−1 for 2′N3-UTP (2,105-fold discrimination). Removing the 3′-OH group from UTP (3′-dUTP) caused a 114-fold loss in catalytic efficiency. For 2′F-2′C-Me-UTP, the catalytic efficiency of 0.0037 ± 0.0018 μM−1 s−1 using the K1/2 method (Table 3) was similar to the kpol/Kd value of 0.0059 μM−1 s−1 that was obtained by measuring kpol and Kd individually (Fig. 3C).
FIG 6.

Differences between substrate efficiency and inhibition potency of nucleotide analogs. (A) Chemical structures of nucleotide analogs. 3′dUTP was used as a control for chain termination. All of the other molecules contain a 3′OH group, with a modification at the 2′ position: OH, F, NH2, C-Me, N3, or ara. (B) Substrate efficiency (kpol/Kd) for each NTP analog was measured from single-nucleotide incorporation experiments, and discrimination levels were calculated as (kpol/Kd,UTP)/(kpol/Kd,UTP analog). Inhibition potency, expressed as IC50, was measured with a long RNA template under steady-state conditions where each UTP analog competes against natural UTP. The discrimination values and IC50s for each nucleotide can be found in Table 3.
TABLE 3.
Comparison between substrate efficiency and inhibition potency of UTP analogs
| UTP analog | kpol/Kda (μM−1 s−1) | Discriminationb | IC50 (μM) |
|---|---|---|---|
| 3′d | 0.0014 ± 0.0002 | 114 | 0.35 ± 0.03 |
| 2′F | 0.029 ± 0.005 | 6 | >1,000 |
| 2′NH2 | 0.0053 ± 0.0017 | 30 | >1,000 |
| 2′N3 | 0.000076 ± 0.000010 | 2105 | >1,000 |
| 2′ara | 0.00024 ± 0.00001 | 667 | 2.8 ± 0.6 |
| 2′C-Me | 0.0027 ± 0.0002 | 59 | 0.25 ± 0.04 |
| 2′F-2′C-Me | 0.0037 ± 0.0018 | 45 | 0.21 ± 0.05 |
The values of kpol/Kd for UTP and UTP analog incorporation were calculated from experimentally obtained K1/2 values using equation 2, and the means ± standard deviations from more than two repeats are reported.
Calculated as (kpol/Kd,UTP)/(kpol/Kd,UTP analog).
The inhibition potency (IC50) of each UTP analog was also determined in a radiometric assay under steady-state kinetics in the presence of competing UTP and with [3H]CTP as the tracer. In this assay, a long RNA template allows multiple incorporations of each NTP. The incorporation of an obligate chain terminator such as 3′-dUTP resulted in potent inhibition of the reaction, with an IC50 of 0.35 ± 0.03 μM (Table 3). Under these conditions, 2′F-2′C-Me-UTP and 2′C-Me-UTP also inhibited NS5B with similar IC50s of 0.21 ± 0.05 and 0.25 ± 0.04 μM, respectively. By comparison, 2′ara-UTP was significantly less potent, with an IC50 of 2.8 ± 0.6 μM. The other molecules were inactive (2′F-, 2′NH2-, and 2′N3-UTP had IC50s of >1,000 μM). Notably, we could not establish any correlation between the efficiency of incorporation of 2′-modified UTP analogs and their inhibition potency (Fig. 6B). We investigated further if changes in the efficiency of chain termination could be responsible for this lack of correlation.
Chain termination with 2′-modified nucleotides and efficiency of incorporation of the next correct nucleotide.
Similar to what was originally done with 2′F-2′C-Me-UTP (Fig. 4A), the NS5B EC was first stalled as the 11-mer in the presence of each UTP analog before adding the next correct nucleotides, GTP and ATP, for 20 s. As expected, the incorporation of 3′-dUMP fully prevented the extension of the stalled 11-mer product in the presence of the next correct nucleotides (Fig. 7). Similarly, both 2′ara-UMP and 2′C-Me-UMP fully inhibited the formation of the 20-mer product. However, 2′F-, 2′NH2-, and 2′N3-UMP all supported at least 90% extension of the 11-mer stalled primer.
FIG 7.

