

Summary
Protein with tau-like repeats (PTL-1) is the sole Caenorhabditis elegans homolog of tau and MAP2, which are members of the mammalian family of microtubule-associated proteins (MAPs). In mammalian neurons, tau and MAP2 are segregated, with tau being mainly localised to the axon and MAP2 mainly to the dendrite. In particular, tau plays a crucial role in pathology, as elevated levels lead to the formation of tau aggregates in many neurodegenerative conditions including Alzheimer's disease. We used PTL-1 in C. elegans to model the biological functions of a tau-like protein without the complication of functional redundancy that is observed among the mammalian MAPs. Our findings indicate that PTL-1 is important for the maintenance of neuronal health as animals age, as well as in the regulation of whole organism lifespan. In addition, gene dosage of PTL-1 is crucial because variations from wild-type levels are detrimental. We also observed that human tau is unable to robustly compensate for loss of PTL-1, although phenotypes observed in tau transgenic worms are dependent on the presence of endogenous PTL-1. Our data suggest that some of the effects of tau pathology result from the loss of physiological tau function and not solely from a toxic gain-of-function due to accumulation of tau.
Key words: Tau, Protein with tau-like repeats, PTL-1, Caenorhabditis elegans, Neurodegenerative diseases, Alzheimer's disease, Aging
Introduction
Tau is a neuronal microtubule-associated protein (MAP) that is predominantly localised to axons (Weingarten et al., 1975; Kosik and Finch, 1987). In mammals, tau has a role in regulating microtubule assembly, stability, and organisation (Cleveland et al., 1977b; Chen et al., 1992; Harada et al., 1994). Mutations in the MAPT locus, which encodes tau, are associated with an increased risk of several neuronal disorders including Parkinson's disease and corticobasal degeneration (reviewed by Wade-Martins, 2012). Furthermore, the pathological significance of tau is demonstrated by the presence of neurofibrillary tangles (NFTs), composed of fibrillar aggregates of this protein, in the brains of individuals suffering from neurodegenerative disorders such as Alzheimer's disease (Lee et al., 2001; Iqbal et al., 2010). It is only partly understood how tau exerts its toxic effects, whether in fibrillar form (Ittner et al., 2008), or before the formation of tangles due to the loss of some important physiological functions (Gómez-Isla et al., 1997; Lee and Leugers, 2012). Despite the significant advances made using tau transgenic and knockout models (reviewed by Götz and Ittner, 2008; Götz et al., 2010; Ittner et al., 2011; Ke et al., 2012), these studies are complicated by the presence of other neuronal MAPs such as MAP2, which may share several physiological roles. In fact, tau knockout mice do not display overt defects in neuronal development or function (Harada et al., 1994; Dawson et al., 2001; Tucker et al., 2001).
Caenorhabditis elegans has one putative homolog in the tau/MAP2/MAP4 family of MAPs, called protein with tau-like repeats (PTL-1) (Goedert et al., 1996; McDermott et al., 1996). PTL-1 contains a high level of sequence homology to tau/MAP2/MAP4 within the microtubule binding repeat (MBR) domain in the carboxyl (C)-terminus, and has been shown to regulate microtubule assembly in vitro (Goedert et al., 1996; McDermott et al., 1996). Immunohistochemistry and analysis of a ptl-1 transcriptional reporter line demonstrate that PTL-1 has a neuronal expression pattern in adult worms (Goedert et al., 1996; Gordon et al., 2008), and henceforth we refer to PTL-1 as the tau/MAP2 homolog as these are the neuronal MAPs in mammals (Dehmelt and Halpain, 2005). PTL-1 has also been shown to have neuronal functions in worms, as it has been implicated in the regulation of microtubule-based motility in several neurons (Tien et al., 2011) and for the optimal functioning of touch receptor neurons in the response to gentle touch (Gordon et al., 2008). Therefore, C. elegans is a convenient in vivo model in which to study the physiological roles of a tau/MAP2-like protein without having to consider compensatory effects produced by other closely-related MAPs.
Tau is implicated in several neurodegenerative conditions called tauopathies, including Alzheimer's disease, and these have an increased incidence with age. Thus, it is of particular interest to understand any age-dependent neuronal effects of perturbing tau or tau-like proteins. A novel aging phenotype was recently observed in C. elegans, where neurons display abnormal structures such as branching or blebbing from the cell body or axon, and these changes progressively accumulate as the organism ages (Pan et al., 2011; Tank et al., 2011; Toth et al., 2012). In a wild-type animal, these morphological changes occur at a low frequency in early adulthood and gradually increase in frequency with age, however, the appearance of these aberrant structures can be delayed or accelerated when mutations are introduced in either tubulin genes (Pan et al., 2011), components of the insulin signalling pathway (Pan et al., 2011; Tank et al., 2011; Toth et al., 2012), or stress-response effectors (Pan et al., 2011; Tank et al., 2011; Toth et al., 2012). We sought to examine if loss of PTL-1 in C. elegans would affect the incidence of these neuronal phenotypes.
We examined aging neurons in ptl-1 mutant strains and observed that these animals show an increased incidence of abnormal structures compared with wild-type animals, and moreover, displayed a reduced lifespan. We were able to rescue accelerated neuronal aging and shortened organismal lifespan by re-expressing PTL-1 in a ptl-1 null mutant. Interestingly, we found that gene dosage of PTL-1 is critical, since either increasing or decreasing the number of copies of ptl-1 is detrimental to the organism. Our findings demonstrate a key role of PTL-1 in maintaining neuronal health with age and in regulating whole organism lifespan.
Results
ptl-1(tm543) and ptl-1(ok621) mutant strains display allelic differences in touch sensitivity
PTL-1 in C. elegans displays high sequence similarity to mammalian tau/MAP2/MAP4 in the C-terminal MBR domain. It is encoded by the gene ptl-1, which is composed of eight coding exons, with exons 5–7 encoding the MBRs (Fig. 1A). Two deletion alleles have been generated for ptl-1, both of which were used in this study: the ok621 mutation generated by the OMRF arm of the C. elegans Knockout Consortium (WormBase ID: WBVar00091905), and the tm543 mutation by the National Bioresource Project, Japan (WormBase ID: WBVar00249582) (Fig. 1A). ptl-1(ok621) is a null mutation (Gordon et al., 2008), whereas ptl-1(tm543) is a shorter deletion encompassing the exons encoding the MBR region, and may result in a truncated protein that has retained the N-terminal projection domain. There are several putative isoforms of ptl-1, with transcripts labelled F42G9.9a–d (http://www.wormbase.org). As a reference for our experiments, we used the sequence of isoform a (F42G9.9a) as this is a confirmed gene that generates a protein with five MBRs (Goedert et al., 1996; Gordon et al., 2008), whereas the other confirmed isoform of PTL-1 (isoform b) has only four putative MBRs (Goedert et al., 1996; McDermott et al., 1996).
Fig. 1.
