

Abstract
Candidemia is the fourth most common kind of microbial bloodstream infection, with Candida albicans being the most common causative species. Echinocandins are employed as the first-line treatment for invasive candidiasis until the fungal species is determined and confirmed by clinical diagnosis. Echinocandins block the FKS glucan synthases responsible for embedding β-(1,3)-d-glucan in the cell wall. The increasing use of these drugs has led to the emergence of antifungal resistance, and elevated MICs have been associated with single-residue substitutions in specific hot spot regions of FKS1 and FKS2. Here, we show for the first time the caspofungin-mediated in vivo selection of a double mutation within one allele of the FKS1 hot spot 1 in a clinical isolate. We created a set of isogenic mutants and used a hematogenous murine model to evaluate the in vivo outcomes of echinocandin treatment. Heterozygous and homozygous double mutations significantly enhance the in vivo resistance of C. albicans compared with the resistance seen with heterozygous single mutations. The various FKS1 hot spot mutations differ in the degree of their MIC increase, substance-dependent in vivo response, and impact on virulence. Our results demonstrate that echinocandin EUCAST breakpoint definitions correlate with the in vivo response when a standard dosing regimen is used but cannot predict the in vivo response after a dose escalation. Moreover, patients colonized by a C. albicans strain with multiple mutations in FKS1 have a higher risk for therapeutic failure.
INTRODUCTION
Fungal infections have emerged over the last few decades as a consequence of increasing cohorts of at-risk individuals. Candida albicans is the most important opportunistic fungal pathogen in humans (1). Mortality rates from C. albicans infection are estimated to be as high as 45% (2), partly due to delayed or inaccurate diagnosis and inappropriate antifungal therapies.
Echinocandin drugs are recommended as the first-line treatment for invasive candidemia (3, 4). Anidulafungin (ANI), caspofungin (CAS), and micafungin (MICA) are lipopeptidic antifungal agents that inhibit the synthesis of the fungal wall component β-(1,3)-d-glucan by noncompetitively blocking the β-(1,3)-d-glucan synthase. Drug resistance is emerging in patients with C. albicans, C. glabrata, C. tropicalis, and C. krusei infections (5–7), which complicates disease monitoring and patient management (8). Presently, echinocandin resistance in C. albicans occurs with an incidence of <1% (9). Single-residue substitutions located in two hot spot regions in each of the genes FKS1 and FKS2 (10), but not FKS3, have been associated with elevated echinocandin MICs (11) in Candida species.
So far in C. albicans, only amino acid substitutions within FKS1 encoding the β-(1,3)-d-glucan synthase complex were found in echinocandin-resistant isolates (12). The most frequent amino acid changes reported were at positions F641, L644, S645, R647, D648, P649 (10), W1358, and R1361 (13) within hot spot 1 (HS1) (amino acid positions 641 to 649, FLTLSLRDP) (12) and hot spot 2 (HS2) (amino acid positions 1357 to 1364, DWIRRYTL) (14), respectively. HS1 and HS2 alterations leading to changes in F641, S645, and R1361 were associated with pronounced MIC elevations. Other HS mutations (in L644, R647, D648, P649, and W1358) confer a discrete MIC increase (14). C. albicans is a diploid fungus carrying a pair of each chromosome and at least two alleles of each gene locus (15). An amino acid substitution in one allele (heterozygous) affects only half of the total cellular glucan synthase pool that can be targeted by echinocandins (5), while substitutions occurring in both alleles (homozygous) affect the whole glucan synthase pool.
Here, we identify and report the first in vivo-selected HS1 FKS1 double mutation (R647R/G and P649P/L) in one allele in C. albicans after long-term caspofungin (CAS) therapy (including a detailed case report and therapeutic regimes). The treatment response of a clinical wild-type (WT) strain and the corresponding sequential heterozygous derivative mutants (R647R/G and P649P/L) are compared using a well-validated murine model of disseminated candidiasis. ANI, CAS, or MICA was applied at a standard dose (designated as AUC100, where AUC is the area under the concentration-time curve) or elevated dose (AUC500 [five times the standard dose]) and compared with each other and the placebo control group.
The impacts of heterozygous single (R647R/G or P649P/L) and double mutations (R647R/G and P649P/L), as well as homozygous double mutations (R647G and P649L), in FKS1 on virulence and in vitro and in vivo susceptibilities were tested in isogenic (identical genetic background) mutant strains.
The hypothesis addressed in this study was that heterozygous double mutations, as well as homozygous double mutations within one allele, enhance the in vivo therapeutic resistance of C. albicans strains even with a dose escalation. We were able to (i) confirm this hypothesis, (ii) show significant differences in the activities of the three echinocandins, and (iii) demonstrate that the reported single nucleotide polymorphisms (SNPs) have no impact on virulence.
MATERIALS AND METHODS
Patient case.
A 27-year-old female patient was reported to have chronic mucocutaneous candidiasis (CMC), suffering from persistent and recurrent infections of the skin, nails, and oral mucosa. In 2005, a C. albicans (azole-resistant isolate) infection was successfully treated with 12 months of CAS (50 mg/day) (16). In 2007, the patient presented with a relapse of CMC. Caspofungin (50 mg/day) was resumed in combination with terbinafine (250 mg/day) and topical ciclopirox cream for another 12 months, resulting in a marked improvement.
During 2007 to 2010, various azole regimens consisting of voriconazole (VOR) (2 × 200 mg/day) or posaconazole (POS) (2 × 800 mg/day) were attempted, with partial effects. In March 2010, the patient began retreatment with intravenous (i.v.) CAS (50 mg/day) for 2 years. Onychomycosis improved slightly (Fig. 1C). In 2012, while still on CAS, a fulminant infection appeared with perioral/periorbital erythematous and erosive lesions accompanied by plaques of pustules at the forehead that were clinically significant (Fig. 1A), in addition to nail-destructive onychomycosis (Fig. 1B). In vitro susceptibility testing according to Etest and EUCAST classified C. albicans as CAS resistant (Table 1). Antifungal treatment was immediately changed to ANI (100 mg/day), and the infections improved significantly, with full healing achieved by October 2012.
FIG 1.
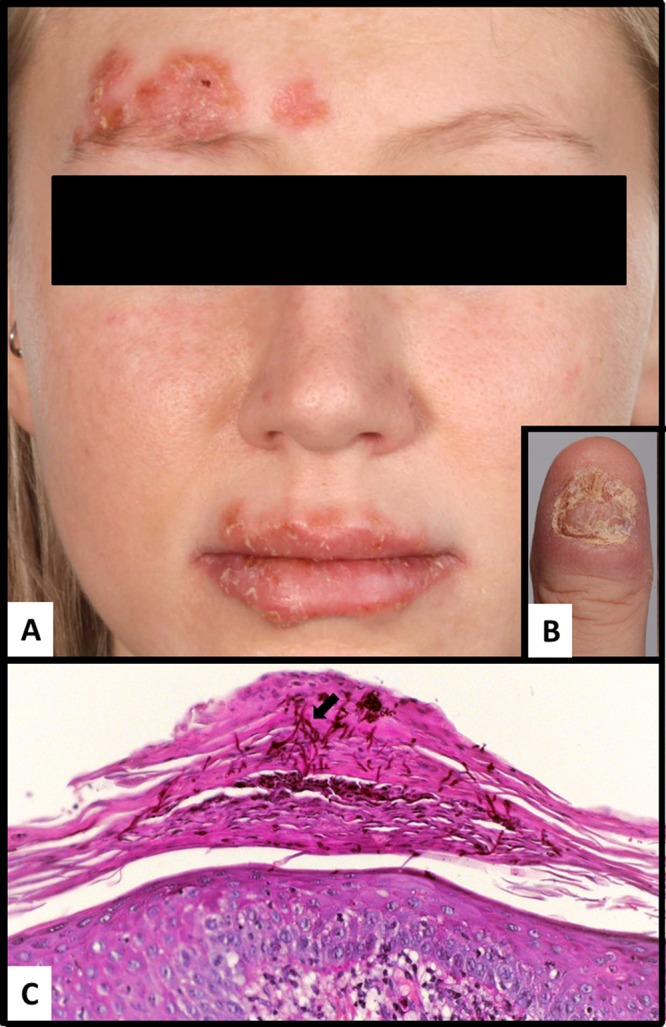
Presentation of the clinical features of the female 27-year-old patient. (A) At the right side of the forehead and the lips, erythematous plaques topped with crusts and pustules are visible. (B) Paronychia and destruction of the nail by C. albicans. (C) Periodic acid-Schiff stain showing parakeratosis with multiple hyphae (arrow) present in the cornified layers.