Chain termination in the presence of the next correct nucleotide. High-resolution Tris-borate-EDTA–urea gel electrophoresis showing the extension of 10- to 11-mer RNA products with UTP or UTP analogs, followed by the addition of the next correct nucleotides (+), following the same scheme as that explained in the legend to Fig. 4A. Formation of RNA products longer than the 11-mer was expressed as the percentage of the initial amount of 11-mer formed for each UTP analog in the absence of GTP and ATP (−). “/” indicates that no product was detected.
In order to uncover subtle differences in chain termination properties of the nucleotide analogs, we also developed a quantitative assay aimed at measuring the efficiency of incorporation of the next correct nucleotides. Each UTP analog (or natural UTP) was used to stall the NS5B EC as an 11-mer product (Fig. 8A). GTP then was added at various concentrations for up to 20 min, and the amount of 12- and 13-mer formation was monitored. The incorporation of the next correct GMP to the RNA following natural UMP was efficient, as supported by the (kpol/Kd)GTP after UMP value of 1.2 ± 0.5 μM−1 s−1 (Fig. 8B). By comparison, the efficiency of incorporation of GMP following 2′F-UMP did not decrease significantly, with a modest 1.6-fold discrimination from GMP incorporation following a natural UMP (Fig. 8B and Table 4). Interestingly, the 2′N3 and 2′NH2 substitutions resulted in a 4.1- and 16-fold, respectively, discrimination of the incorporation of the next correct GMP (Table 4). As expected, 2′ara-UMP prevented the incorporation of the next GMP, with a (kpol/Kd)GTP after 2′ara-UMP value of 0.00009 ± 0.00001 μM−1s−1. Of all the 2′-modified nucleotide analogs we tested, 2′F-2′C-Me-UMP and 2′C-Me-UMP caused the most dramatic effect: no GMP incorporation was detected at up to 1 mM GTP for up to 20 min, resulting in a discrimination level of >2 × 106 for both compounds compared to that of natural UMP (Fig. 8C and Table 4).
FIG 8.

Kinetics of next-correct-nucleotide incorporation. (A) Principle of the reaction. Once the stalled 10-mer EC was isolated, adding either the natural UTP (40 s) or a UTP analog (5 min) enabled the formation of the 11-mer product. After this step, GTP at various concentrations was added and reacted for a fixed time. The boldfaced “C” represents the radiolabeled nucleoside. The box around “A” represents the opposing nucleoside to the incoming nucleotide. (B) Gel image showing the GTP concentration-dependent incorporation of one or two consecutive GMPs into the 11-mer RNA containing a UMP or 2′F-UMP at its 3′ end. The sum of the 12- and 13-mer RNA products was expressed as a percentage of the initial amount of 11-mer. The percentage of GMP incorporation product was plotted as a function of GTP concentration after UMP (left) and 2′F-UMP (right). The data were fit to a sigmoid dose-response equation (equation 1) to obtain K1/2 values. The kpol/Kd values for GMP incorporation after UMP and 2′F-UMP were calculated from K1/2 values using equation 2, and the means ± standard deviations from more than two repeats are reported in Table 4. (C) GTP incorporation after 2′F-2′C-Me-UMP and 2′C-Me-UMP terminated RNA. Once the stalled 10-mer EC was isolated, it was mixed with 100 μM UTP analogs for 5 min to form the 11-mer product. After this step, GTP at various concentrations was added and reacted for 20 min. The gel image shows that no GMP incorporation (12-mer) was detected up to 1 mM GTP reaction for 20 min, resulting for both compounds in a discrimination level of >2 × 106 compared to GTP incorporation after natural UMP (Table 4).
TABLE 4.
Incorporation efficiency of the next correct GMPs after incorporated UMP analogs
| Incorporated UMP analogs | kpol/Kda (μM−1 s−1) | Discriminationb |
|---|---|---|
| 2′F | 0.74 ± 0.3 | 1.6 |
| 2′NH2 | 0.076 ± 0.02 | 16 |
| 2′N3 | 0.29 ± 0.1 | 4.1 |
| 2′ara | 0.00009 ± 0.00001 | 13,000 |
| 2′C-Me | <5.8 × 10−7 | >2 × 106 |
| 2′F-2′C-Me | <5.8 × 10−7 | >2 × 106 |
The values of kpol/Kd for GTP incorporation after UMP or UMP analogs were calculated from experimentally obtained K1/2 values using equation 2, and the means ± standard deviations from more than two repeats are reported.