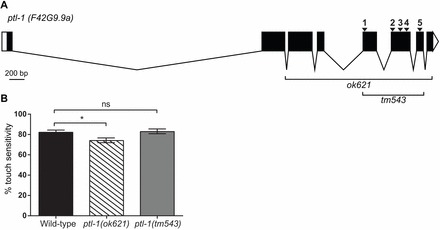
ptl-1 alleles ok621 and tm543 display allelic differences in touch sensitivity. (A) Genomic locus of ptl-1 (transcript F42G9.9a) indicating regions comprising the ok621 and tm543 deletions. Exons are shown as black boxes, introns as solid lines and untranslated regions at the 5′ and 3′ as white boxes; 5′ is on the left. Regions encoding putative microtubule binding repeats are marked 1–5 with arrowheads. (B) Data for sensitivity to gentle touch, shown for ptl-1(ok621) and ptl-1(tm543) mutant strains and wild-type controls (n = 45 in total for two biological replicates). Assays were conducted on 1-day-old adults. Bars indicate mean±s.e.m. One-way ANOVA, Bonferroni post-test; ns, no significance; *P<0.05.
Mammalian MAPs such as tau and MAP2 are known to execute a range of physiological functions in neurons (Drubin and Kirschner, 1986; Avila et al., 2004; Dehmelt and Halpain, 2005; Ittner et al., 2010). As two alleles of ptl-1 are now available, we investigated the role of PTL-1 in C. elegans neurons by assaying both ptl-1(ok621) and ptl-1(tm543) mutant strains. Although PTL-1 is expressed in several neuronal subtypes in adult worms (Goedert et al., 1996; Gordon et al., 2008), the highest levels are detected in the touch receptor neurons, which regulate the response to gentle touch (Chalfie and Sulston, 1981; Chalfie et al., 1985). Previously published data indicate that ptl-1(ok621) mutants are mildly touch insensitive compared with wild-type worms (Gordon et al., 2008). In line with this, we found that ptl-1(ok621) worms were less touch sensitive (touch sensitivity = 74±2%) compared with wild-type (82±2%) (Fig. 1B). We also tested the ptl-1(tm543) C-terminal deletion strain, and observed that there was no difference in touch sensitivity between this mutant (83±2%) and wild-type (Fig. 1B). These findings suggest that loss of full-length PTL-1, but not of the MBR domain alone, results in defective touch receptor neurons.
ptl-1(ok621) and ptl-1(tm543) mutant strains show a high frequency of abnormal neuronal structures in touch receptor and GABAergic neurons in early adulthood
Recently, a novel age-related phenotype was reported in touch receptor neurons where in wild-type animals, axons accumulated branches and blebs, and cell bodies displayed branching as the animals aged (Pan et al., 2011; Tank et al., 2011; Toth et al., 2012). The appearance of these abnormal neuronal structures was accelerated in the presence of mutations in mec-7 and mec-12 tubulin genes (Pan et al., 2011), both of which are highly expressed in touch neurons (Mitani et al., 1993; Savage et al., 1994; Fukushige et al., 1999). We investigated whether mutations in ptl-1 would produce a similar effect. Initially, we studied the appearance of abnormal neuronal structures in wild-type and ptl-1(ok621) animals by imaging individual worms until the animals died (Fig. 2A,B; supplementary material Fig. S1). By assaying 15 worms of each genotype every day over their entire lifespan, we observed a progressive phenotype as previously reported, where wild-type neurons accumulated branching and blebbing structures with age. Interestingly, our data indicate that the accumulation of morphological changes is significantly accelerated in the touch receptor neurons in ptl-1(ok621) mutants. We observed that the proportion of ptl-1(ok621) animals with neurons displaying abnormal structures reached 50% at day 4, compared with wild-type animals where this level was only reached at day 8 (Fig. 2C).
Fig. 2.
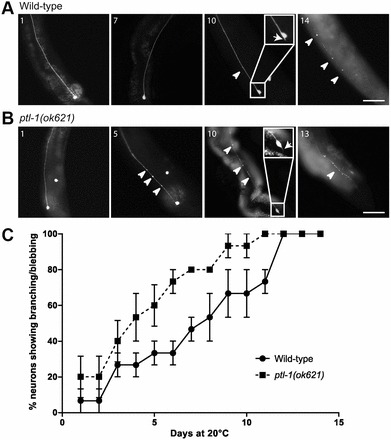
ptl-1(ok621) mutant strain displays an accelerated onset of appearance of abnormal neuronal structures in touch receptor neurons. A representative animal is shown for each genotype (n = 15 total). Neurons were visualised using the Pmec-4::gfp reporter. Worms were imaged every day until death. Representative time points are shown, with arrowheads indicating blebbing and arrows indicating branching phenotypes. For images taken at all time points of a representative worm, see supplementary material Fig. S1. Insets on day 10 show a close-up of the cell body for the respective animal. (A) ALM neuron of a wild-type worm. The worm died on day 15. (B) ALM neuron of a ptl-1(ok621) mutant worm. The worm died on day 14. (C) Percentage of branching/blebbing observed in the complete data set of wild-type and ptl-1(ok621) animals (n = 15). Values are mean±s.e.m. A paired t-test was applied to the means of each strain at each time point and the average of the entire curve compared; P<0.001. Scale bars: 50 µm.
To complement this experiment, we used a transverse approach, observing synchronised populations of wild-type and ptl-1(ok621) animals on alternate days from day 1 to 15 of adulthood. In this experiment, we recorded data for the touch receptor neurons at the anterior and posterior halves of the animal separately, as it became apparent that the posterior touch neurons accumulate branches and blebs at a much faster rate compared with the anterior touch neurons, showing a high incidence of these abnormal structures even in early adulthood. Additionally, since PTL-1 is not expressed in one of the three posterior touch neurons (PVM) (Goedert et al., 1996; Gordon et al., 2008), only the morphology of the two PLM neurons was considered in this analysis. Although we were able to observe similar trends when comparing the morphology of both posterior and anterior touch neurons of wild-type versus ptl-1 mutant strains, we observed high variability in the incidence of abnormal structures in the posterior touch neurons in populations of wild-type animals and therefore we restrict the discussion to findings obtained in anterior touch neurons. Data obtained from scoring posterior touch neurons can be found in supplementary material Fig. S2. For both ptl-1 mutant strains, we separately scored for cell body branching, axon blebbing, and axon branching (Fig. 3A–C). Consistent with our earlier findings, we observed a higher incidence of abnormal structures in the ptl-1(ok621) null mutant compared with wild-type, and in addition we observed the same phenotype in the MBR-deficient ptl-1(tm543) strain. In general, these structural abnormalities occur in a particular order, with cell body branching occurring first, followed by axon blebbing and axon branching. Specifically, on day 5 of the assay we observe cell body branching in 81% of ptl-1(ok621) (n = 37) and 73% of ptl-1(tm543) (n = 37) animals assayed compared with 36% of wild-type controls (n = 39) (Fig. 3A). At the same time point, blebbing can be seen in 19% of ptl-1(ok621) and 22% of ptl-1(tm543) animals compared with 5% in wild-type controls (Fig. 3B), whereas axon branching is observed in 3% of both ptl-1 mutant strains assayed compared with 0% of wild-type control animals (Fig. 3C). Data obtained from scoring branching and blebbing together in anterior touch neurons in ptl-1 mutant strains can be found in supplementary material Fig. S2. Our results suggest that the ptl-1(tm543) mutation could be hypomorphic, such that the protein product in this mutant is able to sustain wild-type levels of touch sensitivity, but is not sufficient to protect the organism from susceptibility to age-dependent loss of structural integrity. Taken together, these observations demonstrate that PTL-1 is important in maintaining neuronal morphology in touch receptor neurons, as a complete loss of this protein or expression of a protein lacking the MBR domain results in a higher incidence of abnormal neuronal structures in younger animals.