TABLE 1.
In vitro susceptibilities for clinical and mutant isolates included in the study and for quality-control strains, as determined by EUCAST or Etesta
| Isolate type and mutant or strain | EUCAST MIC (mg/liter) (susceptibility classification) forb: |
Etest MIC (mg/liter) (susceptibility classification) forc: |
|||||
|---|---|---|---|---|---|---|---|
| ANI | MICA | VOR | ISA | FLU | CAS | AMB | |
| Clinical isolates or QCd strain | |||||||
| SCL 1130.04e | <0.008 (S) | <0.008 (S) | 0.5 (R) | 0.06 | >16 (R) | 0.125 (S) | 0.5 (S) |
| RRCL 110.1f | 0.03 (S) | >1 (R) | 4 (R) | 0.5 | >16 (R) | 2 (R) | 0.5 (S) |
| ATCC 22019g | 0.125 (I) | >1 (I) | <0.03 (S) | <0.03 | 2 (S) | 0.5 (S) | 0.38 (S) |
| Mutant isolates or QC strain | |||||||
| SSWT SC5314h | 0.015 (S) | <0.008 (S) | <0.03 (S) | <0.03 | 0.25 (S) | 0.19 (S) | 0.25 (S) |
| RRMH2h | 0.06 (R) | 0.03 (R)i | <0.03 (S) | <0.03 | 0.25 (S) | 0.75 (R) | 0.19 (S) |
| RSMH1h | 0.015 (S) | 0.03 (R)j | <0.03 (S) | <0.03 | <0.125 (S) | 0.25 (S) | 0.25 (S) |
| SRMH1h | 0.015 (S) | 0.008 (S) | <0.03 (S) | <0.03 | 0.25 (S) | 0.25 (S) | 0.25 (S) |
| RRMHO2h | 0.06 (R) | 1 (R)k | <0.03 (S) | <0.03 | <0.125 (S) | 1 (R) | 0.38 (S) |
| ATCC 6258g | 0.06 (S) | 0.125 | 0.125 (S) | <0.03 | 16 (R) | 0.38 (I) | 0.75 (S) |
Susceptibility classifications were performed according to the EUCAST breakpoints (BP), except for CAS and Etest, for which CLSI BP were adopted, as recommended by the manufacturer.
ANI, anidulafungin; MICA, micafungin; VOR, voriconazole; ISA, isavuconazole; FLU, fluconazole; S, susceptible; R, resistant; I, intermediate.
CAS, caspofungin; AMB, amphotericin B.
QC, quality control.
The clinical isolates 952.04 and 5104.04 revealed the same MICs within ±1 dilution step difference; these strains do not carry coding FKS1 HS1 or HS2 mutations.
The clinical isolate 111.12 revealed the same MIC within ±1 dilution step difference; these strains carry the mutations R647R/G and P649P/L.
Quality-control strains.
SC5314 is the wild-type parental strain; mutations carried by the strains are as follows: RRMH2, R647R/G and P649P/L; RSMH1, R647R/G; SRMH1, P649P/L; and RRMHO2, R647G and P649L.
Micafungin trailing 0.03 to 1 but stays at <50% endpoint in this range.
Micafungin creeping increase 0.03 to 0.25.
Micafungin trailing 0.06 to 0.25 and just >50% endpoint in this range.
Random amplified polymorphic DNA typing of clinical isolates.
Random amplified polymorphic DNA (RAPD) typing of the clinical isolates (n = 5) was performed using the published primer pairs M13, OPA18, OPE18, CA2, RSD10, and RSD12 together with already published PCR conditions (17–20). For DNA extractions, all strains were cultured on Sabouraud 2% glucose agar for 24 h at 37°C. DNA was extracted from pure cultures using the UltraClean microbial DNA isolation kit (Mo Bio Laboratories, Inc.) according to manufacturer's instructions. The DNA concentration of the DNA extract was determined using NanoVue (GE Healthcare Life Sciences), according to the manufacturer's instructions. Genomic DNA extracts were diluted to 50 ng/μl for PCR. A volume of 18 μl of all PCR products was loaded together with 3 μl of orange (6×) gel loading dye (New England BioLabs, Inc.) on 1.8% agar (Rotigarose; Roth) gel with ethidium bromide at a concentration of 0.5 μg/ml. As a reference, the SimplyLoad 100-bp extended range DNA ladder (Lonza Cologne GmbH) was used. The electrophoresis conditions were 3 h at 80 V and 100 mA. The electrophoresis gels were visualized in a GelDoc EZ system (Bio-Rad). The typing assay was performed twice and double-blinded by two different investigators. The C. albicans strains ATCC 90028, ATCC 64550, and ATCC 64548 were used as population controls.
In vitro susceptibility testing.
The MICs for azoles (voriconazole, isavuconazole, and fluconazole) and echinocandins (ANI and MICA) were tested for the echinocandin-susceptible strains isolated in 2004 (C. albicans SCL 1130.04, SCL 952.04, SCL 5104.04, and SCL 111.12) and the echinocandin-resistant (RRCL 110.12) strain carrying the mutations R647R/G and P649P/L, the parental strain, C. albicans SSWT SC5314, the isogenic mutants, and the mutants C. albicans RRMH2 (R647R/G and P649P/L), RSMH1 (R647R/G), SRMH1 (P649P/L), and RRMHO2 (R647G and P649L), according to EUCAST methodology (EDEF 7.2) (21) (see Table 1). In addition, CAS and amphotericin B were tested using CLSI methodology and Etest (bioMérieux). C. krusei strain ATCC 6258 or Candida parapsilosis strain ATCC 22019 (21, 22) was used as a quality-control strain.
Sequencing of FKS1 gene regions hot spot 1 and hot spot 2.
Genomic DNA was extracted using the UltraClean microbial DNA isolation kit (Mo Bio Laboratories, Inc.), according to the manufacturer's instructions. Sequencing was performed using the same primers and conditions as those published by Garcia-Effron et al. (23). In short, the Candida universal FKS1 primers 1HS1F (AAT GGG CTG GTG CTC AAC AT) and 1HS1F (CCT TCA ATT TCA GAT GGA ACT TGA TG) were used for amplifying FKS1 HS1, while for FKS1 HS2, the primers 1HS2F (AAG ATT GGT GCT GGT ATG GG) and 1HS2R (TAA TGG TGC TTG CCA ATG AG) were used. As a PCR master mix, the Qiagen LongRange PCR kit and conditions were as follows: initial denaturation at 96°C for 4 min, followed by 40 cycles of 94°C for 30 s, 55° for 30 s, 74°C for 90 s, and a final elongation step of 74°C for 4 min. For cleaning the PCR products, ExoSAP-IT was used. For sequencing, the BigDye Terminator version 3.1 cycle sequencing kit was used in combination with the 3500 Genetic Analyzer (Applied Biosystems), according to the manufacturer's instructions.
Construction of isogenic C. albicans FKS1 mutants.
The generated isogenic mutant plasmids used are shown in Table S1 in the supplemental material, and the primers used are shown in Table S2 in the supplemental material. An FKS1 deletion cassette was constructed using the SAT1 flipper technology (24) with the primers FKS1_55 and FKS1KO_53 for the upstream homologous region and FKS1_35 and FKS1_33 for the downstream homologous region. The homologous regions were cloned into pSFS2a (24) using the restriction sites ApaI/XhoI and SacII/SacI, respectively. The resulting plasmid was linearized with PvuI and transformed into C. albicans SC5314 (25) to replace the FKS1 coding sequence and get a heterozygous FKS1/fks1 knockout (see Table S3 in the supplemental material). The SAT1 marker was recycled by overnight cultivation in maltose-containing medium.