Calculated as (kpol/Kd, GTP after UMP = 1.2 ± 0.5 μM−1 s−1)/(kpol/Kd, GTP after UMP analog).
DISCUSSION
The ability to design nucleotide analogs as substrates for viral polymerases has been the cornerstone to successful development of many antiviral therapeutics. Perhaps the best examples are historically found with HIV reverse transcriptase, an enzyme that is potently inhibited by the triphosphate form of dideoxycytidine, 3′-azidothymidine, 2′,3′-dideoxy-3′-thiacytidine (3TC), or 9-(2-phosphonomethoxypropyl)adenine (PMPA). In these cases, inhibition potency is conferred by the lack of the hydroxyl group at the 3′ position on the sugar moiety of the nucleotide, thereby preventing further DNA primer extension once the nucleotide analog is incorporated. By definition, these molecules are obligate chain terminators. More recently, the emergence of novel RNA polymerase inhibitors revealed an important shift in the paradigm of chain termination: nucleotide analogs containing a 3′-OH group can also act as bona fide chain terminators. For example, inhibition of HCV RNA polymerase can be achieved with 3′-hydroxyl ribonucleotides containing an azido group at the 4′ position (14, 27) or a methyl at the 2′-C position (13, 20, 22, 28). The combination of a C-methyl and a fluoro group at the 2′ position on the sugar, perhaps best exemplified by the 2′-deoxy-2′-fluoro-2′-C-methyl (2′F-2′C-Me) UMP prodrug sofosbuvir, so far has provided some of the most potent and least toxic NS5B polymerase inhibitors (23). However, the ability to introduce substitutions at the 2′ position while retaining substrate recognition has been constrained by the need to maintain an important hydrogen bond formed between the 2′ group and amino acid Asp225 in the active site of NS5B (29). The second limitation to the design of more potent HCV polymerase inhibitors is the lack of understanding of the detailed mechanism of inhibition of ribonucleotide analogs that have been, for the most part, empirically discovered. In spite of the recent high-resolution crystal structure of HCV NS5B in complex with the RNA primer-template duplex (24), the exact positioning of the incoming nucleotide and its interactions with key amino acids during catalysis remain elusive, which has prevented the use of structure-based drug design for this class of inhibitors. Another limitation to the design of ribonucleotide analogs resulted from the lack of a robust in vitro assay system to conduct pre-steady-state kinetics with NS5B-RNA elongation complex (EC), thereby preventing detailed mechanistic studies of nucleotide incorporation. The difficulty in assembling a stable NS5B-RNA EC in vitro stems primarily from the fact that the RdRp, with its β-hairpin loop, adopts a closed conformation that prevents direct binding of a double-stranded RNA as the substrate. In contrast, similar RNA polymerases, such as the ones from poliovirus, norovirus, and foot-and-mouth disease virus, can readily accommodate preformed primers because they do not harbor any equivalent autoinhibitory secondary structure (30–32). This technical limitation was recently solved by optimizing the conditions to trap the stable EC containing HCV NS5B with a primer template and separating the EC from the abortive short RNA products and unincorporated nucleotides (9). Being able to remove the unreacted species (RNA and NTPs) from the EC in order to avoid potential interference in the assay was paramount in order to conduct single-nucleotide incorporation experiments; therefore, this methodology was used as the basis for our study (Fig. 2).
