

Abstract
Efavirenz is commonly used to treat patients coinfected with human immunodeficiency virus and tuberculosis. Previous clinical studies have observed paradoxically elevated efavirenz plasma concentrations in patients with the CYP2B6*6/*6 genotype (but not the CYP2B6*1/*1 genotype) during coadministration with the commonly used four-drug antituberculosis therapy. This study sought to elucidate the mechanism underlying this genotype-dependent drug-drug interaction. In vitro studies were conducted to determine whether one or more of the antituberculosis drugs (rifampin, isoniazid, pyrazinamide, or ethambutol) potently inhibit efavirenz 8-hydroxylation by CYP2B6 or efavirenz 7-hydroxylation by CYP2A6, the main mechanisms of efavirenz clearance. Time- and concentration-dependent kinetics of inhibition by the antituberculosis drugs were determined using genotyped human liver microsomes (HLMs) and recombinant CYP2A6, CYP2B6.1, and CYP2B6.6 enzymes. Although none of the antituberculosis drugs evaluated at up to 10 times clinical plasma concentrations were found to inhibit efavirenz 8-hydroxylation by HLMs, both rifampin (apparent inhibition constant [Ki] = 368 μM) and pyrazinamide (Ki = 637 μM) showed relatively weak inhibition of efavirenz 7-hydroxylation. Importantly, isoniazid demonstrated potent time-dependent inhibition of efavirenz 7-hydroxylation in both HLMs (inhibitor concentration required for half-maximal inactivation [KI] = 30 μM; maximal rate constant of inactivation [kinact] = 0.023 min−1) and recombinant CYP2A6 (KI = 15 μM; kinact = 0.024 min−1) and also formed a metabolite intermediate complex consistent with mechanism-based inhibition. Selective inhibition of the CYP2B6.6 allozyme could not be demonstrated for any of the antituberculosis drugs using either recombinant enzymes or CYP2B6*6 genotype HLMs. In conclusion, the results of this study identify isoniazid as the most likely perpetrator of this clinically important drug-drug interaction through mechanism-based inactivation of CYP2A6.
INTRODUCTION
Efavirenz is currently the preferred nonnucleoside reverse transcriptase inhibitor for treatment of human immunodeficiency virus (HIV) in patients who are coinfected with tuberculosis (1, 2). Although highly effective, when used at the standard adult dose of 600 mg efavirenz per day, there is high interindividual variability in efavirenz plasma concentrations leading to adverse clinical effects (3, 4). Furthermore, this variability in plasma concentrations appears to be enhanced when efavirenz is coadministered with standard first-line antituberculosis therapy, thereby greatly complicating rational efavirenz dosing recommendations (5–8).
A combination of rifampin, isoniazid, pyrazinamide, and ethambutol is typically administered during the initial 2 months of tuberculosis treatment, followed by 4 months of therapy with rifampin and isoniazid. Reductions in plasma concentrations of drugs coadministered with the antituberculosis drugs are frequently observed and are often attributed to the induction of drug clearance pathways (metabolic enzymes and transporters) by rifampin (9). Increases in drug concentrations can also result from inhibition of drug clearance by coadministered antituberculosis drugs. For example, isoniazid has been identified as a mechanism-based inactivator of the cytochrome P450 (CYP) 1A2, 2A6, 2C9, 2C19, and 3A enzymes (10, 11).
Efavirenz is cleared primarily through metabolism by CYP2B6 (8-hydroxylation), as well as by CYP2A6 (7-hydroxylation) and UDP-glucuronosyltransferase (UGT) 2B7 (direct N-glucuronidation) (12–15). There is currently no evidence that efavirenz is substantially cleared by other mechanisms. A common CYP2B6 variant allele (CYP2B6*6) accounts for a significant proportion of the observed variability in efavirenz plasma concentrations in treated patients (15–17). This variant CYP2B6*6 allele includes two nonsynonymous single nucleotide polymorphisms (c.516G>T and c.785A>G), which result in two amino acid changes (Q172H and K262R) with direct effects on enzyme catalysis (18, 19). The c.516G>T polymorphism was also shown to disrupt normal CYP2B6 gene splicing, resulting in substantial decreases in enzyme levels (20).
Recent studies indicate that the effect of 4-drug antituberculosis therapy on efavirenz plasma concentrations depends on the CYP2B6 genotype of the patient (6, 21, 22). In a cross-sectional study of 56 adult Ghanaian patients, we showed that efavirenz concentrations were significantly higher in homozygous CYP2B6 c.516TT patients receiving antituberculosis therapy than in those not receiving this therapy, while there was no effect of the antituberculosis drugs in patients with the c.516GG or c.516GT genotypes (6). This result was subsequently confirmed in two different longitudinal studies, including a study of 32 coinfected South African children (21) and a study of 307 coinfected Cambodian adults (22) that measured efavirenz concentrations during and following discontinuation of antituberculosis therapy. In both studies, efavirenz concentrations were significantly higher during the initial antituberculosis treatment phase in patients with CYP2B6 slow-metabolizer genotypes (primarily c.516TT), but there was no difference in CYP2B6 intermediate or fast-metabolizer patients. Although it was speculated that this effect might be the result of inhibition of efavirenz metabolism by isoniazid, the mechanism underlying this interaction is currently unknown.
In this study, we used genotyped human liver microsomes (HLMs) and recombinant CYP enzymes to evaluate the potential for rifampin, isoniazid, pyrazinamide, and/or ethambutol to inhibit CYP-mediated efavirenz metabolism. We hypothesized that the observed CYP2B6 genotype-dependent drug interaction might result either from inhibition of CYP2A6-mediated 7-hydroxylation of efavirenz, which is quantitatively important in individuals with decreased CYP2B6 activity (23), or from increased susceptibility of the variant CYP2B6.6 allozyme (H172 and R262) to antituberculosis drug inhibition.
MATERIALS AND METHODS
Reagents.