For reintegration of the FKS1 gene at its endogenous locus, the FKS1 coding sequence was cloned into pSFS2a-FKS1dr (pSFS2a containing the downstream homologous region of the deletion cassette) with the primers FKS1_55 and FKS1clo_53 and using the restriction sites ApaI/XhoI. The resulting plasmid (pSFS2a-FKS1reint) was linearized with PvuI and transformed into C. albicans strain CA-MT424 (FKS1/fks1). Correct integration was checked by colony PCR.
For the mutagenesis of FKS1, PCRs were performed using pSFS2a-FKS1reint with the primers FKS1_55 and the corresponding mutagenesis reverse primer, as well as with the corresponding mutagenesis forward primer and FKS1_33, respectively (see Table S3 in the supplemental material). The purified PCR products were mixed, and the whole reintegration cassette was amplified using the primers FKS1_55_2 and FKS1_33_2. The cassette was transformed into CA-MT424 to integrate the mutated FKS1 allele at the deleted locus. Correct integration was checked by colony PCR, and the SAT1 marker was recycled.
For the construction of homozygous mutants, the PCRs were performed with primers FKS1_55 and the corresponding mutagenesis reverse primer, as well as with the corresponding mutagenesis forward primer and FKS1clo_53, respectively. The purified PCR products were mixed, the mutated FKS1 coding sequence was amplified using the primers FKS1_55 and FKS1clo_53; this was cloned into pSFS2a-FKS1dr as described above for the reintegration plasmid. Prior to transformation, the plasmids were linearized with PvuI. For the construction of the homozygous double mutant, pSFS2a-FKS1_2mut_reint was used to replace the wild-type allele in C. albicans strain CA-MT441 (see Table S2 in the supplemental material). Correct integration was checked by colony PCR, and the SAT1 marker was recycled. For the construction of the homozygous single mutants, the corresponding plasmids containing mutated FKS1 alleles were used for integration at the deleted alleles of C. albicans strains CA-MT457 and CA-MT458, respectively. In these two strains, the wild-type allele in CA-MT424 had been directly replaced before by the corresponding mutated allele. The presence of the introduced point mutations was confirmed by sequencing the corresponding region in all mutants.
Hematogenous mouse model. (i) Pilot study for inoculum determination.
To evaluate the in vivo virulence of isogenic mutant strains, a total of 24 NRMI mice were challenged in groups of three with four isogenic mutant strains (C. albicans SSMH2, RSMH1, SRMH1, and SSMHO2; detailed information is shown in Table 1; see also Table S1 in the supplemental material) and two inoculum amounts (5 × 105 CFU/mouse and 5 × 106/mouse). The aim was to achieve sufficient infection without causing mortality. Using a C. albicans cell concentration for infection of 5 × 106/mouse, nine out of 12 mice died. Challenging the mice with a dose of 5 × 105 CFU/mouse led to one out of three mice per group dying, and the kidney burden in the surviving mice was found to be between 104 and 105 CFU/ml kidney homogenate. Hence, an inoculum of 1 × 105/mouse was used for the treatment studies.
(ii) In vivo treatment studies.
Evaluation of in vivo resistances of C. albicans clinical isolates (SCL and RRCL strains; Tables 1 and 2) against ANI, CAS, MICA, and the placebo (0.09% NaCl) was performed in a hematogenous mouse model. Eighty-four NMRI mice (weight, 26.0 g to 30.0 g; Harlan Scandinavia, Allerød, Denmark) were kept with free access to food and water. On day 0, the mice were challenged by intravenous injection (200 μl administered with a 25-gauge syringe) of a C. albicans (5 × 105 CFU/ml; final concentration per mouse, 1 × 105) suspension. The mice were challenged with either of the two strains SCL (n = 42 mice) or RRCL (n = 42 mice) and treated by the intraperitoneal (i.p.) route on days 1 to 3 with 0.5 ml of either ANI, CAS, or MICA at standard (AUC100) and high-dose (AUC500) or with a placebo (see below).
TABLE 2.
Overview of FKS mutations found in clinical isolates gained between 2004 (x.04) and 2012 (x.012) from the index/case patient
| Isolate |
FKS1 hot spot 1 mutation by positiona: |
FKS1 hot spot 2 mutation by positiona: |
||||||
|---|---|---|---|---|---|---|---|---|
| 1641 (A) | 1653 (A) | 1662 (T) | 1929 (A) | 1939 (A) | 1946 (C) | 4215 (C) | 4230 (T) | |
| SCL 1130.04 | A/T | A/G | T/C | T | A | C | C/T | T/C |
| 952.04 | A/T | A/G | T/C | T | A | C | C/T | T/C |
| 5104.04 | A/T | A/G | T/C | T | A | C | C/T | T/C |
| RRCL 110.12 | A/T | A/G | T/C | T | A/G | C/T | C/T | T/C |
| 111.12 | A/T | A/G | T/C | T | A/G | C/T | C/T | T/C |
| aab | P547 | T551 | I554 | T643 | R647R/Gc | P649P/Ld | I1375 | A1410 |
Nucleic acid mutation. Boxheads indicate position (wild-type nucleic acid).
Regular type, amino acid change: nucleic acid change does not result in a change of amino acid (regular type); bold type, wild-type amino acid/position/heterozygous mutation.
Amino acid change, arginine (R) to glycine (G); codon change, AGA to GGA.
Amino acid change, proline (P) to leucine (L); codon change, CCU to CUU.
The treatment studies of isogenic mutant strains and their parental strain were divided into three animal experiments (per antifungal compound tested), as a total of 312 NMRI mice (weight between 26.0 g and 30.0 g) were used. The inoculation of mice and treatment scheme were performed as outlined above, and the dosing was as described below. The treatment groups consisted of six mice and control groups of eight mice. As a backup, four additional mice were available to replace mice that were inoculated outside the tail vein and therefore excluded; if the backup mice were not needed, they were allocated to the control groups (hence, some control groups included nine mice).
The echinocandin treatment doses used in the murine model were the standard dose (AUC100), which was the dose that resulted in human equivalent exposure and was calculated using our echinocandin pharmacokinetic-pharmacodynamic studies in this exact mouse model reported previously (26), ANI100 (2.16 mg/kg of body weight), CAS100 (0.76 mg/kg), and MICA100 (2.71 mg/kg). The high dose (AUC500) was five times the human-equivalent exposure dose and was included in order to examine if a dose escalation would be useful for overcoming the resistance of C. albicans.
For determining the fungal burden (CFU/ml tissue homogenate), kidneys were aseptically removed at day 4. The organ weights were determined, and the kidneys were placed in sterile physiological saline (750 μl per pair of kidneys); the organs were stored at −80°C prior to counting CFU. The fungal burden was determined using the spot technique, which involved plating two 20-μl spots of 10-fold dilutions of tissue homogenate (homogenization performed with a homogenizer [RW 16 Basic; IKA Labortechnik, Bie & Berntsen, Copenhagen, Denmark]). The CFU count was expressed as the log10 of CFU/ml kidney homogenate. The lower limit of detectable CFU was 25 CFU/ml tissue homogenate. All murine experiments reported in this study were approved by the Danish Animal Experimentation Committee under the Ministry of Justice (2009/561-1637).
Statistical analyses.
To check for normal distribution of the fungal burden data, the Shapiro-Wilk normality test was performed. The Kruskal-Wallis test was then applied, as the data were found to be not normally distributed. Outliers (maximum one per control group with <100 CFU/ml kidney homogenate) in the control groups (each eight placebo-treated mice) were removed for statistical significance calculation, as these mice did not successfully establish a manifest C. albicans infection or clear the infection. The outliers were identified by Prism GraphPad version 5. To guarantee the transparency of all data and to provide the readers with a complete image, the outliers are included in all tables and figures. In all analyses, P values of ≤0.05 were regarded as statistically significant.