The first goal of the study was to determine the maximum rate of nucleotide incorporation (kpol) and the dissociation equilibrium constant (Kd) for 2′F-2′C-Me-UTP, the active metabolite of sofosbuvir. We used 2′F-2′C-Me-UTP as a benchmark, because it is a clinically relevant molecule that is known to inhibit HCV polymerase (15–17). We found that the NS5B EC only moderately discriminates 2′F-2′C-Me-UTP, with Kd and kpol values of 113 ± 28 μM and 0.67 ± 0.05 s−1 (Fig. 3), respectively, compared to 490 μM and 27 s−1 for natural UTP (10). Under conditions of single-turnover kinetics, we demonstrated that the efficiency of nucleotide incorporation (kpol/Kd) of 2′F-2′C-Me-UMP can be accurately determined from a single reaction time point (0.0037 ± 0.0018 μM−1 s−1) (Table 3). This value is similar to the catalytic efficiency obtained by measuring kpol and Kd individually from time course experiments (0.0059 ± 0.0015 μM−1s−1) (Fig. 3). Using this new method, we also determined the catalytic efficiency of natural UMP and GMP incorporation to be (kpol/Kd)UTP = 0.16 ± 0.017 μM−1s−1 and (kpol/Kd)GTP = 0.20 ± 0.043 μM−1s−1, respectively (Fig. 5 and Tables 1 and 2). These values are within 3-fold of those obtained with conventional pre-steady-state kinetic experiments, further validating our method (9, 10). Our single-reaction-time-point assay enabled us not only to conveniently measure the incorporation efficiency of nucleotides under pre-steady-state kinetics with the NS5B EC but also apply it to other systems where enzyme and compound supply are limited (33).
The second goal of our study was to use transient kinetics to understand the relationship between substrate efficiency and inhibition potency of nucleotide analogs toward HCV NS5B. We chose a series of 2′-modified UTP containing a hydroxyl group at the 3′ position in order to measure the contribution of the 2′ moiety (i) to the extent of discrimination by the EC and (ii) to its ability to cause chain termination. Nucleotide analogs containing the 2′F and 2′NH2 moieties were the most efficient substrates, where discrimination compared to natural UTP was only 6- and 30-fold, respectively. However, none of these molecules were able to inhibit HCV NS5B (IC50, >1 mM) (Table 3). In contrast, 2′F-2′C-Me-, 2′C-Me-, and 2′ara-UTP were less efficiently incorporated. However, these compounds were potent against NS5B, with IC50s of 0.21 ± 0.05 μM for 2′F-2′C-Me-UTP, 0.25 ± 0.04 μM for 2′C-Me-UTP, and 2.8 ± 0.6 μM for 2′ara-UTP (Table 3). Between these three molecules, incorporation efficiency correlated well with inhibition potency (Fig. 6). However, the fact that a very efficient substrate such as 2′F-UTP was unable to inhibit NS5B led us to consider that the nature of the 2′ moiety can also affect efficiency of incorporation of the next incoming nucleotide. This was confirmed by our qualitative and quantitative chain termination assays, where 2′F, 2′NH2, and 2′N3 substitutions in the last nucleotide of the nascent RNA were unable to prevent formation of longer extension products in the presence of the next correct nucleotides, suggesting they are not chain terminators for the EC (Fig. 7 and 8). The fact that 2′F-UTP did not cause any chain termination was surprising, given that 2′F-cytidine had previously been shown to inhibit HCV replication (34). In our hands, 2′F-CTP did not act as an inhibitor against HCV polymerase (see Fig. S2 in the supplemental material). When incorporated by other polymerases, 2′-fluoro-modified nucleotides could significantly reduce the efficiency of polymerization (35, 36). These different results highlight that the 2′-fluoro moiety by itself might cause some level of chain termination. However, given the results presented in this study, we can conclude that the 2′-fluoro substitution contributes less to chain termination than the 2′-C-methyl moiety.