Efavirenz, 7-hydroxyefavirenz, 7-hydroxyefavirenz-D4, 8-hydroxyefavirenz, and 8-hydroxyefavirenz-D4 were purchased from Toronto Research Chemicals (Toronto, Canada). Clopidogrel, pyrazinamide, rifampin, and isoniazid were from Sigma-Aldrich (St. Louis, MO). Ethambutol was from MP Biomedicals, LLC (Solon, OH). Pooled HLMs (all CYP2B6*1/*1; n = 10 from 2 females and 8 males, ages 36 to 74, all European-Americans) were from the frozen liver bank maintained at Tufts University, Boston, MA (24). Recombinant CYP2A6, CYP2B6.1, and CYP2B6.6 containing coexpressed CYP reductase and cytochrome B5 were from BD Biosciences (Woburn, MA). Individual liver microsomes from white German donors with CYP2B6*1/*1 (n = 7, 4 females and 3 males, ages 37 to 77) and CYP2B6*6/*6 (n = 8, 5 females and 3 males, ages 37 to 77) genotypes were from the human liver bank maintained at the Dr. Margarete Fischer-Bosch Institute of Clinical Pharmacology, Stuttgart, Germany. All liver donors were also shown not to carry the common CYP2A6*9b slow-metabolizer allele previously shown to influence efavirenz plasma concentrations (16). The use of human liver tissues was approved by the local ethical committees of Washington State University (Pullman, WA), the Charité, Humboldt University (Berlin, Germany), and the University Clinic (Tuebingen, Germany). Written informed consent was obtained from liver donors, and the study was conducted in accordance with the Declaration of Helsinki.
Efavirenz hydroxylation assays.
An assay to measure rates of formation of 8-hydroxy-efavirenz and 7-hydroxy-efavirenz by HLMs and recombinant CYPs was developed based on previously published methods with some modifications (13, 25). Briefly, 100-μl incubations of enzymes (HLMs or CYPs), substrate, inhibitor, and NADPH-regenerating system were performed for the times specified below in a water bath at 37°C. Final enzyme concentrations were 0.1 mg protein/ml for HLMs and 10 pmol P450/ml for CYP2A6 and CYP2B6. The reaction was stopped by adding two volumes of ice-cold acetonitrile containing 10% acetic acid and internal standards (200 ng 7-hydroxy-D4-efavirenz and 50 ng 8-hydroxy-D4-efavirenz), vortexed, and centrifuged at 13,000 relative centrifugal force (RCF) for 5 min, and the supernatant was analyzed by high-performance liquid chromatography (HPLC) with mass spectrometry detection.
The HPLC-mass spectrometry system consisted of an Agilent 1100 pump (Agilent Technologies, Santa Clara, CA), a CTC-PAL autosampler (Leap Technologies, Carrboro, NC), and an API 4000 mass spectrometer (Applied Biosystems, Framingham, MA). An isocratic mobile phase of 56% acetonitrile with 44% 20 mM ammonium formate (pH 4.75) was pumped at 0.35 ml per minute through a Synergi Hydro-RP 150- by 2-mm column (Phenomenex, Torrance, CA) to achieve separation of 8-hydroxy-efavirenz and 7-hydroxy-efavirenz peaks. Mass spectrometer settings included a source temperature of 450°C and a source voltage of 1.5 kV. Negative ion transitions monitored were m/z 330 → 258 for 7- and 8-hydroxy-efavirenz and m/z 334 → 258 for D4-labeled 7- and 8-hydroxy-efavirenz. The amount of metabolite formed per minute per milligram of HLMs (or per pmol of CYP) was calculated using a standard curve generated using samples with known concentrations of 7- and 8-hydroxy-efavirenz and internal standards dissolved in a blank matrix.
Inhibition screening assays with and without preincubation.
For assays with inhibitor preincubation, 250-μl (final volume) incubations were prepared in 1.5 ml polypropylene containing inhibitor (no substrate), NADPH-regenerating mixture, and HLMs and incubated for 20 min at 37°C, and 100 μl was transferred to each of two new incubation tubes containing efavirenz (30 μM final concentration, dried down from a methanolic solution). After a further 20 min, the reaction was stopped with internal standard in solvent and processed as described above for 7- and 8-hydroxyefavirenz concentration measurements. For assays without inhibitor preincubation, the inhibitor was added directly to a tube containing efavirenz. Positive-control inhibitors included clopidogrel at a 0.5 μM concentration for CYP2B6 and 8-methoxypsoralen at a 0.5 μM concentration for CYP2A6.
Time- and concentration-dependent inactivation kinetics.
Polypropylene tubes containing enzyme (HLMs at 1 mg protein/ml or CYP2A6 at 100 pmol P450/ml), isoniazid (10 to 250 μM; 1.4 to 34 mg/liter), NADPH-regenerating mixture, and phosphate buffer to a total volume of 150 μl were prepared and incubated at 37°C. After specified intervals, 10-μl aliquots were diluted 10-fold by transferring to tubes containing 90 μl of efavirenz (100 μM final concentration) in NADPH-regenerating mixture and incubated at 37°C for a further 20 min. The reaction was stopped by adding internal standard in solvent, and hydroxy-efavirenz concentrations were measured. The maximal rate constant of inactivation (kinact) and the inhibitor concentration required for half-maximal inactivation (KI) were estimated from Kitz-Wilson plots of inactivation half-life compared to the reciprocal of isoniazid concentration using linear regression as previously described (26).
Metabolite intermediate complex formation.
Pooled HLMs, CYP2A6, CYP2B6.1, and CYP2B6.6 were diluted in 50 mM phosphate buffer (pH 7.4) with an NADPH-regenerating mixture and isoniazid (250 μM final concentration), and 200 μl was added to each well of a 96-well flat-bottomed clear polystyrene plate on ice. HLM concentration was 0.5 mg protein/ml, while CYP concentration was 25 pmol P450/ml. Matched control reactions lacked the NADPH-regenerating mixture. UV absorbance spectra were measured over the range of 400 to 500 nm before and after incubation for 15 min at 37°C in a plate reader (SpectraMax i3; Molecular Devices, Sunnyvale, CA).
Rifampin and pyrazinamide inhibition kinetics.
Polypropylene tubes were prepared containing NADPH-regenerating mixture, efavirenz (10 to 100 μM; 3.2 to 32 mg/liter), and either rifampin (1 to 250 μM; 0.8 to 200 mg/liter) or pyrazinamide (25 to 5,000 μM; 3.1 to 625 mg/liter). The reaction was started by adding HLMs (0.1 mg protein/ml final concentration) and incubated for 20 min at 37°C, and 7- and 8-hydroxyefavirenz concentrations were measured. Nonlinear regression using the mixed competitive-noncompetitive inhibition model was used as described previously (25) to estimate the apparent inhibition constant (Ki), Michaelis-Menten constant (Km), maximum velocity (Vmax), and alpha (α) values. The value of α was used to indicate the relative “mix” of noncompetitive (α = 1.0) compared to competitive (α = infinity) inhibition (25).
Statistical analyses.
Effects of inhibitor preincubation and the CYP2B6 genotype on HLM activities were evaluated by analysis of variance with multiple-comparison testing using the Student-Newman-Keuls test (Sigmaplot v.12; Systat Software, San Jose, CA). A P value of less than 0.05 was considered statistically significant.
RESULTS
Antituberculosis drugs are inhibitors of 7- but not 8-hydroxyefavirenz formation in HLMs.