Filamentation ability tests.
All studied C. albicans strains were grown overnight on Sabouraud 2% glucose agar (Carl-Roth) at 37°C. The cells were harvested in 0.9% NaCl. The cell number was adjusted to 5 × 105 cells/ml using the Neubauer CE chamber hemocytometer. A volume of 500 μl was centrifuged at 13,000 rpm for 5 min in a Mikro 20 (Hettrich) centrifuge, the supernatant was discarded, and the cell pellet was suspended in 500 μl fetal bovine serum (FBS) (Gibco, Invitrogen). FBS with cells was transferred into a Nikon BioStation IM (Nikon) cell incubator and monitoring system. The cells were evaluated by microscopy after 0.5 h, 1 h, 2 h, and 3 h incubation at 37°C in the Nikon BioStation IM using BioStation software version 2.1. In addition, for a long-time (4 h) comparison of the germ tube formation experiment, movies were captured for SC5314, RRMH2, SCL, and RRCL in four individual experiments per strain. The images were captured every 10 min at ×200 and ×400 magnification.
Chitin content measurement by FACS analyses.
All studied C. albicans strains were overnight grown on Sabouraud 2% glucose agar at 37°C. The cells were harvested in 0.9% NaCl. The cell number was adjusted to 5 × 106 cells/ml using the Neubauer CE chamber hemocytometer. The cells were inoculated into fresh Sabouraud 2% glucose agar and were harvested after 4 h of incubation at 37°C with shaking at 200 rpm. The samples were fixed in 10% (vol/vol) neutral-buffered formalin for 15 min. The cells were washed with sterile 0.9% NaCl and stained with 25 μg/ml calcofluor white (Sigma-Aldrich) for 15 min at room temperature. The cells were washed twice with 0.9% NaCl to remove excessive stain, and they were filtered using a CellTrics filter (5 μm pore size; Partec) to remove cell clumps. The cells were prepared using the standard procedure for fluorescence-activated cell sorter (FACS) analysis. The measurement of 10,000 events was carried out using the FACSVerse flow cytometer (BD Biosciences). The results were given as the fluorescence intensity (calcofluor-A). Unstained SSWT (SC5314) was used as the reference strain for gaiting the cell population. For statistical analyses, the minimum, median, and maximal fluorescence signals were determined. SSWT stained with calcofluor white served as a wild-type reference. The fluorescence peaks of the clinical isolates (strains SCL and RRCL) were compared to the fluorescence peak of the SSWT population. All FACS experiments were performed independently in triplicate.
RESULTS
In vivo emergence of heterozygous double mutants of C. albicans and CAS medication.
Five clinical isolates obtained between 2004 and 2012 from a female patient suffering from chronic mucocutaneous candidiasis (CMC) (case report available in Materials and Methods) were tested for their in vitro susceptibilities against ANI, CAS, MICA, fluconazole (FLU), isavuconazole, voriconazole (VOR), and amphotericin B. The isolates from 2004 were found to be FLU and VOR resistant but echinocandin susceptible according to the EUCAST breakpoints (using ANI as a marker for ANI and CAS susceptibility, as is currently recommended) and CAS susceptible according to the CLSI breakpoints (22, 27). In contrast, the isolates gained in 2012 were micafungin resistant and showed an elevated ANI MIC compared to that of the initial isolate but not exceeding the EUCAST breakpoint (MIC, 0.03 mg/liter; ANI breakpoint, susceptible [S] ≤ 0.03 mg/liter); they were therefore classified as anidulafungin and caspofungin susceptible using the EUCAST breakpoint, with anidulafungin as a marker for caspofungin. In contrast, these isolates were classified as caspofungin resistant using the Etest and CLSI breakpoints. Detailed data on the MICs are given in Table 1.
To investigate the underlying molecular resistance mechanism, FKS1 HS1 and HS2 were sequenced. Notably, strains isolated in 2004 carried four silent mutations (no amino acid [aa] change, also called synonymous substitution), one within the FKS1 HS1 A1929T (T643) and five outside the FKS1 HS regions, located at nucleic acid positions A1641A/T (P547), A1653A/G (T551), T1662T/C (I554), C4125C/T (I1375), and T4230T/C (A1410). In addition, two missense (i.e., having an aa change, also called nonsynonymous) mutations in the same allele (heterozygous) were found at nucleic acid positions A1939A/G and C1946C/T, leading to the residue changes R647R/G and P649P/L, respectively (Table 2). Identical silent mutations found in FKS1 HS1 and HS2 were shared by all isolates, strongly suggesting their clonal origin and in vivo development of resistance. To verify this hypothesis, randomly amplified polymorphic DNA typing (RAPD typing) was performed with five markers (M13, CA2, OPA18, OPE18, and RSD10). As displayed in Fig. 2, the RAPD genotype was identical for the clinical isolates. The data herein support the development of acquired CAS resistance in C. albicans during a 6-year treatment period with CAS.
FIG 2.

Genotyping of clinical isolates of C. albicans isolated between 2004 and 2012 using RAPD. PCR products for the five markers, M13 (A), CA2 (B), OPA18 (C), OPE18 (D), and RSD10 (E) (17–20) were separated on 1.8% agar gel for a run time of 3 h at 80 V and 100 mA. Electrophoresis gels were stained with ethidium bromide. Lane 0, marker; lane 1, isolate 952/04; lane 2, isolate 1130/04; lane 3, isolate 5104/04; lane 4, isolate 110/12; and lane 5, isolate 111/12.
To evaluate the impact of the double mutations on the therapeutic response in vivo, the in vitro echinocandin-resistant clinical isolate carrying the R647R/G and P649P/L mutations (RRCL) and the susceptible wild-type clinical isolate (SCL) were compared in a murine hematogenous candidiasis model. ANI, CAS, and MICA were administered at two doses (AUC100 and AUC500). The fungal burden in the kidney homogenates of animals infected with the susceptible wild-type strain were significantly reduced (Fig. 3). Comparing the control mouse group with each of the treatment groups, the reduction in kidney burden was found to be highly significant in all treated groups (P < 0.01), with the exception of that for MICA100, for which the reduction was less pronounced, albeit still statistically significant (P < 0.05) (see Table S4 in the supplemental material).
FIG 3.
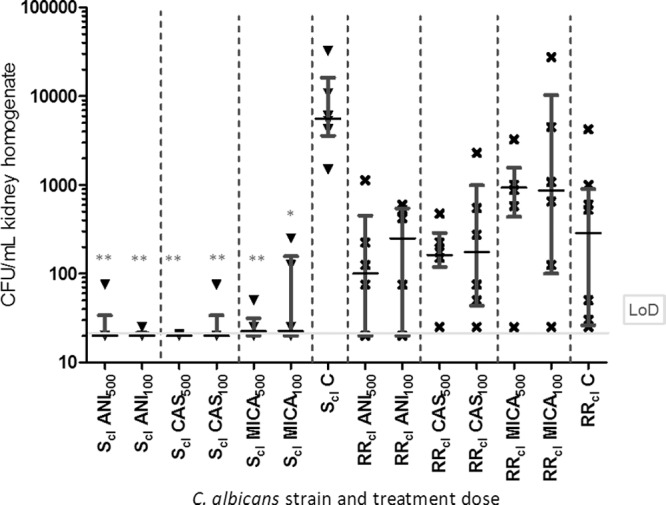
Evaluation of in vivo susceptibilities of the two clinical strains 1130.04 (SCL, no mutation in FKS1) and 110.12 (RRCL, heterozygous double mutation in FKS1 at positions R647R/G and P649P/L) and high dose (AUC500) or standard dose (AUC100) of anidulafungin (ANI), caspofungin (CAS), or micafungin (MICA). The treatment groups consisted of six mice and control groups of eight mice. Infected mice treated with placebo served as a control (C). CFU per ml kidney homogenate is depicted at the end of treatment. The limit of detection (LoD) and median with interquartile range are indicated. Statistical significance was calculated using the Kruskal-Wallis test and Dunnett's multiple comparison test: *, P < 0.5; **, P < 0.01; and ***, P < 0.001. Outliers (maximum of one outlier per control group) were removed for calculation only (CFU ≤ 100 in the placebo-treated groups).