How much reduction of incorporation efficiency for the next nucleotide would be required to cause potent overall inhibition? The incorporation of 2′ara-UMP did not completely prevent the incorporation of the next nucleotide, but its efficiency was reduced by 13,000-fold compared to that of natural UMP (Table 4). At this level of discrimination, we can consider 2′ara-UTP to be a pseudo-obligate chain terminator. Likewise, 2′F-2′C-Me-UMP and 2′C-Me-UMP behaved exactly like 3′d-UMP, with no detectable levels of incorporation of the next correct nucleotide. To our knowledge, this represents the first attempt to provide quantitative analysis of chain termination by measuring the efficiency of incorporation of the next correct nucleotide. It is generally assumed that, in the absence of any proofreading mechanism, nucleotide incorporation by RNA polymerases is irreversible. However, it has recently been reported that HCV NS5B was able to excise the 3′-terminal nucleotide from the RNA (25). A suggested extension of this finding could be to measure the effect of NTP-mediated excision on the stability of incorporation and the inhibition potency of chain terminators, such as those we just characterized. Moreover, studies will be needed to understand at the structural level how subtle modifications on the sugar moiety can influence substrate recognition and chain termination efficiency. The current model provided by crystal structures of free nucleotides shows that although 2′-deoxyribonucleotides typically adopt a 2′-endo conformation, the 2′-fluoro group seems to lock the nucleotide into a 3′-endo (north) position that is similar to that of ribonucleotides (27, 37). However, it remains to be seen how these modifications at the 2′-OH position affect the orientation of the sugar in the context of the ternary complex composed of NS5B, the double-stranded RNA, and the incoming nucleotide. Additional kinetic studies combined with structural analysis would help us understand the mechanism of inhibition of RNA polymerases of important human pathogens.
Supplementary Material
ACKNOWLEDGMENT
We thank Chris Sneeringer for critical reading of the manuscript.
Footnotes
Published ahead of print 14 April 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02666-14.
REFERENCES
- 1.Moradpour D, Penin F, Rice CM. 2007. Replication of hepatitis C virus. Nat. Rev. Microbiol. 5:453–463. 10.1038/nrmicro1645 [DOI] [PubMed] [Google Scholar]
- 2.Alter HJ, Houghton M. 2000. Clinical medical research award. Hepatitis C virus and eliminating post-transfusion hepatitis. Nat. Med. 6:1082–1086. 10.1038/80394 [DOI] [PubMed] [Google Scholar]
- 3.Simmonds P, Bukh J, Combet C, Deleage G, Enomoto N, Feinstone S, Halfon P, Inchauspe G, Kuiken C, Maertens G, Mizokami M, Murphy DG, Okamoto H, Pawlotsky JM, Penin F, Sablon E, Shin IT, Stuyver LJ, Thiel HJ, Viazov S, Weiner AJ, Widell A. 2005. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology 42:962–973. 10.1002/hep.20819 [DOI] [PubMed] [Google Scholar]
- 4.Lesburg CA, Cable MB, Ferrari E, Hong Z, Mannarino AF, Weber PC. 1999. Crystal structure of the RNA-dependent RNA polymerase from hepatitis C virus reveals a fully encircled active site. Nat. Struct. Biol. 6:937–943. 10.1038/13305 [DOI] [PubMed] [Google Scholar]
- 5.Ago H, Adachi T, Yoshida A, Yamamoto M, Habuka N, Yatsunami K, Miyano M. 1999. Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Structure 7:1417–1426. 10.1016/S0969-2126(00)80031-3 [DOI] [PubMed] [Google Scholar]