Ethambutol (100 μM), pyrazinamide (1,000 μM), rifampin (100 μM), isoniazid (100 μM), and their combination were evaluated for inhibition of 7- and 8-hydroxylation of efavirenz (30 μM) in pooled HLMs (Fig. 1). Assays were conducted both with and without preincubation of putative inhibitors for 20 min in the presence of microsomes and an NADPH-regenerating system. Results were compared with positive-control mechanism-based inhibitors of the major CYPs mediating efavirenz hydroxylation in human liver, including clopidogrel (CYP2B6-selective inhibitor) and 8-methoxypsoralen (CYP2A6-selective inhibitor).
FIG 1.
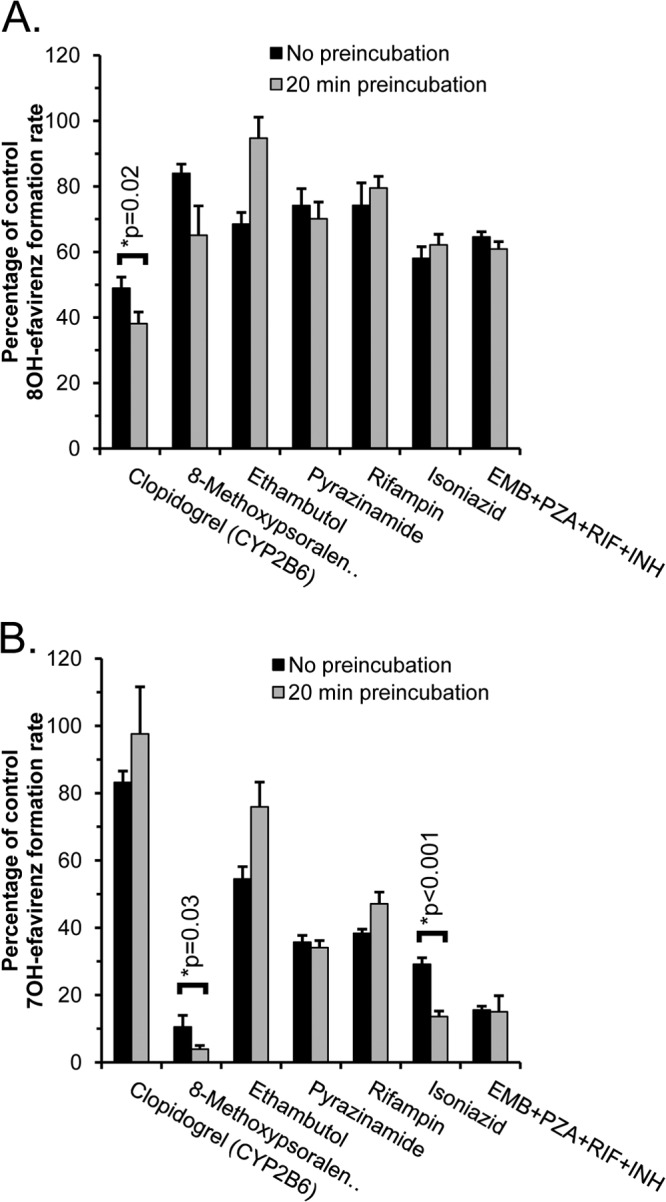
The effect of antituberculosis drugs on 8-hydroxyefavirenz (A) and 7-hydroxyefavirenz (B) formation in human liver microsomes (n = 10 pooled). Antituberculosis drugs evaluated included ethambutol (100 μM), pyrazinamide (1,000 μM) rifampin (100 μM), isoniazid (100 μM), and their combination (EMB+PZA+RIF+INH). Clopidogrel (0.5 μM) and 8-methoxypsoralen (0.5 μM) were included as positive-control selective inhibitors of CYP2B6 and CYP2A6, respectively. Efavirenz concentration was 30 μM. Inhibitors were either preincubated for 20 min with microsomes and NADPH and transferred to a tube containing efavirenz or added directly to a tube containing efavirenz. Results are the means and standard deviations from triplicate determinations of pooled microsomes and are expressed as a percentage of control reactions performed without any inhibitor. Also shown are the P values for pairwise comparisons (analysis of variance [ANOVA] with Student-Newman-Keuls test) evaluating effects of preincubation for all inhibitors that decreased activity by more than 50%.
As shown in Fig. 1A, none of the antituberculosis drugs or their combination decreased 8-hydroxyefavirenz formation by more than 50%, while the CYP2B6 inhibitor (clopidogrel) decreased 8-hydroxyefavirenz formation by 51% ± 3% (mean ± standard deviation [SD]) without inhibitor preincubation and even further with inhibitor preincubation by 62% ± 4% (P = 0.02 for the effect of preincubation). However, as shown in Fig. 1B, three of the four antituberculosis drugs individually decreased 7-hydroxyefavirenz formation by at least 50%, including rifampin (62% ± 1%), pyrazinamide (64% ± 2%), and isoniazid (71% ± 2%), while the mixture of all antituberculosis drugs decreased formation by 84% ± 2%. Preincubation significantly enhanced the extent of inhibition by isoniazid to 86% ± 2% (P < 0.001) but had no effect on inhibition by pyrazinamide and decreased inhibition by rifampin. As expected, the CYP2A6 inhibitor (8-methoxypsoralen) substantially decreased 7-hydroxyefavirenz formation by 89% ± 3% without preincubation, and the extent of inhibition was enhanced further by preincubation to 96% ± 1% (P = 0.03).
Isoniazid is a mechanism-based inhibitor of CYP2A6 but not of CYP2B6.1 or CYP2B6.6.
Since there was significant enhancement of inhibition of efavirenz 7-hydroxylation in HLMs from preincubation with isoniazid, a more detailed analysis of the effects of time and isoniazid concentration was conducted using HLMs and recombinant CYP2A6, including derivation of kinetic parameters for enzyme inactivation (Fig. 2). Both HLMs (Fig. 2A) and CYP2A6 (Fig. 2B) showed saturable inhibition of 7-hydroxyefavirenz formation with increasing preincubation time, consistent with mechanism-based inhibition. Kitz-Wilson plots of these data (Fig. 2C and D) revealed kinact values of 0.023 and 0.024 min−1 and KI values of 31 and 15 μM for 7-hydroxyefavirenz formation by HLMs and CYP2A6, respectively. Isoniazid also inhibited 8-hydroxyefavirenz formation by recombinant CYP2A6 in a time-dependent manner, although with a somewhat slower kinact value of 0.015 min−1 and a KI of 10 μM. However, isoniazid did not inhibit 8-hydroxyefavirenz formation by HLMs at concentrations up to 250 μM or with preincubation times up to 30 min.