In contrast, the reduction in kidney burden failed to reach statistical significance (between the different treatment groups and between the placebo with the treatment groups) in mice infected with RRCL (MICs of ≤0.03 μg/ml [ANI], >1 μg/ml [MICA], and >2 μg/ml [CAS]). By comparing the median values of different treatments with those of the placebo groups, it is obvious that the fungal burden is lowest in ANI500 and CAS500, followed by ANI100 and CAS100 and the placebo control (see Table S5 in the supplemental material). Significant differences in the CFU/g kidney within the control groups of mice infected with either SCL or RRCL were observed, as well as differences between those receiving standard and high-dose medications (see Table S4 in the supplemental material). By comparing the median values of SCL (MICs of ≤0.008 μg/ml [ANI and MICA] and 0.125 μg/ml [CAS]) and RRCL, a reduction in that for RRCL is obvious even if not statistically significant (Fig. 3; see also Table S5 in the supplemental material). These in vivo data fully support and correlate with the in vitro data, as ANI was found to be the most active echinocandin against RRCL in vivo.
Influence of heterozygous single and double mutations and homozygous double mutations on virulence and in vivo treatment response.
To evaluate the influence of both mutations individually and in combination, both in vitro and in vivo, isogenic mutants (identical genetic background) were generated (see Table S1 in the supplemental material). An overview of the in vitro susceptibility results of the parental strain and isogenic mutants is given in Table 1. The parental strain and SRMH1 (heterozygous P649P/L) were classified as susceptible in vitro against all echinocandins. RSMH1 (heterozygous R647R/G) was classified as resistant to MICA only, since it was only one dilution step above the breakpoint. Strain RRMH2 (heterozygous double mutation; P649P/L and R647R/G) and RRMHO2 (homozygous double mutation; P649L and R647G) were classified as panechinocandin resistant in vitro. Notably, the ANI and MICA MICs were only one dilution above the respective breakpoints for the heterozygous double mutant RRMH2. But the homozygous double mutant RRMHO2 had a MICA MIC that was six dilutions above the breakpoint. Thus, a stepwise increase in MICs and development from single-echinocandin to multiple-echinocandin resistance was observed comparing single to double codon alterations and hetero- to homozygous mutations, respectively. Comparing the clinical heterozygous double mutant RRCL with the heterozygous isogenic mutant RRMH2, it is obvious that these two mutations apparently affect ANI susceptibility to a much lesser extent than they affect susceptibility to MICA and CAS, as the MICs for MICA and CAS were several times higher. However, caspofungin testing was performed using the commercial Etest and therefore may not be directly comparable (28), and an interpretation of the MICA susceptibility data should be done with caution, as both isolates showed a trailing phenotype, the implication of which has not been elucidated for the echinocandins (Table 1).
To evaluate the differences in the in vitro responses toward the different echinocandins in vivo, a murine model was again used (Fig. 4). A statistically significant reduction in the fungal burdens in kidneys was observed for ANI, CAS, and MICA (AUC100 and AUC500) and mutant strains compared to those of the placebo-treated controls. The median of fungal burden per treatment arm and placebo group prove that the parental strain (SSWT) and the two strains carrying a heterozygous single mutation have the best in vivo treatment responses (see Table S6 in the supplemental material). This is in agreement with in vitro data, as all strains were categorized as ANI susceptible (Table 1). Dose escalation markedly reduced the fungal burden of the heterozygous double mutant RRMH2, whereas the burden reduction for the homozygous double mutant RRMHO2 was less prominent (see Table S6). All CAS-treated groups showed statistically significant lower fungal burdens than the mice in the placebo control groups. By comparing the median fungal burden, the treatment efficacy against the heterozygous double mutant RRMH2 was limited at the standard dose but improved with dose escalation (see Table S6). While in vitro resistance to CAS was confirmed in vivo for the RRMH2 mutant, resistance in RRMHO2 was not observed in vivo. Based on their fungal burdens, SSWT, RSMH1, and SRMH1 were found to be CAS susceptible both in vivo and in vitro (Table 1 and Fig. 4).
FIG 4.
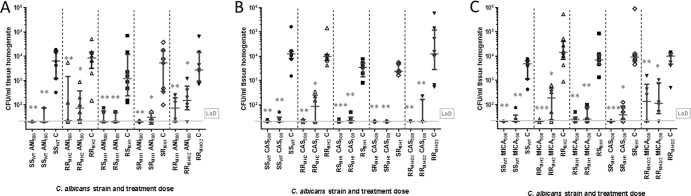
In vivo susceptibilities of isogenic mutants derived from parental strain (SSCL). The parental strain and the four isogenic mutant strains (RRMH2, RSMH1, SRMH1, and RRMHO2) were evaluated for high (AUC500) and standard (AUC100) doses of anidulafungin (ANI) (A), caspofungin (CAS) (B), and micafungin (MICA) (C) and compared to placebo control. The treatment groups consisted of six mice and the control groups consisted of eight mice. The CFU counts per ml kidney homogenate are depicted after the end of treatment. The limit of detection (LoD) and median with interquartile range are indicated in each graph. The statistic is based on the Kruskal-Wallis test and Dunnett's multiple comparison tests, with statistical significances given as P < 0.5 (*), P < 0.01 (**), and P < 0.001 (***); outliers (maximum of one per control group) were removed for calculation only (CFU ≤ 100 in the placebo-treated groups). The strains carry the following mutations in their FKS gene: RRMH2 heterozygous double mutation (R647R/G and P649P/L), RSMH1 (R647R/G) and SRMH1 (P649P/L) heterozygous single mutations, and RRMHO2 homozygous double mutation (R647G and P649L).
The micafungin-treated groups displayed statistically significant lower fungal burdens than those of the placebo-treated mice (P < 0.05; see Fig. 4). The two double mutants RRMH2 and RRMHO2 showed only limited responses against MICA100 treatment. In contrast to RRMHO2, RRMH2 responded to dose escalation. Both strains were resistant toward MICA in vitro. For all other strains, standard dosing was sufficient to clear the infections (Fig. 4), even though RSMH1 was found to be MICA resistant according to the EUCAST breakpoints (Table 1). However, we would like to mention that the clinical isolates and the mutants constructed in the laboratory represent different background strains, and therefore, comparisons have to be made with caution.
The clinical isolates (SCL and RRCL) were found to differ in virulence in terms of kidney CFU count and treatment responses (Fig. 3). On the contrary, isogenic mutants that exclusively carry FKS1 HS1 mutations did not vary in their virulence (Fig. 4). The major difference between RRMH2 and RRCL was that RRMH2 responded to dose escalation of ANI, CAS, and MICA, whereas RRCL did not. Thus, a clear trend between standard and escalated dosing was found for RRMH2 and RRMHO2 and ANI and MICA (Fig. 4; see also Table S5 in the supplemental material). In contrast to the clinical isolates, no differences in virulence were observed in a comparison of the parental strain and the isogenic mutants (Fig. 4; see also Table S6 in the supplemental material).
Fluorescence-activated cell sorting (FACS) analyses on the chitin content of clinical isolates (SCL and RRCL), isogenic mutants (SRMH2, RSMH2, RRMH2, and RRMHO2), and SSWT (used as a reference) resulted in highly similar fluorescence signals for all cell populations (see Fig. S1 and S2 in the supplemental material). In vitro germination tests in fetal bovine serum showed that both clinical isolates SCL and RRCL showed decreased filamentation efficacy. After 1 h at 37°C, about 60% of all cells formed germ tubes, while all isogenic mutants (RSMH1, SRMH1, RRMH2, and RRMHO2) behaved like the parental wild-type strain SC5314 (after 1 h at 37°C, 100% of all cells formed germ tubes) (Fig. 5). A decreased filamentation ability was most pronounced in RRCL.