Fig. 3.
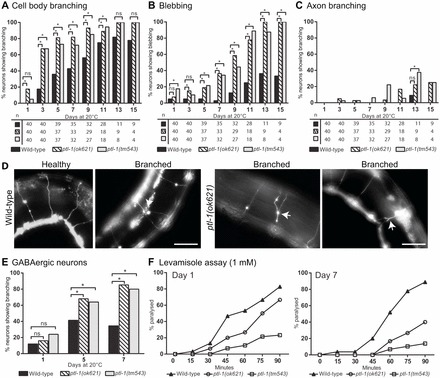
ptl-1(ok621) and ptl-1(tm543) mutant strains show defects in maintaining neuronal integrity with age in touch receptor and GABAergic neurons. Neurons were visualised using the Pmec-4::gfp reporter for touch receptor neurons or the Punc-47::gfp reporter for GABAergic neurons. Worms were imaged every second day from day 1 to day 15. For imaging assays, the χ2 test for independence was used to analyse differences between genotypes. (A–C) Anterior touch neurons in ptl-1 mutants scored for (A) cell body branching, (B) blebbing along the axon and (C) axon branching. Sample sizes are indicated below graphs. (D) Representative image of the phenotype scored in GABAergic neurons, showing healthy neurons and a branched commissure in wild-type, and representative images of branched commissures in ptl-1(ok621) worms. Arrows indicate branching. Scale bars: 50 µm. (E) Data for the incidence of branching in GABAergic neurons for ptl-1 mutant strains. (F) Assay for paralysis after levamisole exposure. Worms were scored for paralysis over 90 minutes on drug plates on day 1 and day 7 of adulthood (n = 30). Statistical analysis, one-way ANOVA; ptl-1(ok621) and ptl-1(tm543) mutant strains, but not wild-type controls, show significant decreases in drug sensitivity between day 1 and day 7. ns, no significance; *P<0.05.
We next investigated if these effects are specific for selected neuronal populations or whether they reflect a general consequence of perturbing PTL-1 function. To this end, we examined a second neuronal cell type in which PTL-1 is also expressed (McKay et al., 2003; Gordon et al., 2008). Using a Punc-47::gfp reporter line, we visualised the 25 GABAergic neurons, which consist of neurons mainly in the ventral nerve cord, as well as in the head and the tail (McIntire et al., 1997). These GABAergic neurons were previously found to also display an age-related phenotype, which is the incidence of branching along commissures that extend to the dorsal side of the animal (Tank et al., 2011). Representative images of the branching phenotype in wild-type and ptl-1(ok621) animals are shown in Fig. 3D. We assayed this phenotype in ptl-1 mutant animals and observed an accelerated accumulation of branching structures in both ptl-1(ok621) and ptl-1(tm543) animals compared with wild-type (Fig. 3E). In particular at day 5, 68% of ptl-1(ok621) and 64% of ptl-1(tm543) animals displayed branching phenotypes compared with 41% in wild-type controls. Data collected for each ptl-1 mutant over 15 days can be seen in supplementary material Fig. S2. We also found that ptl-1 mutant strains show significantly lower sensitivity to the cholinergic agonist levamisole compared with wild-type controls at early and mid adulthood, with the ptl-1(tm543) strain appearing to have lower sensitivity compared with ptl-1(ok621) (Fig. 3F). This suggests that ptl-1 mutant animals are defective in cholinergic/GABAergic transmission (Lewis et al., 1980). Additionally, we observed a significant decrease in levamisole sensitivity for both ptl-1 mutant strains between day 1 and day 7, which correlates with the higher incidence of branching commissures in GABAergic neurons at later time points (Fig. 3E,F). In conclusion, ptl-1 mutant strains also display defects in a neuronal subtype other than touch receptor neurons, suggesting that PTL-1 has a broader role in the maintenance of neuronal structural integrity.
Lifespan reduction in ptl-1(ok621) and ptl-1(tm543) mutant strains
Having found an age-related phenotype in strains carrying mutations in ptl-1, we performed a lifespan assay to determine if differences exist between wild-type and ptl-1 mutant strains. We found that both ptl-1(ok621) (Fig. 4A) and ptl-1(tm543) (Fig. 4B) mutants had a shorter lifespan compared with wild-type. Analysis of the two survival curves indicates that this difference is statistically significant; however, it appears that lifespan reduction is more severe in the ptl-1(ok621) null mutant strain that has a 37% shorter median lifespan compared with wild-type, whereas the median lifespan in the ptl-1(tm543) mutant strain is 10% shorter than wild-type.
Fig. 4.
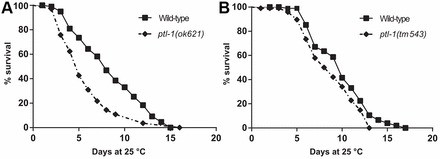
ptl-1 mutant strains display a reduction in lifespan. (A,B) Survival curves for (A) ptl-1(ok621) and (B) ptl-1(tm543) mutant strains. Log rank and Gehan-Breslow-Wilcoxon tests; P<0.05 for both ptl-1(ok621) and ptl-1(tm543) compared with wild-type.
Re-expressing PTL-1 under the control of the ptl-1 promoter rescues the age-dependent neuronal phenotype and lifespan reduction in the ptl-1 null mutant line
To confirm that the neuronal aging and lifespan phenotypes found in ptl-1 mutant worms are attributable to the loss of PTL-1 function, we generated a transgenic line expressing the ptl-1 cDNA under the control of the ptl-1 promoter and the PTL-1 3′UTR control element. As no commercial antibody against PTL-1 is available, we tagged PTL-1 at the C-terminus with the short peptide V5 in order to enable detection of PTL-1 expressed from the transgene. Transgenic worms were generated by biolistic transformation, and an integrated line was obtained after selection using the dual antibiotic method (Semple et al., 2012). We confirmed that our transgene was expressed by immunoblotting (Fig. 5A) and immunofluorescence (Fig. 5B). The size of the band observed on the immunoblot is consistent with previous experiments that have shown that PTL-1 runs at ∼75 kDa on an SDS-PAGE gel (Goedert et al., 1996) (Fig. 5A). Furthermore, using a V5-specific antibody for immunofluorescence staining, we detected PTL-1::V5 localising to axons and cell bodies in neurons, including those comprising the nerve ring in the head, as well as in the tail (Fig. 5B).
Fig. 5.