FIG 2.

Time- and concentration-dependent kinetics for inhibition by isoniazid (10 to 250 μM) of the 7-hydroxylation of efavirenz (100 μM) in pooled (n = 10) human liver microsomes (A) and recombinant CYP2A6 (B). Kitz-Wilson plots of these data are shown in panels C and D. Inactivation half-life values were obtained by linear regression of the log-linear plots (see fitted curves in panels A and B) and plotted against the reciprocal of inhibitor concentration in panels C and D. Estimates of the maximal rate constant of inactivation (kinact) and the inhibitor concentration required for half-maximal inactivation (KI) were derived by linear regression of Kitz-Wilson plot data.
Although we observed no significant inhibition of 8-hydroxyefavirenz formation by isoniazid in pooled HLMs regardless of preincubation time, all liver donors in that pool were CYP2B6*1/*1 genotype, and so it is possible that the variant CYP2B6.6 enzyme containing 2 amino acid substitutions is more susceptible to inhibition by isoniazid than the common CYP2B6.1 enzyme. Consequently, we evaluated the effects of isoniazid with or without 20 min of preincubation on 8-hydroxyefavirenz formation by recombinant CYP2B6.1 and CYP2B6.6. However, as shown in Fig. 3, isoniazid concentrations up to 250 μM failed to inhibit enzyme activity by more than 50%.
FIG 3.
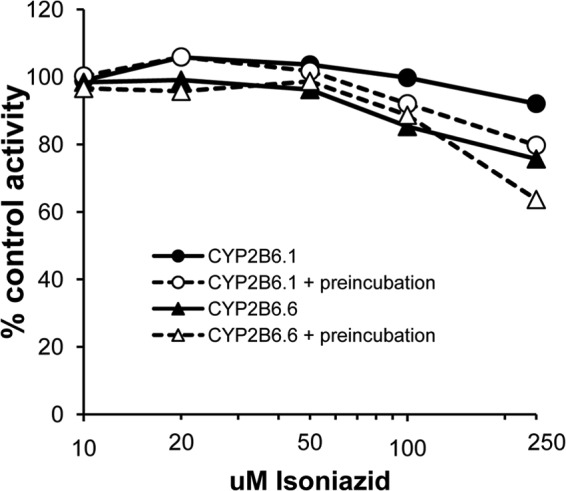
Effect of increasing isoniazid concentration on 8-hydroxyefavirenz formation by CYP2B6.1 (wild-type enzyme) and CYP2B6.6 (H172 and R262 variant allozyme) either with or without 20 min of preincubation of isoniazid with enzyme and NADPH.
The ability of isoniazid to form a spectrally detectable metabolic complex was next evaluated using pooled HLMs, CYP2A6, CYP2B6.1, and CYP2B6.6 (Fig. 4). Both HLMs (Fig. 4A) and CYP2A6 (Fig. 4B) showed evidence for spectral complex formation after 15 min of incubation, with a UV differential absorbance peak spanning 450 to 490 nm. Neither CYP2B6.1 nor CYP2B6.6 showed evidence for metabolite intermediate complex formation with isoniazid, consistent with the lack of effect of isoniazid on efavirenz hydroxylation by CYP2B6.1 and CYP2B6.6.
FIG 4.
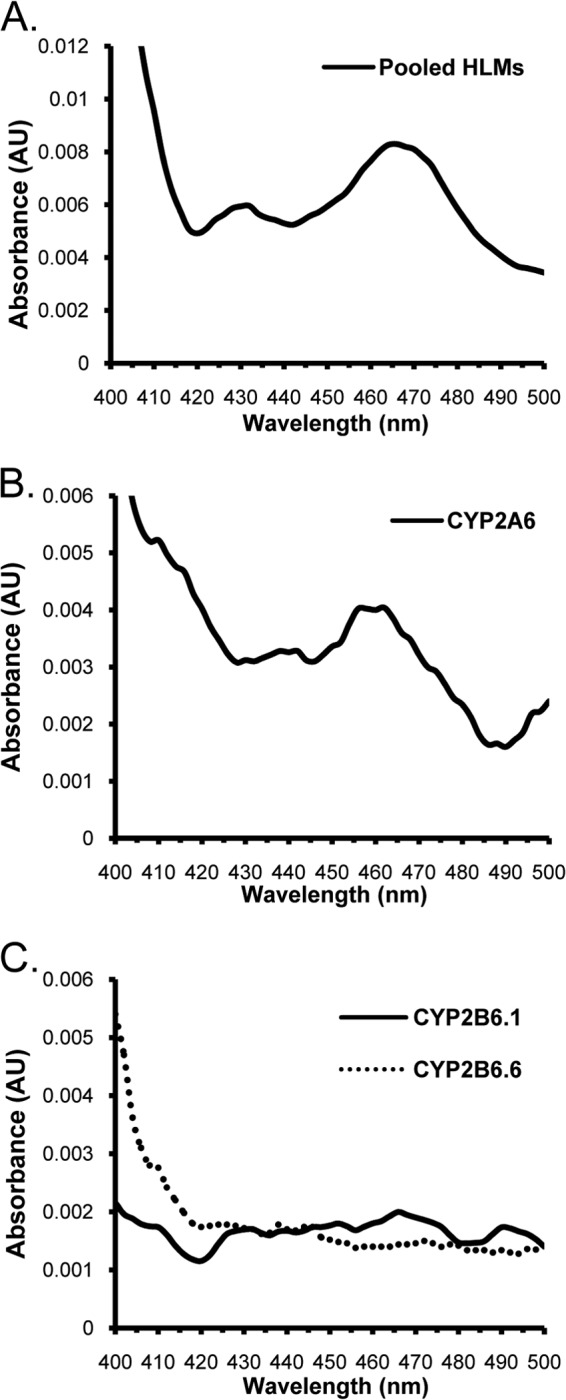
UV absorbance spectral scans (400 to 500 nm) showing the effect of NADPH and 15 min of incubation on metabolite intermediate complex formation of isoniazid (250 μM) with pooled (n = 10) human liver microsomes (A), recombinant CYP2A6 (B), and recombinant CYP2B6.1 and CYP2B6.6 (C).
Rifampin and pyrazinamide are relatively weak inhibitors of 7-hydroxyefavirenz formation.
We next determined the type of inhibition and Ki values for inhibition of efavirenz hydroxylation in pooled HLMs by rifampin and pyrazinamide. Rifampin showed relatively weak noncompetitive inhibition of 7-hydroxyefavirenz formation with a Ki of 368 μM, while pyrazinamide showed somewhat weaker mixed competitive-noncompetitive inhibition of 7-hydroxyefavirenz formation with a Ki of 637 μM. Neither compound inhibited 8-hydroxyefavirenz formation by more than 50% using rifampin concentrations up to 250 μM or pyrazinamide concentrations up to 2,500 μM.