FIG 5.

Filamentation ability of the clinical strains SCL (A) and RRCL (B) and the corresponding laboratory strains SSWT (C) and RRMH2 (D) in fetal bovine serum after 60 min at 37°C. Delayed germination of approximately 40% of all Candida albicans cells is observed in panels A and B, but 100% of all cells in panels C and D are germinated.
For the first time, it was demonstrated that these SNPs in FKS1 HS1 do not necessarily have an impact on the (i) chitin content, (ii) filamentation ability, and (iii) virulence of C. albicans strains when only these SNPs are integrated into a wild-type strain.
DISCUSSION
In the current study, a female patient suffering from CMC due to MICA- and CAS-resistant C. albicans (Table 1) failed to be cured by long-term CAS medication but was cured by ANI. To determine whether strain RRCL evolved from SCL or replaced SCL under selection pressure of CAS, RAPD genotyping of the clinical isolates was performed. The efficacy of RAPD as a typing method for clinical C. albicans isolates was previously demonstrated (17, 18). RAPD revealed a clonal relationship between the isolates (Fig. 2), which was additionally supported by the presence of six identical synonymous substitutions, one of which was located in the FKS1 HS1 (Table 2). Based on this data, we conclude that echinocandin resistance of C. albicans emerged in the patient.
Molecular analyses demonstrated that RRCL carried the amino acid substitutions R647R/G and P649P/L in FKS1 HS1 in the same allele (Table 2); the occurrence of such a double amino acid substitution in FKS1 HS1 is new, and its impact on in vitro and in vivo echinocandin susceptibility is demonstrated for the first time.
Recently, it was shown that isolated heterozygous (29) and homozygous (23) point mutations at the amino acid position P649 refer to echinocandin resistance. In contrast to the isolates studied by Arendrup et al. (29) and Garcia-Effron et al. (23), our clinical isolate carries a heterozygous P649P/L substitution that is accompanied by a second amino acid substitution (R647R/G). Also, the isolated homozygous amino acid substitution R647G was published previously by Dannaoui et al. (30). This mutation was found to influence the MICs of only CAS and MICA but not ANI. In general, mutations at positions P649 and P647 in C. albicans were associated with discrete elevations of echinocandin MICs compared with those of FKS1 HS1 alterations involving codons S645 and F641 (22). In our study, only the simultaneous presence of both mutations leads to high-level MICs for CAS and MICA (Table 1). Infections caused by isolates carrying an FKS1 mutation at either position S645 or F641 were not cleared by standard doses (10 mg/kg) of MICA and CAS (31, 32). This is similar to our data, as strain RRCL was demonstrated in vivo to be resistant in a hematogenous murine model against all echinocandins at a standard dose (Fig. 3). However, in our case study, CMC was cured successfully with ANI treatment. This discrepancy may be explained by various factors. First, the hematogenous infection in the animal model may be more difficult to cure than the less-severe superficial infection in this patient. Second, the lower virulence and growth rate of the clinical isolate may lead to a slower response to treatment, as a certain number of multiplication rounds are needed for activity. Thus, efficacy might have been obtained if the treatment duration had been extended beyond the standard 3 days in the mouse model. Moreover, comparing the in vitro susceptibility of RRCL with that of the corresponding isogenic mutant strain RRMH2, it is obvious that ANI susceptibility is less influenced by these mutations than is the case for CAS and MICA. The MICs of the clinical isolate are remarkably higher for CAS and MICA. This finding correlated with the clinical response, as the patient failed caspofungin but not anidulafungin treatment. Since strain RRMH2 displays pronounced trailing when tested according to the EUCAST breakpoints, however, the micafungin MIC data interpretation may be affected.
A fundamental finding was that the clinical isolate RRCL and the corresponding RRMH2 differ in their in vivo responses, particularly with dose escalation (AUC500) (Fig. 3 and 4). The efficacy in mice challenged with RRCL was not improved with high doses of ANI, MICA, and CAS, in contrast to what was observed for mice challenged with the isogenic mutant RRMH2. Such differences may be explained by the different genetic backgrounds of the clinical and the laboratory-derived mutant strains or by additional molecular mechanisms that were acquired by RRCL during long-term exposure to CAS. Such additional adaptations associated with echinocandin resistance in C. albicans were published previously; among them are the overexpression of HSP90 (33), the overexpression of Cdr2p ATP-binding cassette (ABC) transporter (34), and elevated chitin levels in fungal cell wall (35). Besides the FKS1 HS1 mutations, additional simultaneously operating mechanisms might contribute to the altered echinocandin resistance of the clinical C. albicans strain RRCL. To fully address the question of alternative mechanisms that enhance echinocandin resistance, a whole-genome comparison and RNA sequencing will be performed on the clinical isolates SCL and RRCL.
Interestingly, others have reported that some FKS1 mutations are associated with reduced virulence and fitness in clinical and laboratory-derived mutant strains (35–37). In the current study, the fungal loads of tissue homogenates of mice infected with either the parental strain SC5314 or isogenic mutant strains were identical, suggesting that mutations do not affect virulence or fitness in vivo. Notably, the clinical isolates SCL and RRCL show reduced filamentation in vitro, in contrast to those of the parental strain SC5314 and the derived isogenic mutants (Fig. 1 and 5). Ben-Ami et al. (36) explained the loss of fitness in homozygous FKS1 HS mutants (F641S, S645F, and S645P) by a reduced maximum catalytic capacity of glucan synthase complex and increased cell wall chitin that comes along with thickened cell walls hampered by filamentation capacities (37). The isogenic mutants investigated herein (carrying either R647R/G and/or P649P/L or R649G and P649L in FKS1 HS1) lacked any change in cell wall chitin (see Fig. S1 in the supplemental material). Therefore, mutations at positions R647R/G and/or P649P/L seem not to influence the filamentation of C. albicans, adding to the previous observations that various resistance mutations may or may not influence virulence and fitness (38). The impact of the position of the amino acid substitution in the FKS1 gene on the fitness of C. albicans needs to be further investigated using competition experiments with bar-coded strains. Moreover, increased or decreased fitness of organisms may depend on the immune status of the host, as significant differences between immunocompetent and neutropenic mice were found (37).
Slater et al. (31) reported that infections due to homozygous FKS S645 mutants were not cleared with MICA at standard or elevated (AUC400) doses. The finding of Slater et al. (31) might be explained by the fact that S645 is the most dominant resistance mutation described yet, and in comparison to our study dose, it was escalated only four times. In our study, infections with RRMHO2 failed treatment with MICA100, ANI100, CAS100, MICA500, and ANI500 (Fig. 4), even though a dose response was observed, as the CFU count decreased for escalated doses (AUC500). Compared with strains in the study of Slater et al. (31), our strains carry weaker mutations and therefore show a partial dose-dependent response. However, only CAS500 was able to clear the infection with RRMHO2 below the detection level. Hence, dose escalation may not be sufficient to fully overcome infections with double-mutant strains.
Translated into clinical practice, our data suggest that the EUCAST MICs of echinocandins interpreted by the associated EUCAST breakpoints correlate with in vivo responses using a standard dosing regimen, but they cannot predict the in vivo response to dose escalation according to the murine model applied. Of note, EUCAST and CLSI have abstained from setting caspofungin epidemiological cutoff values for Candida species because of unacceptable high variation in the MIC ranges obtained over time and between centers (27). Also, commercial systems, such as Etest, do not overcome these variation problems (28). Therefore, it is currently not recommended to perform caspofungin susceptibility testing in routine laboratories; instead, anidulafungin and micafungin should be tested and reported as echinocandin markers (27). Nevertheless, caspofungin susceptibility testing is performed for research purposes to evaluate the degree of increase in the MIC. The presence of simultaneous mutations within the FKS1, as well as the history of long-term echinocandin therapy and the occurrence of homozygous FKS1 mutations, should be treated with caution in terms of ongoing treatment with ANI, CAS, or MICA. The presence of an FKS1 HS mutation in a clinical isolate does not necessarily imply that the isolate is less virulent than a wild-type C. albicans strain. C. albicans strains carrying either a heterozygous/homozygous double mutation in an allele or homozygous single mutations are more likely to be therapy refractory in vivo not only at standard doses but also with dose escalation.