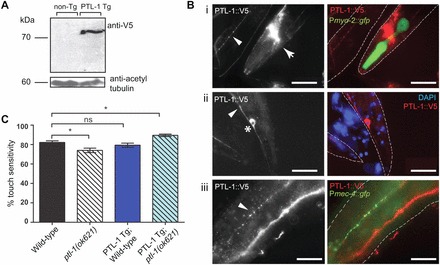
Re-expression of PTL-1 under the regulation of the ptl-1 promoter can be detected in neurons of transgenic animals and rescues touch sensitivity in a null mutant. ‘PTL-1 Tg’ refers to the PTL-1::V5 transgene; the terms ‘wild-type’ and ‘ptl-1(ok621)’ following this refer to the genotype at the genomic ptl-1 locus, whether wild-type or ok621 mutant, respectively. (A) Immunoblot showing the presence of a band corresponding to PTL-1::V5 expression from the transgene, probed using an anti-V5 antibody. Non-Tg, non-transgenic wild-type animals. (B) Immunofluorescence micrographs showing the expression of the PTL-1::V5 transgene (anti-V5 antibody) in neurons. Grayscale images on the left show the red channel only, the nerve ring is indicated by arrows, axons by arrowheads, and cell bodies by asterisks. (Bi) The pharynx is shown and gfp expression can be observed from the Pmyo-2::gfp transformation reporter. (Bii) A tail neuron is shown with staining in both the cell body and axon. (Biii) Co-localisation with a reporter line for touch receptor neurons (Pmec-4::gfp) is shown, demonstrating that the transgene is expressed in touch neurons. Dotted lines indicate the outline of the animal as determined by phase-contrast microscopy. Ventral is down. Scale bars: 50 µm. (C) Data for sensitivity to gentle touch, shown for PTL-1 transgenic animals (n>45 total for two biological replicates). Control strains are the same as those shown in Fig. 1B. Assays were conducted on 1-day-old adults. Bars indicate mean±s.e.m. One-way ANOVA, Bonferroni post-test; ns, no significance; *P<0.05.
We crossed these PTL-1 transgenic worms with both the null mutant ptl-1(ok621) and the Pmec-4::gfp reporter line, and assayed for age-related morphological changes in touch receptor neurons as described above. We confirmed expression of the transgene in touch neurons by staining for PTL-1::V5 in transgenic animals crossed with the Pmec-4::gfp reporter line and observing co-localisation between signals from anti-V5 immunofluorescence and GFP (Fig. 5Biii). We also observed that PTL-1 transgenic animals are touch sensitive both on a wild-type and ptl-1(ok621) null mutant genetic background (Fig. 5C). For assays on neuronal morphology, data are presented only for anterior touch receptor neurons as we have shown that the percentage of animals showing abnormal structures in these neurons is low at early time points and that this is less variable compared with the same phenotype in posterior touch receptor neurons, making it easier to monitor changes in neuron morphology with time. We found that re-expressing PTL-1 in ptl-1(ok621) animals appeared to delay the accumulation of abnormal neuronal structures such as branching and blebbing to almost wild-type levels (Fig. 6Ai), such that when these animals were assayed from day 5 onwards, the proportion of animals showing these phenotypes in anterior touch neurons was not significantly different from wild-type. For example, at day 5 of this assay 20% of PTL-1 transgenic; ptl-1(ok621) worms (n = 40) displayed abnormal structures in anterior touch neurons compared with 32% in wild-type (n = 40). Thus we were able to rescue the accelerated accumulation of abnormal structures in touch neurons of ptl-1(ok621) mutants by re-expressing PTL-1 in these animals. Somewhat surprisingly, we also observed that animals expressing the PTL-1 transgene on a wild-type background accumulated branches and blebbing structures with increased frequency compared with non-transgenic wild-type controls, and that these levels were comparable with that observed in the strain carrying only the ptl-1(ok621) mutation (Fig. 6Aii). For example, at day 5 of this assay, 55% of ‘PTL-1 transgenic; wild-type’ worms (n = 38) displayed abnormal neuronal structures, compared with 32% in wild-type controls (n = 40). It therefore appears that modulation of PTL-1 levels by either a null mutation or increasing the gene copy number (by introducing copy(ies) of a PTL-1 transgene in addition to the endogenous gene locus) negatively affects neuronal integrity.
Fig. 6.
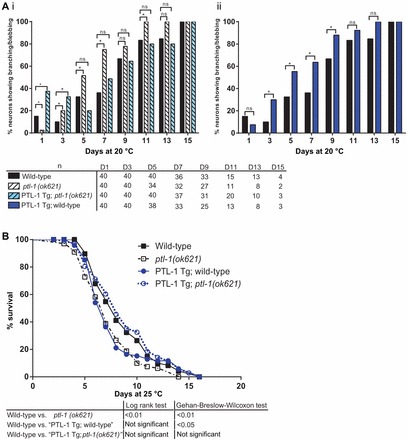
Re-expression of PTL-1 rescues age-related abnormal neuron morphology and lifespan reduction in the ptl-1(ok621) mutant. ‘PTL-1 Tg’ refers to the PTL-1::V5 transgene, and the terms ‘wild-type’ and ‘ptl-1(ok621)’ following this refer to the genotype at the genomic ptl-1 locus, whether wild-type or ok621 mutant, respectively. (A) Neuron imaging time course of PTL-1 transgenic worms, showing data for anterior touch receptor neurons visualised using the Pmec-4::gfp reporter. (Ai) Control strains together with the PTL-1 Tg; ptl-1(ok621) rescue strain. (Aii) Effect of the PTL-1 Tg alone, compared with wild-type controls. Worms were imaged every second day from day 1 to day 15. Sample sizes are indicated below the graph. The χ2 test for independence was used to analyse differences between genotypes; ns, no significance; *P<0.05. (B) Survival curves for PTL-1 transgenic worms. n = 100 for each strain at the start of the assay. Results of statistical analysis are shown in the table below.
Furthermore, we conducted a lifespan assay on PTL-1 transgenic worms that were either wild-type at the genomic ptl-1 locus or that carried the ptl-1(ok621) mutation (Fig. 6B). We found that re-expressing PTL-1 in a ptl-1 null mutant background resulted in a survival curve that was very similar to non-transgenic wild-type worms. Therefore, re-expressing PTL-1 in a ptl-1 null mutant is able to rescue both the lifespan reduction and the premature neuronal aging observed in ptl-1(ok621) animals (Fig. 6A). Interestingly, expressing the PTL-1 transgene together with endogenous PTL-1 led to a reduction in lifespan. This reduction in lifespan for the ‘PTL-1 transgenic; wild-type’ strain is statistically significant according to the Gehan-Breslow-Wilcoxon test for significance. Increasing the number of copies of ptl-1 thus appears to produce a detrimental effect both in terms of neuronal health and whole organism lifespan, despite displaying wild-type touch sensitivity.
Human tau does not robustly rescue for loss of PTL-1
As noted above, PTL-1 in C. elegans and tau in humans have high sequence similarity in the C-terminal domain that is important for microtubule binding, and also have a similar overall charge distribution (Goedert et al., 1996; McDermott et al., 1996; Gordon et al., 2008). To determine if there is functional conservation between these two proteins, we generated transgenic C. elegans strains expressing a cDNA of the longest isoform of human tau (htau40, referred to here as htau). The htau cDNA was expressed under the control of the ptl-1 promoter and PTL-1 3′ UTR as described above. htau transgenic animals were similarly generated by biolistic transformation and an integrated line was isolated. Immunoblotting analysis showed that htau is expressed in the transgenic line but not in non-transgenic wild-type worms (Fig. 7A).
Fig. 7.