Inhibition by isoniazid is similar in CYP2B6*6 HLMs and CYP2B6*1 HLMs.
Next, donor gender-, age-, and race/ethnicity-matched HLMs with either CYP2B6*1/*1 (n = 7) or CYP2B6*6/*6 (n = 8) genotype were used to evaluate genotype-dependent inhibitor effects (Fig. 5). As shown in Fig. 5A, none of the antituberculosis drugs inhibited 8-hydroxyefavirenz formation in CYP2B6*1 or CYP2B6*6 HLMs by more than 50%, except for isoniazid in CYP2B6*6 HLMs at the highest concentration (500 μM). However, the greater inhibition by 500 μM isoniazid in CYP2B6*6 HLMs (53% ± 5% decrease) than in CYP2B6*1 HLMs (42% ± 12% decrease) did not achieve statistical significance (P > 0.05). Interestingly, there was significantly less inhibition of 8-hydroxyefavirenz formation by the CYP2B6 inhibitor (clopidogrel) in CYP2B6*6 HLMs (41% ± 6% decrease) compared to that in CYP2B6*1 (57% ± 7% decrease) HLMs (P = 0.006).
FIG 5.
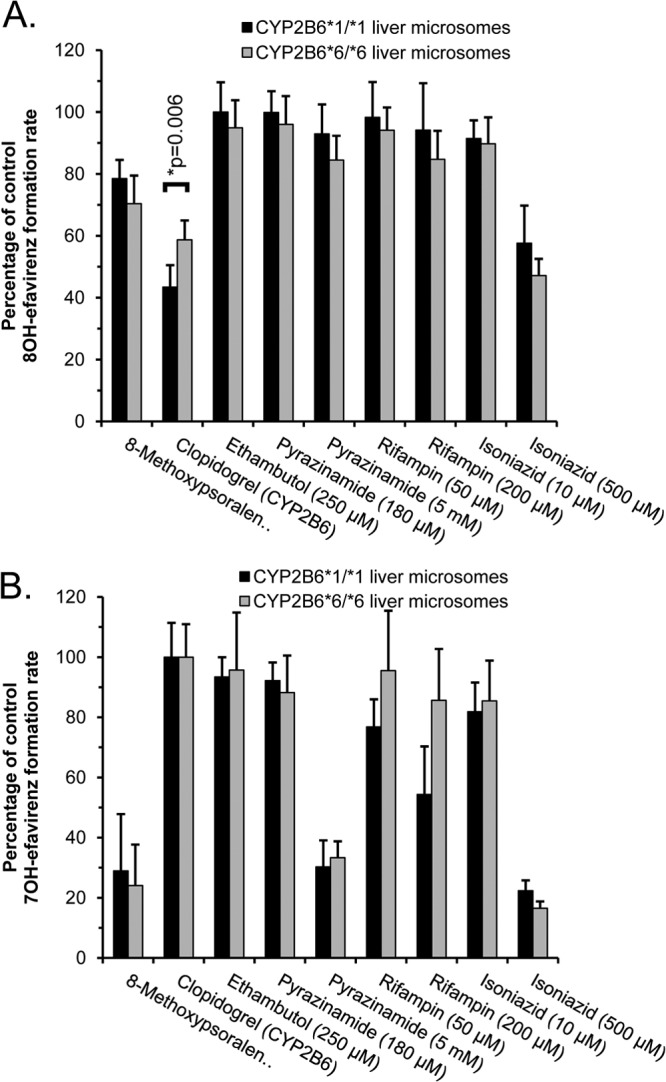
The effect of antituberculosis drugs on 8-hydroxyefavirenz (A) and 7-hydroxyefavirenz (B) formation in human liver microsomes with either CYP2B6*1/*1 (n = 7 individuals) or CYP2B6*6/*6 (n = 8 individuals) genotypes. Clopidogrel (1 μM) and 8-methoxypsoralen (0.5 μM) were included as positive-control selective inhibitors of CYP2B6 and CYP2A6, respectively. Efavirenz concentration was 30 μM. Results are the means and standard deviations from activities determined for individual liver microsomes expressed as a percentage of control reactions performed without any inhibitor. Also shown are the P values for pairwise comparisons (ANOVA with Student-Newman-Keuls test) evaluating effects of the CYP2B6 genotype for all inhibitors that decreased activity by more than 50%.
Pyrazinamide (at 5 mM), isoniazid (at 500 μM), and the CYP2A6 inhibitor (8-methoxypsoralen) inhibited 7-hydroxyefavirenz formation in both CYP2B6*1 and CYP2B6*6 HLMs by at least 50% (Fig. 5B). However, there were no differences in the extent of inhibition by pyrazinamide, isoniazid, or 8-methoxypsoralen in CYP2B6*6 HLMs compared with that in CYP2B6*1 HLMs (P > 0.05).
Finally, time- and isoniazid concentration-dependent inhibition studies of efavirenz hydroxylation were also conducted using pooled HLMs from 7 donors with CYP2B6*1/*1 and from 8 donors with CYP2B6*6/*6 genotypes. kinact and KI values (from 3 independent measurements) for inhibition of 7-hydroxyefavirenz formation were similar (P > 0.05) in CYP2B6*6 HLMs (0.016 ± 0.004 min−1 and 31 ± 7 μM, respectively) and CYP2B6*1 HLMs (0.012 ± 0.001 min−1 and 21 ± 2 μM, respectively). kinact and KI values for the inhibition of 8-hydroxyefavirenz formation by isoniazid could not be estimated.
DISCUSSION
The results of this study indicate that paradoxical increases in efavirenz concentrations in CYP2B6*6/*6 genotype patients being treated with 4-drug first-line antituberculosis therapy are most likely a consequence of mechanism-based inactivation of CYP2A6 by isoniazid. The main evidence supporting this includes time- and isoniazid concentration-dependent inhibition of 7-hydroxyefavirenz formation in both HLMs and recombinant CYP2A6 coincident with formation of a spectrally detectable metabolite intermediate complex and a lack of significant inhibition of 8-hydoxyefavirenz formation by HLMs by any of the studied antituberculosis drugs, including isoniazid. Inactivation by isoniazid also appears to be relatively potent with inhibitor concentration at half-maximal rate of inactivation (KI) values for both HLMs and CYP2A6, ranging from 15 to 31 μM, which are lower than average peak plasma isoniazid concentrations, reported to range from 36 to 79 μM (5 to 11 mg/liter) (27–29).