In conclusion, the adoption of EUCAST breakpoints for the clinical strains resulted in a classification that correlated with the clinical response in the patient. In the animal model, the clinical strains with heterozygous double mutations within the FKS1 gene and the laboratory-generated strain with a homozygous double mutation failed to respond to dose escalation. C. albicans strains carrying heterozygous double mutations have higher in vitro MICs for echinocandins than do the parental and heterozygous single-mutation strains. Neither the heterozygous single mutations (R647R/G and P649P/L), the heterozygous double mutations (R647R/G and P649P/L), nor the homozygous double mutations (R647G and P649L) in the FKS1 HS1 had an impact on (i) the virulence of isogenic C. albicans with respect to CFU/ml kidney homogenate, (ii) the filamentation capacity, and (iii) chitin content. Therefore, any loss of fitness and/or virulence cannot be provided based on the presence of FKS1 HS mutations, but it probably depends on the nature and position of the specific alteration. The various FKS1 HS mutations differ in MIC elevations, in their response to ANI, CAS, and MICA, and in their impact on virulence.
Supplementary Material
ACKNOWLEDGMENTS
K.K. was supported by grants from the FWF (FWF-DAC AP-I0125–B22 and AP-25333–B22).
We thank Jytte Mark Andersen and Birgit Brandt for their excellent technical assistance. We also thank Markus Nagl for statistical support and D. Hnisz for mutant generation design.
Footnotes
Published ahead of print 14 April 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00123-14.
REFERENCES
- 1.Delaloye J, Calandra T. 2013. Invasive candidiasis as a cause of sepsis in the critically ill patient. Virulence 5:154–162. 10.4161/viru.26187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang CH, He XS, Chen J, Ouyang B, Zhu XF, Chen MY, Xie WF, Chen L, Zheng DH, Zhong Y, Chen XX, Guan XD. 2012. Fungal infection in patients after liver transplantation in years 2003 to 2012. Ann. Transplant. 17:59–63. 10.12659/AOT.883695 [DOI] [PubMed] [Google Scholar]
- 3.Pappas PG, Kauffman CA, Andes D, Benjamin DK, Jr, Calandra TF, Edwards JE, Jr, Filler SG, Fisher JF, Kullberg BJ, Ostrosky-Zeichner L, Reboli AC, Rex JH, Walsh TJ, Sobel JD, Infectious Diseases Society of America 2009. Clinical practice guidelines for the management of candidiasis: 2009 update by the Infectious Diseases Society of America. Clin. Infect. Dis. 48:503–535. 10.1086/596757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ullmann AJ, Akova M, Herbrecht R, Viscoli C, Arendrup MC, Arikan-Akdagli S, Bassetti M, Bille J, Calandra T, Castagnola E, Cornely OA, Donnelly JP, Garbino J, Groll AH, Hope WW, Jensen HE, Kullberg BJ, Lass-Flörl C, Lortholary O, Meersseman W, Petrikkos G, Richardson MD, Roilides E, Verweij PE, Cuenca-Estrella M, ESCMID Fungal Infection Study Group 2012. ESCMID* guideline for the diagnosis and management of Candida diseases 2012: adults with haematological malignancies and after haematopoietic stem cell transplantation (HCT). Clin. Microbiol. Infect. 18(Suppl 7):53–67. 10.1111/1469-0691.12041 [DOI] [PubMed] [Google Scholar]
- 5.Jensen RH, Johansen HK, Arendrup MC. 2013. Stepwise development of a homozygous S80P substitution in Fks1p, conferring echinocandin resistance in Candida tropicalis. Antimicrob. Agents Chemother. 57:614–617. 10.1128/AAC.01193-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lewis JS, Jr, Wiederhold NP, Wickes BL, Patterson TF, Jorgensen JH. 2013. Rapid emergence of echinocandin resistance in Candida glabrata resulting in clinical and microbiologic failure. Antimicrob. Agents Chemother. 57:4559–4561. 10.1128/AAC.01144-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pfaller MA, Moet GJ, Messer SA, Jones RN, Castanheira M. 2011. Candida bloodstream infections: comparison of species distributions and antifungal resistance patterns in community-onset and nosocomial isolates in the SENTRY Antimicrobial Surveillance Program, 2008–2009. Antimicrob. Agents Chemother. 55:561–566. 10.1128/AAC.01079-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pfaller MA. 2012. Antifungal drug resistance: mechanisms, epidemiology, and consequences for treatment. Am. J. Med. 125(1 Suppl):S3–S13. 10.1016/j.amjmed.2011.10.001 [DOI] [PubMed] [Google Scholar]
- 9.Pfaller MA, Messer SA, Woosley LN, Jones RN, Castanheira M. 2013. Echinocandin and triazole antifungal susceptibility profiles for clinical opportunistic yeast and mold isolates collected from 2010 to 2011: application of new CLSI clinical breakpoints and epidemiological cutoff values for characterization of geographic and temporal trends of antifungal resistance. J. Clin. Microbiol. 51:2571–2581. 10.1128/JCM.00308-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perlin DS. 2007. Resistance to echinocandin-class antifungal drugs. Drug Resist. Updat. 10:121–130. 10.1016/j.drup.2007.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Niimi K, Monk BC, Hirai A, Hatakenaka K, Umeyama T, Lamping E, Maki K, Tanabe K, Kamimura T, Ikeda F, Uehara Y, Kano R, Hasegawa A, Cannon RD, Niimi M. 2010. Clinically significant micafungin resistance in Candida albicans involves modification of a glucan synthase catalytic subunit GSC1 (FKS1) allele followed by loss of heterozygosity. J. Antimicrob. Chemother. 65:842–852. 10.1093/jac/dkq073 [DOI] [PubMed] [Google Scholar]
- 12.Katiyar SK, Alastruey-Izquierdo A, Healey KR, Johnson ME, Perlin DS, Edlind TD. 2012. Fks1 and Fks2 are functionally redundant but differentially regulated in Candida glabrata: implications for echinocandin resistance. Antimicrob. Agents Chemother. 56:6304–6309. 10.1128/AAC.00813-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park S, Kelly R, Kahn JN, Robles J, Hsu MJ, Register E, Li W, Vyas V, Fan H, Abruzzo G, Flattery A, Gill C, Chrebet G, Parent SA, Kurtz M, Teppler H, Douglas CM, Perlin DS. 2005. Specific substitutions in the echinocandin target Fks1p account for reduced susceptibility of rare laboratory and clinical Candida sp. isolates. Antimicrob. Agents Chemother. 49:3264–3273. 10.1128/AAC.49.8.3264-3273.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arendrup MC. 2013. Candida and candidaemia. Susceptibility and epidemiology. Dan. Med. J. 60:B4698.. [PubMed] [Google Scholar]
- 15.Butler G, Rasmussen MD, Lin MF, Santos MA, Sakthikumar S, Munro CA, Rheinbay E, Grabherr M, Forche A, Reedy JL, Agrafioti I, Arnaud MB, Bates S, Brown AJ, Brunke S, Costanzo MC, Fitzpatrick DA, de Groot PW, Harris D, Hoyer LL, Hube B, Klis FM, Kodira C, Lennard N, Logue ME, Martin R, Neiman AM, Nikolaou E, Quail MA, Quinn J, Santos MC, Schmitzberger FF, Sherlock G, Shah P, Silverstein KA, Skrzypek MS, Soll D, Staggs R, Stansfield I, Stumpf MP, Sudbery PE, Srikantha T, Zeng Q, Berman J, Berriman M, Heitman J, Gow NA, Lorenz MC, Birren BW, Kellis M, Cuomo CA. 2009. Evolution of pathogenicity and sexual reproduction in eight Candida genomes. Nature 459:657–662. 10.1038/nature08064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jayasinghe M, Schmidt S, Walker B, Röcken M, Schaller M. 2006. Successful treatment of azole-resistant chronic mucocutaneous candidosis with caspofungin. Acta Derm. Venereol. 86:563–564. 10.2340/00015555-0165 [DOI] [PubMed] [Google Scholar]