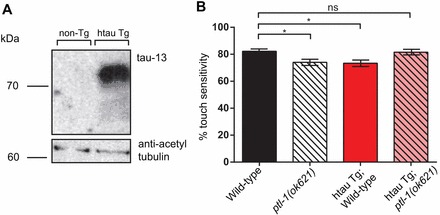
Expression of human tau rescues touch sensitivity in the ptl-1(ok621) null mutant but not in a wild-type background. ‘htau Tg’ refers to the human tau transgene, and the terms ‘wild-type’ and ‘ptl-1(ok621)’ immediately following this refer to the genotype at the ptl-1 locus, whether wild-type or ok621 mutant, respectively. (A) Immunoblot showing the presence of a band corresponding to the htau40 transgene, probed using the tau 13 antibody. Non-Tg, non-transgenic wild-type animals. (B) Data for sensitivity to gentle touch, shown for htau transgenic animals (n>45 total for two biological replicates). Control strains are the same as those shown in Fig. 1B. Assays were conducted on 1-day-old adults. Bars indicate mean±s.e.m. One-way ANOVA, Bonferroni post-test; ns, no significance; *P<0.05.
Interestingly, we observed that htau expression in a wild-type background resulted in touch insensitivity (Fig. 7B), and was also detrimental to worms both in terms of neuronal aging and whole organism lifespan (Fig. 8A,B). In particular, the transgenic line expressing htau in a wild-type background displayed a higher incidence of branching and blebbing phenotypes in anterior touch receptor neurons compared with non-transgenic wild-type worms at day 3, 5, 7 and 9 of the assay (Fig. 8Aii). In addition, this transgenic line also showed a significant reduction in lifespan compared with the wild-type control (Fig. 8B). Our data are consistent with previous reports indicating that wild-type htau expression under the control of a pan-neuronal promoter results in defects in motility, cholinergic neuron transmission, and lifespan (Kraemer et al., 2003). It is notable that htau transgenic lines reported in the study conducted by Kraemer et al. were generated by microinjection (Kraemer et al., 2003), whereas the transgenic lines in our study were generated by biolistic transformation, which we would expect to introduce a lower transgene copy number than by microinjection (Praitis et al., 2001). Therefore it appears that wild-type htau, even at a low expression level, is detrimental to C. elegans when expressed in addition to endogenous PTL-1.
Fig. 8.
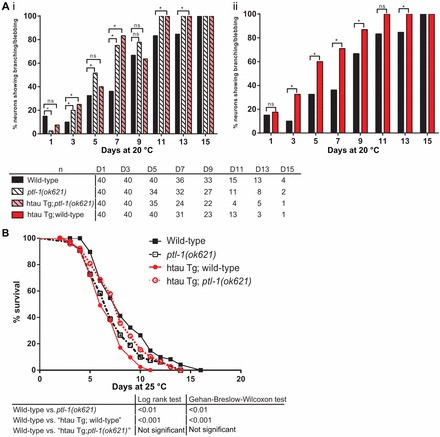
Expression of human tau does not robustly rescue defects observed in the ptl-1 null mutant, but phenotypes observed in tau transgenic lines are dependent on endogenous PTL-1. ‘htau Tg’ refers to the human tau transgene, and the terms ‘wild-type’ and ‘ptl-1(ok621)’ immediately following this refer to the genotype at the ptl-1 locus, whether wild-type or ok621 mutant, respectively. (A) Neuron imaging time course of htau transgenic worms showing data for anterior touch receptor neurons visualised using the Pmec-4::gfp reporter. (Ai) Control strains together with the htau Tg; ptl-1(ok621) rescue strain. (Aii) Effect of the htau Tg alone, compared with wild-type controls. Worms were imaged every second day from day 1 to day 15. Sample sizes are indicated below the graph. Control strains, wild-type and ptl-1(ok621), are the same as those shown for the PTL-1 transgenics (Fig. 6A). The χ2 test for independence was used to analyse differences between genotypes; ns, no significance; *P<0.05. (B) Survival curves for htau transgenic worms. n = 100 for each strain at the start of the assay. Results of statistical analysis are shown in the table below. Control strains are the same as those shown for the PTL-1 transgenics (Fig. 6B).
We next investigated whether htau would rescue the defects observed in the ptl-1 null mutant. When htau is expressed in ptl-1(ok621) null mutant worms, touch sensitivity is rescued to wild-type levels (Fig. 7B). However, the frequency of abnormal structures observed in anterior touch receptor neurons is not significantly different compared with the non-transgenic ptl-1 null mutant strain on days 3, 5, 7 and 9 of the assay (Fig. 8Ai). This finding indicates that htau cannot compensate for loss of PTL-1 in terms of protecting animals from premature neuronal aging, but is able to rescue the functional defect in touch neurons. Interestingly, we also observed that the transgenic line expressing htau in a ptl-1 null mutant has a lifespan that is intermediate between that of non-transgenic wild-type and ptl-1(ok621) null mutant strains, and is not significantly different from these control strains (Fig. 8B). While this latter observation suggests that htau may compensate in part for the absence of PTL-1 in the regulation of whole organism lifespan, overall our data indicate that htau expression does not robustly rescue for the loss of PTL-1.
Curiously, comparing htau transgenic lines that are either wild-type or null for endogenous PTL-1 indicates that the presence of the ptl-1(ok621) null mutation improved some of the detrimental effects of htau expression. For example, in the touch assay, the expression of htau resulted in reduced touch sensitivity in the wild-type background but rescued touch responsiveness in the null mutant (Fig. 7B). Furthermore, the htau transgenic line in a wild-type background had a higher percentage of animals displaying abnormal neuronal structures compared with the htau transgenic line in a ptl-1 null mutant background at days 1, 3, 5 and 9 of the assay (Fig. 8A). In particular, on day 5, the percentage of ‘htau transgenic; wild-type’ worms displaying branching or blebbing was 60% (n = 40), compared with 40% in ‘htau transgenic; ptl-1(ok621)’ worms (n = 35) and 32% in non-transgenic wild-type worms (n = 40). In terms of lifespan, the strain carrying the htau transgene on a wild-type background has a significantly shorter lifespan compared with non-transgenic wild-type worms, but this effect is ameliorated in ‘htau transgenic; ptl-1(ok621)’ strain (Fig. 8B). These data suggest some degree of functional conservation between htau and PTL-1.
Discussion
PTL-1 is important for the maintenance of neuronal integrity and lifespan in C. elegans
We have shown that PTL-1 in C. elegans is involved in the age-associated preservation of neuronal structural integrity as well as in the regulation of lifespan. Moreover, we observed that the severity of phenotypes observed in htau transgenic worms is dependent on the presence or absence of endogenous PTL-1, suggesting some functional conservation between these proteins. Our data indicate that targeting PTL-1 in C. elegans is a useful model to study the physiological roles of neuronal tau/MAP2-like MAPs in vivo.