Isoniazid also inhibited 8-hydroxyefavirenz formation by recombinant CYP2A6 in a time-dependent manner, although with a somewhat lower kinact value than for inhibition of 7-hydroxyefavirenz formation. However, there was no clear evidence for significant inhibition of 8-hydroxyefavirenz formation by isoniazid (or 8-methoxypsoralen) in HLMs except at the highest isoniazid concentration tested (500 μM) in CYP2B6*6/*6 genotype HLMs. This suggests that CYP2A6 probably contributes only a minor proportion of the observed efavirenz 8-hydroxylation activity in HLMs, as reported previously (13), except perhaps in CYP2B6*6 slow metabolizers.
Our results are supported by a recent pharmacogenetic study of treated coinfected Cambodian patients, among whom CYP2B6 516TT patients who also carried slow-metabolizer N-acetyltransferase 2 (NAT2) genotypes showed even higher efavirenz levels than those without these NAT2 variants (22). N-Acetylation by NAT2 is the main mechanism for clearance of isoniazid, and although plasma concentrations of isoniazid were not measured, it was speculated that patients with slow-metabolizer NAT2 genotypes had increased isoniazid concentrations with resultant increased inhibition of efavirenz clearance.
Pyrazinamide and rifampin could potentially contribute to the observed antituberculosis drug interaction through inhibition of efavirenz 7-hydroxylation. A relatively high dose of pyrazinamide is normally used (1.5 to 2 g per day) for tuberculosis treatment, and the average expected peak plasma concentrations of 252 to 746 μM (31 to 92 mg/liter) (28, 29) are close to the Ki value determined here (637 μM). Consequently, inhibition by pyrazinamide is possible, particularly if higher doses are used. On the other hand, rifampin is a relatively weak inhibitor of efavirenz 7-hydroxylation with a Ki value (368 μM) that is 9 to 50 times higher than the expected average peak plasma concentrations of 7 to 40 μM (6 to 33 mg/liter) (29–31). Regardless, none of the putative inhibitors other than isoniazid showed evidence for mechanism-based inactivation, which presents the greatest concern for eliciting clinically important drug-drug interactions (32). However, because of the known limitations of these in vitro models, controlled pharmacokinetic drug-drug interaction studies in healthy human volunteers or patients would be needed to completely exclude pyrazinamide and/or rifampin as inhibitors of efavirenz metabolism.
We found no evidence that the Q172H and K262R amino acid substitutions associated with the CYP2B6*6 allele enhance susceptibility to inhibition by isoniazid or any of the other antituberculosis drugs tested (our alternate hypothesis). Three prior studies have shown altered susceptibility of the expressed CYP2B6.6 allozyme to inhibition by various drugs, including clopidogrel, sertraline, clotrimazole, itraconazole, raloxifene, ticlopidine, and phencyclidine (33–35). However, in all those studies, the amino acid changes resulted in reduced susceptibility to inhibition rather than increased susceptibility to inhibition that would be needed to explain the clinical data. In good agreement with those studies, we also found decreased inhibition of efavirenz 8-hydroxylation by clopidogrel in CYP2B6*6/*6 genotype HLMs compared with that of CYP2B6*1/*1 HLMs. However, this finding contrasts with the results of a recent study that showed increased potency of inhibition of 8-hydroxyefavirenz formation by clopidogrel in CYP2B6*6/*6 genotype HLMs compared with that in CYP2B6*1/*1 HLMs (36). The reason for this discrepancy may be a consequence of differences in inhibition assay. Specifically, in that study (36), clopidogrel was not preincubated with HLMs and NADPH, while preincubation of clopidogrel was performed in this study in order to maximize the inhibitory effect, since clopidogrel is a known mechanism-based inhibitor of CYP2B6 (37). Substantiating this, we observed enhancement of inhibition by clopidogrel of efavirenz 8-hydroxylation in HLMs with inhibitor preincubation, consistent with mechanism-based inhibition.
There are several limitations to the current study. Autoinduction is a well-known feature of continued efavirenz therapy, in large part because of increased expression of CYP2B6 (38, 39). As far as we are aware, none of the donors of the HLMs used in this study had been exposed to efavirenz (or other inducers, such as rifampin), and so it is possible that our results could be different if we had studied microsomes from efavirenz-exposed livers. Efavirenz is also metabolized by direct N-glucuronidation (40), which could be quantitatively important in CYP2B6*6/*6 slow-metabolizer individuals. Consequently, we cannot rule out an additional contribution to the observed drug-drug interaction from inhibition of efavirenz glucuronidation by one or more of the antituberculosis drugs.
In conclusion, the results of this study provide a rational mechanistic explanation for a clinically important genotype-dependent drug-drug interaction. This information should be of considerable value for developing future guidelines for the individualized treatment of patients with HIV-tuberculosis coinfection.
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health, National Institute of General Medical Sciences (grants GM-061834 and GM-102130), to M.H.C., the National Institutes of Allergy and Infectious Diseases (grant AI-091448) and Child Health and Human Development (grant HD-071779) to A.K., the German Federal Ministry of Education and Research (Virtual Liver Network grant 0315755) to U.M.Z., and the National Institute of Diabetes and Digestive and Kidney Diseases (contract N01-DK-7-0004/HHSN267200700004C) to the Liver Tissue and Cell Distribution System, Minneapolis, MN.