- 6.Bressanelli S, Tomei L, Roussel A, Incitti I, Vitale RL, Mathieu M, De Francesco R, Rey FA. 1999. Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Proc. Natl. Acad. Sci. U. S. A. 96:13034–13039. 10.1073/pnas.96.23.13034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caillet-Saguy C, Simister PC, Bressanelli S. 2011. An objective assessment of conformational variability in complexes of hepatitis C virus polymerase with non-nucleoside inhibitors. J. Mol. Biol. 414:370–384. 10.1016/j.jmb.2011.10.001 [DOI] [PubMed] [Google Scholar]
- 8.Scrima N, Caillet-Saguy C, Ventura M, Harrus D, Astier-Gin T, Bressanelli S. 2012. Two crucial early steps in RNA synthesis by the hepatitis C virus polymerase involve a dual role of residue 405. J. Virol. 86:7107–7117. 10.1128/JVI.00459-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jin Z, Leveque V, Ma H, Johnson KA, Klumpp K. 2012. Assembly, purification, and pre-steady-state kinetic analysis of active RNA-dependent RNA polymerase elongation complex. J. Biol. Chem. 287:10674–10683. 10.1074/jbc.M111.325530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Powdrill MH, Tchesnokov EP, Kozak RA, Russell RS, Martin R, Svarovskaia ES, Mo H, Kouyos RD, Gotte M. 2011. Contribution of a mutational bias in hepatitis C virus replication to the genetic barrier in the development of drug resistance. Proc. Natl. Acad. Sci. U. S. A. 108:20509–20513. 10.1073/pnas.1105797108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lohmann V, Roos A, Korner F, Koch JO, Bartenschlager R. 1998. Biochemical and kinetic analyses of NS5B RNA-dependent RNA polymerase of the hepatitis C virus. Virology 249:108–118. 10.1006/viro.1998.9311 [DOI] [PubMed] [Google Scholar]
- 12.Tomei L, Vitale RL, Incitti I, Serafini S, Altamura S, Vitelli A, De Francesco R. 2000. Biochemical characterization of a hepatitis C virus RNA-dependent RNA polymerase mutant lacking the C-terminal hydrophobic sequence. J. Gen. Virol. 81:759–767 [DOI] [PubMed] [Google Scholar]
- 13.Carroll SS, Tomassini JE, Bosserman M, Getty K, Stahlhut MW, Eldrup AB, Bhat B, Hall D, Simcoe AL, LaFemina R, Rutkowski CA, Wolanski B, Yang Z, Migliaccio G, De Francesco R, Kuo LC, MacCoss M, Olsen DB. 2003. Inhibition of hepatitis C virus RNA replication by 2′-modified nucleoside analogs. J. Biol. Chem. 278:11979–11984. 10.1074/jbc.M210914200 [DOI] [PubMed] [Google Scholar]
- 14.Klumpp K, Leveque V, Le Pogam S, Ma H, Jiang WR, Kang H, Granycome C, Singer M, Laxton C, Hang JQ, Sarma K, Smith DB, Heindl D, Hobbs CJ, Merrett JH, Symons J, Cammack N, Martin JA, Devos R, Najera I. 2006. The novel nucleoside analog R1479 (4′-azidocytidine) is a potent inhibitor of NS5B-dependent RNA synthesis and hepatitis C virus replication in cell culture. J. Biol. Chem. 281:3793–3799. 10.1074/jbc.M510195200 [DOI] [PubMed] [Google Scholar]
- 15.Ma H, Jiang WR, Robledo N, Leveque V, Ali S, Lara-Jaime T, Masjedizadeh M, Smith DB, Cammack N, Klumpp K, Symons J. 2007. Characterization of the metabolic activation of hepatitis C virus nucleoside inhibitor beta-d-2′-deoxy-2′-fluoro-2′-C-methylcytidine (PSI-6130) and identification of a novel active 5′-triphosphate species. J. Biol. Chem. 282:29812–29820. 10.1074/jbc.M705274200 [DOI] [PubMed] [Google Scholar]