Our focus in the study of PTL-1 is in the context of neuronal integrity, where tau is understood to play a significant role. Tau is pathologically important in several neurodegenerative disorders in which the progressive formation of NFTs from aggregated forms of tau is a histopathological hallmark of disease (reviewed by Lee et al., 2001). It is increasingly appreciated that tau not only assumes a toxic gain-of-function as its levels are elevated and aggregated forms of tau accumulate (Gómez-Ramos et al., 2006; Ittner et al., 2008; Clavaguera et al., 2009; Avila et al., 2010), but rather also that the loss of normally functioning tau results in disease (Gómez-Isla et al., 1997; Santacruz et al., 2005). To clarify this issue, it is critical to acquire a thorough understanding of the biological roles of tau in vivo in the absence of pathology. As a microtubule-binding protein, tau plays many important physiological functions (Weingarten et al., 1975; Cleveland et al., 1977a; Baas et al., 1991; Chen et al., 1992; Lee et al., 1998; Liao et al., 1998; Reynolds et al., 2008; Ittner et al., 2010; Morris et al., 2011), however, investigating tau in mammalian models is complicated by the presence of other MAPs, which appear to share several biological roles (Dehmelt and Halpain, 2005; Sontag et al., 2012). Due to the complex functional redundancy between mammalian MAPs, many studies aiming to address the biological functions of tau have focussed on overexpression models, with few studies utilising gene-targeting approaches to investigate endogenous tau (reviewed by Götz and Ittner, 2008; Götz et al., 2010). In addition, three tau knockout mouse lines have been generated (Harada et al., 1994; Dawson et al., 2001; Tucker et al., 2001) and in general, these mice do not show defects in neuronal development or function (reviewed by Ke et al., 2012), an exception being in the case of an Alzheimer's disease model, where the loss of tau appeared to exacerbate effects of a mutated β-amyloid precursor protein (Dawson et al., 2010). This general lack of morphological defects is potentially due to compensatory effects by MAP2 (Harada et al., 1994). Therefore, despite the significant contribution of these studies, it would be useful to have a model for tau where a gene-targeted approach can be taken without also having to consider compensatory functions attributable to other MAPs. Investigating PTL-1 in C. elegans has the advantage of PTL-1 being the only homolog of tau/MAP2 in the worm (McDermott et al., 1996; Gordon et al., 2008), meaning that shared physiological functions between tau/MAP2 family members can be addressed.
There are two ptl-1 mutants available: ptl-1(ok621) is a null mutant (Gordon et al., 2008), whereas the ptl-1(tm543) line putatively generates a protein product containing only the N-terminal region (Fig. 1A). The N-terminus of PTL-1 is largely conserved amongst nematodes, is proline-rich, highly acidic, and also contains several serine/threonine-proline motifs that are potential phosphorylation sites (Goedert et al., 1996; McDermott et al., 1996; Gordon et al., 2008). Analysis of these mutant strains allowed us not only to investigate the role of PTL-1 in vivo, but also to dissociate functions of PTL-1 that are attributable solely to microtubule-binding from functions of the full-length protein. We found that both mutants of ptl-1 show a decreased capacity to maintain neuronal integrity with age, as evidenced by a higher frequency of abnormal morphological structures such as branching and blebbing in touch receptor and GABAergic neurons in early adulthood compared with wild-type. Interestingly, this effect was also observed when we increased the copy number of full-length PTL-1 by expression of a stable transgene in addition to the endogenous locus. Therefore, correct gene dosage of PTL-1 is critical for the maintenance of neuronal structures with age, as either increasing or decreasing this level is detrimental to the organism. In addition when we assayed touch sensitivity, a response that requires functional touch receptor neurons, we found that the ptl-1(ok621) null mutant is less responsive to gentle touch compared with wild-type, but that there was no difference in touch sensitivity from wild-type in the ptl-1(tm543) mutant lacking the MBR domain, suggesting that this allele may be hypomorphic, or that the N-terminal region may have functions that are sufficient to maintain touch sensitivity. Interestingly, we observed the opposite effect when assaying for defects in cholinergic/GABAergic transmission, with ptl-1(tm543) animals displaying a more severe phenotype compared with ptl-1(ok621). Importantly, our data indicate that although the microtubule-binding functions of PTL-1 are not necessary for wild-type touch sensitivity, the maintenance of touch neuron structural integrity with age requires full length PTL-1. Our findings also demonstrate that wild-type functioning of neurons may not be sufficient to preserve neuronal health with age. Another C. elegans MAP that is expressed in touch receptor neurons is the EMAP-like protein or ELP-1, which does not display high sequence similarity to PTL-1 but also regulates touch sensitivity (Hueston et al., 2008). In light of this, allelic differences in touch sensitivity observed between ptl-1 mutants could be due to a requirement for both ELP-1 and the N-terminus of PTL-1 for wild-type touch responsiveness. A role of ELP-1 in neuronal aging, however, remains to be investigated.
Having found an age-associated neuronal phenotype in ptl-1 mutant lines, we investigated if these strains also displayed defects in lifespan. We found that both mutants show a lifespan reduction compared with wild-type, and that furthermore, increasing the number of copies of ptl-1 also results in a shortening of lifespan. This indicates that a tight regulation of PTL-1 is required to maintain wild-type lifespan. As discussed above, our observations also indicate a role for PTL-1 in maintaining structural integrity in neurons, which may be related or additional to the role of this protein in regulating lifespan. Previous reports suggest that the factors regulating whole organism lifespan and age-related neuronal integrity may be separable. For example, C. elegans carrying mutations in the daf-2 insulin receptor homolog are long-lived (Kenyon et al., 1993). This lifespan extension requires the forkhead box O (FOXO) transcription factor daf-16, meaning that a daf-2;daf-16 double mutant is not long-lived (Kenyon et al., 1993). Tank and colleagues have shown that a daf-2 mutant displays significantly less neuronal branching in early adulthood compared with wild-type, and that this effect is ameliorated in daf-2;daf-16 double mutants (Tank et al., 2011). Importantly, RNAi-mediated knockdown of daf-16 in a daf-2 mutant in non-neuronal cells results in an animal with wild-type lifespan, but has no effect on the daf-2-mediated delay of neuronal aging (Tank et al., 2011). This and other evidence from previous reports (Pan et al., 2011; Tank et al., 2011) suggests that the processes that influence neuronal aging and whole organism aging can be decoupled from one another. Further investigations are required to determine whether the function of PTL-1 in stabilising neuronal structures with age influences lifespan, or vice versa.
If PTL-1 has biological functions in regulating both aging in neurons and aging in the whole organism, where does this protein exert its effects? In adult worms, it appears that PTL-1 is only expressed in neurons (Goedert et al., 1996; Gordon et al., 2008). Previous studies have also established a role for PTL-1 specifically in touch neurons, in particular with regards to mechanosensation (Gordon et al., 2008) and microtubule-based transport (Tien et al., 2011). In the case of branching and bleb formation in neurons, there is some evidence that these morphological changes are due to cell-autonomous effects (Tank et al., 2011; Toth et al., 2012). For example, expression of DAF-16 only in neurons of a daf-2;daf-16 mutant delays the formation of abnormal neuron structures compared with a non-transgenic daf-2;daf-16 control (Tank et al., 2011). In addition, reduced activity of the heat shock factor-1 transcription factor (hsf-1) in the whole organism resulted in early onset branching and blebbing in touch receptor neurons, but re-expressing hsf-1 only in this subset of neurons was able to rescue this effect (Toth et al., 2012). With regards to the role of PTL-1 in regulating organismal lifespan, previous studies have suggested that signalling events in neurons alone are sufficient to alter lifespan (Apfeld and Kenyon, 1999; Alcedo and Kenyon, 2004). Therefore, it is possible that PTL-1 has a cell-autonomous effect on neuronal integrity as well as whole organism aging. If neuronal functions of PTL-1 regulate whole organism lifespan, it remains to be determined whether this refers to a role in all neurons or in a particular subset of neurons. Ablation of sensory neurons such as thermosensory neurons (Lee and Kenyon, 2009) or gustatory and olfactory neurons (Alcedo and Kenyon, 2004) in C. elegans results in a shortening or extension of lifespan, respectively. In addition, C. elegans mutants defective in sensory cilia in some neurons have been shown to be long-lived (Apfeld and Kenyon, 1999). Studies performed using several mechanosensory defective (mec) mutants predominantly affecting touch receptor neurons, such as mec-1, mec-8 and mec-12, have also demonstrated differences in lifespan compared with wild-type (Apfeld and Kenyon, 1999; Pan et al., 2011). As PTL-1 is expressed in most, if not all, neurons in the worm, the reduced lifespan of ptl-1 mutants may be due to the loss of PTL-1 function in one or more of these neuronal subsets.