Footnotes
Published ahead of print 12 May 2014
REFERENCES
- 1.CDC. 2013. Recommendations for regimens for the concomitant treatment of tuberculosis and HIV infection in adults. CDC, Atlanta, GA: http://www.cdc.gov/tb/TB_HIV_Drugs/default.htm [Google Scholar]
- 2.Lawn SD, Meintjes G, McIlleron H, Harries AD, Wood R. 2013. Management of HIV-associated tuberculosis in resource-limited settings: a state-of-the-art review. BMC Med. 11:253. 10.1186/1741-7015-11-253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marzolini C, Telenti A, Decosterd LA, Greub G, Biollaz J, Buclin T. 2001. Efavirenz plasma levels can predict treatment failure and central nervous system side effects in HIV-1-infected patients. AIDS 15:71–75. 10.1097/00002030-200101050-00011 [DOI] [PubMed] [Google Scholar]
- 4.Stahle L, Moberg L, Svensson JO, Sonnerborg A. 2004. Efavirenz plasma concentrations in HIV-infected patients: inter- and intraindividual variability and clinical effects. Ther. Drug Monit. 26:267–270. 10.1097/00007691-200406000-00008 [DOI] [PubMed] [Google Scholar]
- 5.Kwara A, Lartey M, Sagoe KW, Xexemeku F, Kenu E, Oliver-Commey J, Boima V, Sagoe A, Boamah I, Greenblatt DJ, Court MH. 2008. Pharmacokinetics of efavirenz when coadministered with rifampin in TB/HIV coinfected patients: pharmacogenetic effect of CYP2B6 variation. J. Clin. Pharmacol. 48:1032–1040. 10.1177/0091270008321790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kwara A, Lartey M, Sagoe KW, Court MH. 2011. Paradoxically elevated efavirenz concentrations in HIV/tuberculosis-coinfected patients with CYP2B6 516TT genotype on rifampin-containing antituberculous therapy. AIDS 25:388–390. 10.1097/QAD.0b013e3283427e05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Friedland G, Khoo S, Jack C, Lalloo U. 2006. Administration of efavirenz (600 mg/day) with rifampicin results in highly variable levels but excellent clinical outcomes in patients treated for tuberculosis and HIV. J. Antimicrob. Chemother. 58:1299–1302. 10.1093/jac/dkl399 [DOI] [PubMed] [Google Scholar]
- 8.Matteelli A, Regazzi M, Villani P, De Iaco G, Cusato M, Carvalho AC, Caligaris S, Tomasoni L, Manfrin M, Capone S, Carosi G. 2007. Multiple-dose pharmacokinetics of efavirenz with and without the use of rifampicin in HIV-positive patients. Curr. HIV Res. 5:349–353. 10.2174/157016207780636588 [DOI] [PubMed] [Google Scholar]
- 9.Baciewicz AM, Chrisman CR, Finch CK, Self TH. 2013. Update on rifampin, rifabutin, and rifapentine drug interactions. Curr. Med. Res. Opin. 29:1–12. 10.1185/03007995.2012.747952 [DOI] [PubMed] [Google Scholar]
- 10.Wen X, Wang JS, Neuvonen PJ, Backman JT. 2002. Isoniazid is a mechanism-based inhibitor of cytochrome P450 1A2, 2A6, 2C19 and 3A4 isoforms in human liver microsomes. Eur. J. Clin. Pharmacol. 57:799–804. 10.1007/s00228-001-0396-3 [DOI] [PubMed] [Google Scholar]
- 11.Polasek TM, Elliot DJ, Somogyi AA, Gillam EM, Lewis BC, Miners JO. 2006. An evaluation of potential mechanism-based inactivation of human drug metabolizing cytochromes P450 by monoamine oxidase inhibitors, including isoniazid. Br. J. Clin. Pharmacol. 61:570–584. 10.1111/j.1365-2125.2006.02627.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ward BA, Gorski JC, Jones DR, Hall SD, Flockhart DA, Desta Z. 2003. The cytochrome P450 2B6 (CYP2B6) is the main catalyst of efavirenz primary and secondary metabolism: implication for HIV/AIDS therapy and utility of efavirenz as a substrate marker of CYP2B6 catalytic activity. J. Pharmacol. Exp. Ther. 306:287–300. 10.1124/jpet.103.049601 [DOI] [PubMed] [Google Scholar]
- 13.Ogburn ET, Jones DR, Masters AR, Xu C, Guo Y, Desta Z. 2010. Efavirenz primary and secondary metabolism in vitro and in vivo: identification of novel metabolic pathways and cytochrome P450 2A6 as the principal catalyst of efavirenz 7-hydroxylation. Drug Metab. Dispos. 38:1218–1229. 10.1124/dmd.109.031393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Belanger AS, Caron P, Harvey M, Zimmerman PA, Mehlotra RK, Guillemette C. 2009. Glucuronidation of the antiretroviral drug efavirenz by UGT2B7 and an in vitro investigation of drug-drug interaction with zidovudine. Drug Metab. Dispos. 37:1793–1796. 10.1124/dmd.109.027706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kwara A, Lartey M, Sagoe KW, Kenu E, Court MH. 2009. CYP2B6, CYP2A6 and UGT2B7 genetic polymorphisms are predictors of efavirenz mid-dose concentration in HIV-infected patients. AIDS 23:2101–2106. 10.1097/QAD.0b013e3283319908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kwara A, Lartey M, Sagoe KW, Rzek NL, Court MH. 2009. CYP2B6 (c. 516G–>T) and CYP2A6 (*9B and/or *17) polymorphisms are independent predictors of efavirenz plasma concentrations in HIV-infected patients. Br. J. Clin. Pharmacol. 67:427–436. 10.1111/j.1365-2125.2009.03368.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zanger UM, Klein K. 2013. Pharmacogenetics of cytochrome P450 2B6 (CYP2B6): advances on polymorphisms, mechanisms, and clinical relevance. Front. Genet. 4:24. 10.3389/fgene.2013.00024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jinno H, Tanaka-Kagawa T, Ohno A, Makino Y, Matsushima E, Hanioka N, Ando M. 2003. Functional characterization of cytochrome P450 2B6 allelic variants. Drug Metab. Dispos. 31:398–403. 10.1124/dmd.31.4.398 [DOI] [PubMed] [Google Scholar]
- 19.Zhang H, Sridar C, Kenaan C, Amunugama H, Ballou DP, Hollenberg PF. 2011. Polymorphic variants of cytochrome P450 2B6 (CYP2B6.4-CYP2B6.9) exhibit altered rates of metabolism for bupropion and efavirenz: a charge-reversal mutation in the K139E variant (CYP2B6.8) impairs formation of a functional cytochrome p450-reductase complex. J. Pharmacol. Exp. Ther. 338:803–809. 10.1124/jpet.111.183111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hofmann MH, Blievernicht JK, Klein K, Saussele T, Schaeffeler E, Schwab M, Zanger UM. 2008. Aberrant splicing caused by single nucleotide polymorphism c.516G>T [Q172H], a marker of CYP2B6*6, is responsible for decreased expression and activity of CYP2B6 in liver. J. Pharmacol. Exp. Ther. 325:284–292. 10.1124/jpet.107.133306 [DOI] [PubMed] [Google Scholar]