- 16.Murakami E, Bao H, Ramesh M, McBrayer TR, Whitaker T, Micolochick Steuer HM, Schinazi RF, Stuyver LJ, Obikhod A, Otto MJ, Furman PA. 2007. Mechanism of activation of beta-d-2′-deoxy-2′-fluoro-2′-c-methylcytidine and inhibition of hepatitis C virus NS5B RNA polymerase. Antimicrob. Agents Chemother. 51:503–509. 10.1128/AAC.00400-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murakami E, Niu C, Bao H, Micolochick Steuer HM, Whitaker T, Nachman T, Sofia MA, Wang P, Otto MJ, Furman PA. 2008. The mechanism of action of beta-d-2′-deoxy-2′-fluoro-2′-C-methylcytidine involves a second metabolic pathway leading to beta-d-2′-deoxy-2′-fluoro-2′-C-methyluridine 5′-triphosphate, a potent inhibitor of the hepatitis C virus RNA-dependent RNA polymerase. Antimicrob. Agents Chemother. 52:458–464. 10.1128/AAC.01184-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reddy PG, Bao D, Chang W, Chun BK, Du J, Nagarathnam D, Rachakonda S, Ross BS, Zhang HR, Bansal S, Espiritu CL, Keilman M, Lam AM, Niu C, Steuer HM, Furman PA, Otto MJ, Sofia MJ. 2010. 2′-Deoxy-2′-alpha-fluoro-2′-beta-C-methyl 3′,5′-cyclic phosphate nucleotide prodrug analogs as inhibitors of HCV NS5B polymerase: discovery of PSI-352938. Bioorg. Med. Chem. Lett. 20:7376–7380. 10.1016/j.bmcl.2010.10.035 [DOI] [PubMed] [Google Scholar]
- 19.Smith DB, Martin JA, Klumpp K, Baker SJ, Blomgren PA, Devos R, Granycome C, Hang J, Hobbs CJ, Jiang WR, Laxton C, Le Pogam S, Leveque V, Ma H, Maile G, Merrett JH, Pichota A, Sarma K, Smith M, Swallow S, Symons J, Vesey D, Najera I, Cammack N. 2007. Design, synthesis, and antiviral properties of 4′-substituted ribonucleosides as inhibitors of hepatitis C virus replication: the discovery of R1479. Bioorg. Med. Chem. Lett. 17:2570–2576. 10.1016/j.bmcl.2007.02.004 [DOI] [PubMed] [Google Scholar]
- 20.Standring DN, Benzaria S, Bergelson S, Bichko V, Bridges E, Chung RT, Cretton-Scott E, Gosselin G, La CollaP, Lanford R, Moussa A, Pan-Zhou X, Pierra C, Qu L, Sommadossi JP, Storer R, Tausek M, Wright T. 2003. NM 283 has potent antiviral activity against genotype 1 chronic hepatitis C virus (HCV-1) infection in the chimpanzee. J. Hepatol. 38(Suppl 2):1–3. 10.1016/S0168-8278(03)80421-X12733516 [DOI] [Google Scholar]
- 21.Stuyver LJ, McBrayer TR, Tharnish PM, Clark J, Hollecker L, Lostia S, Nachman T, Grier J, Bennett MA, Xie MY, Schinazi RF, Morrey JD, Julander JL, Furman PA, Otto MJ. 2006. Inhibition of hepatitis C replicon RNA synthesis by beta-d-2′-deoxy-2′-fluoro-2′-C-methylcytidine: a specific inhibitor of hepatitis C virus replication. Antivir. Chem. Chemother. 17:79–87 [DOI] [PubMed] [Google Scholar]
- 22.Tomassini JE, Getty K, Stahlhut MW, Shim S, Bhat B, Eldrup AB, Prakash TP, Carroll SS, Flores O, MacCoss M, McMasters DR, Migliaccio G, Olsen DB. 2005. Inhibitory effect of 2′-substituted nucleosides on hepatitis C virus replication correlates with metabolic properties in replicon cells. Antimicrob. Agents Chemother. 49:2050–2058. 10.1128/AAC.49.5.2050-2058.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sofia MJ, Bao D, Chang W, Du J, Nagarathnam D, Rachakonda S, Reddy PG, Ross BS, Wang P, Zhang HR, Bansal S, Espiritu C, Keilman M, Lam AM, Steuer HM, Niu C, Otto MJ, Furman PA. 2010. Discovery of a beta-d-2′-deoxy-2′-alpha-fluoro-2′-beta-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J. Med. Chem. 53:7202–7218. 10.1021/jm100863x [DOI] [PubMed] [Google Scholar]
- 24.Mosley RT, Edwards TE, Murakami E, Lam AM, Grice RL, Du J, Sofia MJ, Furman PA, Otto MJ. 2012. Structure of hepatitis C virus polymerase in complex with primer-template RNA. J. Virol. 86:6503–6511. 10.1128/JVI.00386-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jin Z, Leveque V, Ma H, Johnson KA, Klumpp K. 2013. NTP-mediated nucleotide excision activity of hepatitis C virus RNA-dependent RNA polymerase. Proc. Natl. Acad. Sci. U. S. A. 110:E348–E357. 10.1073/pnas.1214924110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson KA, Simpson ZB, Blom T. 2009. Global kinetic explorer: a new computer program for dynamic simulation and fitting of kinetic data. Anal. Biochem. 387:20–29. 10.1016/j.ab.2008.12.024 [DOI] [PubMed] [Google Scholar]