Human tau and C. elegans PTL-1 display some functional conservation
We generated tau transgenic worms that expressed the longest isoform of human tau under the control of the ptl-1 promoter, and observed (1) that human tau expression is detrimental to worms, (2) that human tau does not robustly rescue loss of PTL-1, and (3) that touch sensitivity, neuronal structural health and lifespan phenotypes of human tau transgenic lines are dependent on PTL-1. The negative effect of expressing human tau in C. elegans, as also observed in (Kraemer et al., 2003; Miyasaka et al., 2005; Brandt et al., 2009), may either be due to overexpression of any MAP having a detrimental effect, or due to a specific axonal role of tau that evolved with the diversification of neuronal MAPs into mainly axon-localised tau and mainly dendrite-localised MAP2. In addition to some shared functions (Sontag et al., 2012), tau and MAP2 play specific roles in their distinct subcellular compartments (Kosik and Finch, 1987; Chen et al., 1992; Harada et al., 1994; Hirokawa et al., 1996). In C. elegans, no such diversification of neuronal MAPs exists, and as PTL-1 is a homolog of both MAP2 and tau, it presumably has both axon- and dendrite-specific functions. Therefore, the axon-specific effects of human tau could negatively affect the worm when tau is present in addition to endogenous PTL-1, and could also be insufficient to rescue for the loss of PTL-1 in a null mutant with regards to neuronal and whole organismal aging.
Our observation that human tau rescues touch insensitivity in a ptl-1 null mutant, together with the finding that the detrimental effects of expressing tau are ameliorated in the absence of endogenous PTL-1, suggests some functional conservation between tau and PTL-1. Although these proteins do not have high similarity over the entire sequence, the imperfect tandem repeats in the C-terminal microtubule binding domain that constitute the only region of homology between these proteins show high amino acid identity. Therefore, these conserved functions may be those attributable to the microtubule-binding capacity of tau and PTL-1.
Concluding remarks
We have found that C. elegans PTL-1 plays important functions in maintaining neuronal health with age, and that these functions may be related or additional to a role in regulating whole organism aging. This is consistent with the notion that at least some of the effects of tau pathology in neurodegenerative conditions may be attributable to a loss of correctly functioning tau, and not solely to a gain-of-toxic function due to aggregated forms of tau. Furthermore, we have shown that PTL-1 is a useful model for a gene-targeted approach to study the physiological roles of a tau-like protein due to PTL-1 being the sole homolog of tau/MAP2 in C. elegans.
Materials and Methods
Strain information
C. elegans strains were cultured on NGM plates seeded with the Escherichia coli strain OP50. Hermaphrodite animals were used for all experiments. The wild-type strain used for all experiments is N2 (Bristol). Strains N2 (Bristol), RB809 ptl-1(ok621), CZ10175 zdIs5[Pmec-4::gfp + lin-15(+)] I and EG1285 oxIs12[Punc-47::gfp + lin-15(+)] X were obtained from the Caenorhabditis Genetics Centre, and FX00543 ptl-1(tm543) was obtained from the National Bioresource Project, Japan (Dr S. Mitani). ptl-1 mutant lines RB809 and FX00543 were both outcrossed six times to wild-type. For neuron imaging assays, strains involved were crossed with CZ10175 to visualise touch neurons and EG1285 to visualise GABAergic motor neurons.
The strains generated were as follows. APD004: ptl-1(ok621) outcrossed six times to N2; APD009: ptl-1(ok621); oxIs12[Punc-47::gfp + lin-15(+)]; APD010: ptl-1(ok621); zdIs5[Pmec-4::gfp + lin-15(+)]; APD015: ptl-1(tm543) outcrossed six times to N2; APD016: ptl-1(tm543); zdIs5[Pmec-4::gfp + lin-15(+)]; APD017: ptl-1(tm543); oxIs12[Punc-47::gfp + lin-15(+)]; APD025: apdIs4[Pptl-1:htau40:ptl-1_3′UTR; Pmyo-2:mCherry; Prpl-28::PuroR::rpl-16_outron::NeoR::let-858_3′UTR]; APD026: apdIs5[Pptl-1:PTL-1::V5:ptl-1_3′UTR; Pmyo-2:gfp; Prpl-28::PuroR::rpl-16_outron::NeoR::let-858_3′UTR]; APD030: apdIs4[Pptl-1:htau40:ptl-1_3′UTR; Pmyo-2:mCherry; Prpl-28::PuroR::rpl-16_outron::NeoR::let-858_3′UTR]; ptl-1(ok621); zdIs5 [Pmec-4::gfp + lin-15(+)]; APD031: apdIs4[Pptl-1:htau40:ptl-1_3′UTR; Pmyo-2:mCherry; Prpl-28::PuroR::rpl-16_outron::NeoR::let-858_3′UTR]; zdIs5[Pmec-4::gfp + lin-15(+)]; APD034: apdIs4[Pptl-1:htau40:ptl-1_3′UTR; Pmyo-2:mCherry; Prpl-28::PuroR::rpl-16_outron::NeoR::let-858_3′UTR]; ptl-1(ok621); APD035: apdIs5[Pptl-1:PTL-1::V5:ptl-1_3′UTR; Pmyo-2:gfp; Prpl-28::PuroR::rpl-16_outron::NeoR::let-858_3′UTR]; ptl-1(ok621); zdIs5[Pmec-4::gfp + lin-15(+)]; APD036: apdIs5[Pptl-1:PTL-1::V5:ptl-1_3′UTR; Pmyo-2:gfp; Prpl-28::PuroR::rpl-16_outron::NeoR::let-858_3′UTR]; zdIs5[Pmec-4::gfp + lin-15(+)]; APD039: apdIs5[Pptl-1:PTL-1::V5:ptl-1_3′UTR; Pmyo-2:gfp; Prpl-28::PuroR::rpl-16_outron::NeoR::let-858_3′UTR]; ptl-1(ok621).