- 21.McIlleron HM, Schomaker M, Ren Y, Sinxadi P, Nuttall JJ, Gous H, Moultrie H, Eley B, Merry C, Smith P, Haas DW, Maartens G. 2013. Effects of rifampin-based antituberculosis therapy on plasma efavirenz concentrations in children vary by CYP2B6 genotype. AIDS 27:1933–1940. 10.1097/QAD.0b013e328360dbb4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bertrand J, Verstuyft C, Chou M, Borand L, Chea P, Nay KH, Blanc FX, Mentre F, Taburet AM, CAMELIA Study Group 2013. Dependence of efavirenz- and rifampicin-isoniazid-based antituberculosis treatment drug-drug interaction on CYP2B6 and NAT2 genetic polymorphisms: ANRS 12154 study in Cambodia. J. Infect. Dis. 209:399–408. 10.1093/infdis/jit466 [DOI] [PubMed] [Google Scholar]
- 23.di Iulio J, Fayet A, Arab-Alameddine M, Rotger M, Lubomirov R, Cavassini M, Furrer H, Gunthard HF, Colombo S, Csajka C, Eap CB, Decosterd LA, Telenti A, Swiss HIV Cohort Study 2009. In vivo analysis of efavirenz metabolism in individuals with impaired CYP2A6 function. Pharmacogenet. Genomics 19:300–309. 10.1097/FPC.0b013e328328d577 [DOI] [PubMed] [Google Scholar]
- 24.Court MH. 2010. Interindividual variability in hepatic drug glucuronidation: studies into the role of age, sex, enzyme inducers, and genetic polymorphism using the human liver bank as a model system. Drug Metab. Rev. 42:209–224. 10.3109/03602530903209288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Greenblatt DJ, Zhao Y, Venkatakrishnan K, Duan SX, Harmatz JS, Parent SJ, Court MH, von Moltke LL. 2011. Mechanism of cytochrome P450-3A inhibition by ketoconazole. J. Pharm. Pharmacol. 63:214–221. 10.1111/j.2042-7158.2010.01202.x [DOI] [PubMed] [Google Scholar]
- 26.Bertelsen KM, Venkatakrishnan K, Von Moltke LL, Obach RS, Greenblatt DJ. 2003. Apparent mechanism-based inhibition of human CYP2D6 in vitro by paroxetine: comparison with fluoxetine and quinidine. Drug Metab. Dispos. 31:289. 10.1124/dmd.31.3.289 [DOI] [PubMed] [Google Scholar]
- 27.Agrawal S, Singh I, Kaur KJ, Bhade SR, Kaul CL, Panchagnula R. 2002. Bioequivalence assessment of rifampicin, isoniazid and pyrazinamide in a fixed dose combination of rifampicin, isoniazid, pyrazinamide and ethambutol vs. separate formulations. Int. J. Clin. Pharmacol. Ther. 40:474–481. 10.5414/CPP40474 [DOI] [PubMed] [Google Scholar]
- 28.Panchagnula R, Sancheti P, Rungta S, Agrawal S, Kaul CL. 2003. Evaluation of bioequivalence of isoniazid and pyrazinamide in three and four drugs fixed dose combinations using WHO simplified protocol. Pharmacol. Res. 48:383–387. 10.1016/S1043-6618(03)00175-0 [DOI] [PubMed] [Google Scholar]
- 29.McIlleron H, Wash P, Burger A, Norman J, Folb PI, Smith P. 2006. Determinants of rifampin, isoniazid, pyrazinamide, and ethambutol pharmacokinetics in a cohort of tuberculosis patients. Antimicrob. Agents Chemother. 50:1170–1177. 10.1128/AAC.50.4.1170-1177.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pargal A, Rani S. 2001. Non-linear pharmacokinetics of rifampicin in healthy Asian Indian volunteers. Int. J. Tuberc. Lung Dis. 5:70–79 [PubMed] [Google Scholar]
- 31.Chang KC, Leung CC, Yew WW, Kam KM, Yip CW, Ma CH, Tam CM, Leung EC, Law WS, Leung WM. 2008. Peak plasma rifampicin level in tuberculosis patients with slow culture conversion. Eur. J. Clin. Microbiol. Infect. Dis. 27:467–472. 10.1007/s10096-007-0454-6 [DOI] [PubMed] [Google Scholar]
- 32.Orr ST, Ripp SL, Ballard TE, Henderson JL, Scott DO, Obach RS, Sun H, Kalgutkar AS. 2012. Mechanism-based inactivation (MBI) of cytochrome P450 enzymes: structure-activity relationships and discovery strategies to mitigate drug-drug interaction risks. J. Med. Chem. 55:4896–4933. 10.1021/jm300065h [DOI] [PubMed] [Google Scholar]
- 33.Shebley M, Hollenberg PF. 2007. Mutation of a single residue (K262R) in P450 2B6 leads to loss of mechanism-based inactivation by phencyclidine. Drug Metab. Dispos. 35:1365–1371. 10.1124/dmd.107.014985 [DOI] [PubMed] [Google Scholar]
- 34.Bumpus NN, Sridar C, Kent UM, Hollenberg PF. 2005. The naturally occurring cytochrome P450 (P450) 2B6 K262R mutant of P450 2B6 exhibits alterations in substrate metabolism and inactivation. Drug Metab. Dispos. 33:795–802. 10.1124/dmd.105.003749 [DOI] [PubMed] [Google Scholar]
- 35.Talakad JC, Kumar S, Halpert JR. 2009. Decreased susceptibility of the cytochrome P450 2B6 variant K262R to inhibition by several clinically important drugs. Drug Metab. Dispos. 37:644–650. 10.1124/dmd.108.023655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu C, Ogburn ET, Guo Y, Desta Z. 2012. Effects of the CYP2B6*6 allele on catalytic properties and inhibition of CYP2B6 in vitro: implication for the mechanism of reduced efavirenz metabolism and other CYP2B6 substrates in vivo. Drug Metab. Dispos. 40:717–725. 10.1124/dmd.111.042416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Richter T, Murdter TE, Heinkele G, Pleiss J, Tatzel S, Schwab M, Eichelbaum M, Zanger UM. 2004. Potent mechanism-based inhibition of human CYP2B6 by clopidogrel and ticlopidine. J. Pharmacol. Exp. Ther. 308:189. 10.1124/jpet.103.056127 [DOI] [PubMed] [Google Scholar]
- 38.Robertson SM, Maldarelli F, Natarajan V, Formentini E, Alfaro RM, Penzak SR. 2008. Efavirenz induces CYP2B6-mediated hydroxylation of bupropion in healthy subjects. J. Acquir. Immune Defic. Syndr. 49:513–519. 10.1097/QAI.0b013e318183a425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ngaimisi E, Mugusi S, Minzi OM, Sasi P, Riedel KD, Suda A, Ueda N, Janabi M, Mugusi F, Haefeli WE, Burhenne J, Aklillu E. 2010. Long-term efavirenz autoinduction and its effect on plasma exposure in HIV patients. Clin. Pharmacol. Ther. 88:676–684. 10.1038/clpt.2010.172 [DOI] [PubMed] [Google Scholar]
- 40.Cho DY, Ogburn ET, Jones D, Desta Z. 2011. Contribution of N-glucuronidation to efavirenz elimination in vivo in the basal and rifampin-induced metabolism of efavirenz. Antimicrob. Agents Chemother. 55:1504–1509. 10.1128/AAC.00883-10 [DOI] [PMC free article] [PubMed] [Google Scholar]