- 17.Ben Abdeljelil J, Saghrouni F, Emira N, Valentin-Gomez E, Chatti N, Boukadida J, Ben Said M, Del Castillo Agudo L. 2011. Molecular typing of Candida albicans isolates from patients and health care workers in a neonatal intensive care unit. J. Appl. Microbiol. 111:1235–1249. 10.1111/j.1365-2672.2011.05121.x [DOI] [PubMed] [Google Scholar]
- 18.Issa SY, Badran EF, Aqel KF, Shehabi AA. 2011. Epidemiological characteristics of Candida species colonizing oral and rectal sites of Jordanian infants. BMC Pediatr. 11:79. 10.1186/1471-2431-11-79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lockhart SR, Joly S, Pujol C, Sobel JD, Pfaller MA, Soll DR. 1997. Development and verification of fingerprinting probes for Candida glabrata. Microbiology 143:3733–3746. 10.1099/00221287-143-12-3733 [DOI] [PubMed] [Google Scholar]
- 20.Vasdinyei R, Deák T. 2003. Characterization of yeast isolates originating from Hungarian dairy products using traditional and molecular identification techniques. Int. J. Food Microbiol. 86:123–130. 10.1016/S0168-1605(03)00251-4 [DOI] [PubMed] [Google Scholar]
- 21.Arendrup MC, Cuenca-Estrella M, Lass-Flörl C, Hope W, EUCAST-AFST 2012. EUCAST technical note on the EUCAST definitive document EDef 7.2: method for the determination of broth dilution minimum inhibitory concentrations of antifungal agents for yeasts EDef 7.2 (EUCAST-AFST). Clin. Microbiol. Infect. 18:E246–E247. 10.1111/j.1469-0691.2012.03880.x [DOI] [PubMed] [Google Scholar]
- 22.Arendrup MC, Cuenca-Estrella M, Lass-Flörl C, Hope WW, European Committee on Antimicrobial Susceptibility Testing–Subcommittee on Antifungal Susceptibility Testing (EUCAST-AFST) 2013. EUCAST technical note on Candida and micafungin, anidulafungin and fluconazole. Mycoses 17:18–20. 10.1111/myc.12170 [DOI] [Google Scholar]
- 23.Garcia-Effron G, Park S, Perlin DS. 2009. Correlating echinocandin MIC and kinetic inhibition of fks1 mutant glucan synthases for Candida albicans: implications for interpretive breakpoints. Antimicrob. Agents Chemother. 53:112–122. 10.1128/AAC.01162-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reuss O, Vik A, Kolter R, Morschhäuser J. 2004. The SAT1 flipper, an optimized tool for gene disruption in Candida albicans. Gene 341:119–127. 10.1016/j.gene.2004.06.021 [DOI] [PubMed] [Google Scholar]
- 25.Gillum AM, Tsay EY, Kirsch DR. 1984. Isolation of the Candida albicans gene for orotidine-5′-phosphate decarboxylase by complementation of S. cerevisiae ura3 and E. coli pyrF mutations. Mol. Gen. Genet. 198:179–182. 10.1007/BF00328721 [DOI] [PubMed] [Google Scholar]
- 26.Arendrup MC, Perlin DS, Jensen RH, Howard SJ, Goodwin J, Hope W. 2012. Differential in vivo activities of anidulafungin, caspofungin, and micafungin against Candida glabrata isolates with and without FKS resistance mutations. Antimicrob. Agents Chemother. 56:2435–2442. 10.1128/AAC.06369-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Espinel-Ingroff A, Arendrup MC, Pfaller MA, Bonfietti LX, Bustamante B, Canton E, Chryssanthou E, Cuenca-Estrella M, Dannaoui E, Fothergill A, Fuller J, Gaustad P, Gonzalez GM, Guarro J, Lass-Flörl C, Lockhart SR, Meis JF, Moore CB, Ostrosky-Zeichner L, Pelaez T, Pukinskas SR, St-Germain G, Szeszs MW, Turnidge J. 2013. Interlaboratory variability of caspofungin MICs for Candida spp. using CLSI and EUCAST methods: should the clinical laboratory be testing this agent? Antimicrob. Agents Chemother. 57:5836–5842. 10.1128/AAC.01519-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arendrup MC, Pfaller MA, Danish Fungaemia Study Group 2012. Caspofungin Etest susceptibility testing of Candida species: risk of misclassification of susceptible isolates of C. glabrata and C. krusei when adopting the revised CLSI caspofungin breakpoints. Antimicrob. Agents Chemother. 56:3965–3968. 10.1128/AAC.00355-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arendrup MC, Perlin DS, Jensen RH, Howard SJ, Goodwin J, Hope W. 2012. Differential in vivo activities of anidulafungin, caspofungin, and micafungin against Candida glabrata isolates with and without FKS resistance mutations. Antimicrob. Agents Chemother. 56:2435–2442. 10.1128/AAC.06369-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dannaoui E, Desnos-Ollivier M, Garcia-Hermoso D, Grenouillet F, Cassaing S, Baixench MT, Bretagne S, Dromer F, Lortholary O, French Mycoses Study Group 2012. Candida spp. with acquired echinocandin resistance, France, 2004–2010. Emerg. Infect. Dis. 18:86–90. 10.3201/eid1801.110556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Slater JL, Howard SJ, Sharp A, Goodwin J, Gregson LM, Alastruey-Izquierdo A, Arendrup MC, Warn PA, Perlin DS, Hope WW. 2011. Disseminated candidiasis caused by Candida albicans with amino acid substitutions in Fks1 at position Ser645 cannot be successfully treated with micafungin. Antimicrob. Agents Chemother. 55:3075–3083. 10.1128/AAC.01686-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wiederhold NP, Najvar LK, Bocanegra RA, Kirkpatrick WR, Patterson TF. 2011. Caspofungin dose escalation for invasive candidiasis due to resistant Candida albicans. Antimicrob. Agents Chemother. 55:3254–3260. 10.1128/AAC.01750-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singh SD, Robbins N, Zaas AK, Schell WA, Perfect JR, Cowen LE. 2009. Hsp90 governs echinocandin resistance in the pathogenic yeast Candida albicans via calcineurin. PLoS Pathog. 5:e1000532. 10.1371/journal.ppat.1000532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schuetzer-Muehlbauer M, Willinger B, Krapf G, Enzinger S, Presterl E, Kuchler K. 2003. The Candida albicans Cdr2p ATP-binding cassette (ABC) transporter confers resistance to caspofungin. Mol. Microbiol. 48:225–235. 10.1046/j.1365-2958.2003.03430.x [DOI] [PubMed] [Google Scholar]
- 35.Lee KK, Maccallum DM, Jacobsen MD, Walker LA, Odds FC, Gow NA, Munro CA. 2012. Elevated cell wall chitin in Candida albicans confers echinocandin resistance in vivo. Antimicrob. Agents Chemother. 56:208–217. 10.1128/AAC.00683-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ben-Ami R, Garcia-Effron G, Lewis RE, Gamarra S, Leventakos K, Perlin DS, Kontoyiannis DP. 2011. Fitness and virulence costs of Candida albicans FKS1 hot spot mutations associated with echinocandin resistance. J. Infect. Dis. 204:626–635. 10.1093/infdis/jir351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ben-Ami R, Kontoyiannis DP. 2012. Resistance to echinocandins comes at a cost: the impact of FKS1 hotspot mutations on Candida albicans fitness and virulence. Virulence 3:95–97. 10.4161/viru.3.1.18886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Andersson DI, Hughes D. 2010. Antibiotic resistance and its cost: is it possible to reverse resistance? Nat. Rev. Microbiol. 8:260–271. 10.1038/nrmicro2319 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.