- 27.Klumpp K, Kalayanov G, Ma H, Le Pogam S, Leveque V, Jiang WR, Inocencio N, De WA, Rajyaguru S, Tai E, Chanda S, Irwin MR, Sund C, Winqist A, Maltseva T, Eriksson S, Usova E, Smith M, Alker A, Najera I, Cammack N, Martin JA, Johansson NG, Smith DB. 2008. 2′-Deoxy-4′-azido nucleoside analogs are highly potent inhibitors of hepatitis C virus replication despite the lack of 2′-alpha-hydroxyl groups. J. Biol. Chem. 283:2167–2175. 10.1074/jbc.M708929200 [DOI] [PubMed] [Google Scholar]
- 28.Pierra C, Benzaria S, Amador A, Moussa A, Mathieu S, Storer R, Gosselin G. 2005. NM 283, an efficient prodrug of the potent anti-HCV agent 2′-C-methylcytidine. Nucleosides Nucleotides Nucleic Acids 24:767–770. 10.1081/NCN-200060112 [DOI] [PubMed] [Google Scholar]
- 29.Bressanelli S, Tomei L, Rey FA, De Francesco R. 2002. Structural analysis of the hepatitis C virus RNA polymerase in complex with ribonucleotides. J. Virol. 76:3482–3492. 10.1128/JVI.76.7.3482-3492.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferrer-Orta C, Arias A, Perez-Luque R, Escarmis C, Domingo E, Verdaguer N. 2004. Structure of foot-and-mouth disease virus RNA-dependent RNA polymerase and its complex with a template-primer RNA. J. Biol. Chem. 279:47212–47221. 10.1074/jbc.M405465200 [DOI] [PubMed] [Google Scholar]
- 31.Gong P, Peersen OB. 2010. Structural basis for active site closure by the poliovirus RNA-dependent RNA polymerase. Proc. Natl. Acad. Sci. U. S. A. 107:22505–22510. 10.1073/pnas.1007626107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zamyatkin DF, Parra F, Alonso JM, Harki DA, Peterson BR, Grochulski P, Ng KK. 2008. Structural insights into mechanisms of catalysis and inhibition in Norwalk virus polymerase. J. Biol. Chem. 283:7705–7712. 10.1074/jbc.M709563200 [DOI] [PubMed] [Google Scholar]
- 33.Jin Z, Smith LK, Rajwanshi VK, Kim B, Deval J. 2013. The ambiguous base-pairing and high substrate efficiency of T-705 (Favipiravir) ribofuranosyl 5′-triphosphate towards influenza A virus polymerase. PLoS One 8:e68347. 10.1371/journal.pone.0068347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stuyver LJ, McBrayer TR, Whitaker T, Tharnish PM, Ramesh M, Lostia S, Cartee L, Shi J, Hobbs A, Schinazi RF, Watanabe KA, Otto MJ. 2004. Inhibition of the subgenomic hepatitis C virus replicon in huh-7 cells by 2′-deoxy-2′-fluorocytidine. Antimicrob. Agents Chemother. 48:651–654. 10.1128/AAC.48.2.651-654.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Richardson FC, Kuchta RD, Mazurkiewicz A, Richardson KA. 2000. Polymerization of 2′-fluoro- and 2′-O-methyl-dNTPs by human DNA polymerase alpha, polymerase gamma, and primase. Biochem. Pharmacol. 59:1045–1052. 10.1016/S0006-2952(99)00414-1 [DOI] [PubMed] [Google Scholar]
- 36.Tisdale M, Ellis M, Klumpp K, Court S, Ford M. 1995. Inhibition of influenza virus transcription by 2′-deoxy-2′-fluoroguanosine. Antimicrob. Agents Chemother. 39:2454–2458. 10.1128/AAC.39.11.2454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clark JL, Hollecker L, Mason JC, Stuyver LJ, Tharnish PM, Lostia S, McBrayer TR, Schinazi RF, Watanabe KA, Otto MJ, Furman PA, Stec WJ, Patterson SE, Pankiewicz KW. 2005. Design, synthesis, and antiviral activity of 2′-deoxy-2′-fluoro-2′-C-methylcytidine, a potent inhibitor of hepatitis C virus replication. J. Med. Chem. 48:5504–5508. 10.1021/jm0502788 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.