Plasmids
Cloning was performed using the Gateway (Invitrogen, Life Technologies) system according to the manufacturer's instructions. Dual antibiotic selection plasmids pBCN40 and pBCN41, encoding visual markers Pmyo-2::mCherry and Pmyo-2::gfp, respectively, were generously provided by Drs J. Semple and B. Lehner (EMBL Centre for Genomic Regulation Systems Biology Unit, Barcelona). The ptl-1 promoter used is a 2.9 kb sequence upstream of ptl-1 and was cloned from Vancouver fosmid WRM0634bB04 (Source BioScience Lifesciences), using the primers 5′-GGGGACAACTTTGTATAGAAAAGTTGCATTCCGCATGGTTGGAAAGAG-3′ (forward primer including attB4 site) and 5′-GGGGACTGCTTTTTTGTACAAACTTGATTTTTCCTGAAAAATTGAAATTGGGAG-3′ (reverse primer including attB1r site). The ptl-1 cDNA was cloned from a C. elegans cDNA library (RIKEN BioResource Centre, Japan; RDB No. 1864), and the V5 epitope tagged at the C-terminus using the forward primer 5′-TCACGTAGAATCGAGACCGAGGAGAGGGTTAGGGATAGGCTTACCCGCCGCGCGATTGAATATAAAATCAGG-3′. Site attB1 was added using 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTCAATGTCAACCCCTCAATCAGAG-3′, and attB2 using 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTTTACGTAGAATCGAGACCGAGGAG-3′. The PTL-1 3′ UTR was cloned from fosmid WRM0634bB04 using forward primer 5′-GGGGACAGCTTTCTTGTACAAAGTGGGATAACAATCGCTGATGTATACCGCGC-3′ (incorporating attB2r) and reverse primer 5′-GGGGACAACTTTGTATAATAAAGTTGACACTTTTAATTACCACTTTATTGAAGAG-3′ (incorporating attB3). The 5′ entry clone was generated using a BP reaction into pDONRP4P1R, the middle entry clone using a BP reaction into pDONR221, and the 3′ entry clone using a BP reaction into pDONRP2RP3 (pDONR vectors from Invitrogen). The htau40 entry clone (in pENTR-SD-D-Topo) was kindly provided by Dr L. Ittner (Brain and Mind Research Institute, University of Sydney). The Multisite LR reaction (Invitrogen) was used to combine entry clones into destination vector pBCN40 or pBCN41.
Generation of transgenic lines
Transgenic worms for rescue experiments were generated by biolistic transformation using the PDS-1000/He™ particle delivery system (BioRad) according to the manufacturer's instructions. Wild-type worms were bombarded with 7 µg of linearised plasmid DNA using previously established methods (Praitis et al., 2001). Selection post-bombardment was undertaken using the dual antibiotic selection protocol (Semple et al., 2012). Integrated lines expressing htau40 cDNA and PTL-1 cDNA tagged at the carboxyl (C)-terminus with V5 under the control of the ptl-1 promoter and PTL-1 3′UTR were obtained and outcrossed six times to wild-type.
Touch sensitivity assay
Animals were synchronised by hypochlorite treatment and cultured at 25°C, with 1-day-old adults used for all assays. Touch assays were performed according to established methods (Chalfie and Sulston, 1981). A positive response was determined as acceleration of the animal away from touch. The number of positive responses was then expressed as a percentage of total touches (ten) for each animal. Assays were conducted blind to the genotype of the worms.
Neuron imaging assay
Age-matched animals synchronised by egg-laying were cultured on plates at 20°C. Starting animals were 1-day-old adults in all cases. For longitudinal assays where individual animals were monitored every day during their lifetimes, these animals were individually mounted into 0.2% tetramisole (Sigma) on 3% agarose pads prepared on standard microscope slides. These animals were rescued by picking them onto a drop of M9 buffer, and recovered well if incubated in tetramisole for under two minutes. For transverse assays where populations of animals were monitored every second day for 15 days, surviving adult worms on a plate were mounted as described, but were not recovered post-imaging. To separate adult worms from their progeny, adult worms were moved to new NGM plates every second day until the assay was completed. Assays were conducted blind to the genotype of the worms. To score the incidence of aberrant neuronal structures in touch receptor neurons, worms were scored as positive if the neuron displayed branching or blebbing at the cell body or axon. For scoring in GABAergic neurons, worms were scored as positive if at least one of the observed commisures displayed branching. For all transverse assays, the proportion of animals scored as positive was expressed then as a percentage of the sample size observed at that time point.
Lifespan assay
Age-matched animals synchronised by egg-laying were cultured on plates at 25°C and the number of surviving animals recorded every day until death. 100 1-day-old adults per strain were plated at the start of each assay. Animals that were lost or displayed internal hatching or bursting were censored. To separate adult worms from their progeny, adult worms were moved to new NGM plates every second day until the assay was completed. Survival curves were generated using GraphPad Prism 5 (GraphPad Software Inc.).
Immunofluorescence
One-day-old adults synchronised by egg-laying and cultured at 23°C were used for all experiments. Animals were permeabilised using the freeze-crack protocol as previously described (Crittenden and Kimble, 1999). Samples were immediately fixed in ice-cold 4% paraformaldehyde at 4°C for >12 hours. The primary antibody used was mouse monoclonal anti-V5 (R960-25, Life Technologies) [1∶200]. The secondary antibody used was goat anti-mouse Alexa Fluor-568 (Sigma) [1∶500]. All antibody dilutions were made in 30% (v/v) normal goat serum (Life Technologies). Samples were mounted onto microscope slides using VectaShield mounting medium (Vector Labs) containing 4′,6-diamidino-2-phenylindole (DAPI) where applicable. Samples were imaged using a BX51 Microscope (Olympus). Micrographs were captured using AnalySIS software (Olympus).
Immunoblot
Samples for immunoblot were prepared by washing worms off plates in M9 buffer, followed by repeated washes in M9 and a final wash in distilled water. Samples resuspended in Laemmli buffer underwent three freeze-thaw cycles in liquid nitrogen before boiling, and were immediately loaded onto 10% acrylamide gels. Primary antibodies used are mouse monoclonal tau 13 antibody (Abcam) [1∶5000] that binds human tau, mouse monoclonal anti-V5 antibody conjugated to HRP (R961-25, Life Technologies) [1∶1500], and mouse monoclonal anti-acetyl alpha-tubulin (T7451, Sigma) [1∶2000]. The secondary antibody used was goat anti-mouse conjugated to horseradish peroxidase (NA931, GE Healthcare) [1∶10,000]. Blots were developed with Immobilon Western chemiluminescent substrate (Millipore) on film.
Pharmacological assays
Pharmacological assays were performed on unseeded plates spread with levamisole (Sigma) at a final concentration of 1 mM. Levamisole was allowed to equilibrate on the plates for 1–2 hours at room temperature prior to scoring. Animals to be assayed were synchronised by egg-laying and were cultivated at 20°C. Animals were picked onto drug plates and scored at room temperature for paralysis (defined as no response to tapping with a platinum wire) at 15-minute intervals for 90 minutes. Scoring was conducted blind to the genotype of the worms.
Statistical analysis
All statistical analysis was performed using the GraphPad Prism 5/6 software (GraphPad Software Inc.) or Microsoft Excel (Microsoft).
Supplementary Material
Acknowledgments
The authors gratefully acknowledge Dr M. Hilliard for providing reagents, Drs J. Semple and B. Lehner for dual antibiotic selection vectors, and Dr L. Ittner for the htau40 construct. The authors thank members of the Nicholas lab and Götz lab for helpful discussions. Some strains were provided by the Caenorhabditis Genetics Center (CGC), which is funded by the National Institutes of Health (NIH) Office of Research Infrastructure Programs [grant number P40 OD010440]. Deposited in PMC for release after 12 months.
Footnotes
Author contributions
Y.L.C., J.G. and H.R.N. designed research, Y.L.C. and X.F. performed research, Y.L.C., X.F., J.G. and H.R.N. analysed data, and Y.L.C., J.G., and H.R.N. wrote the paper.
Funding
H.R.N. is supported by a University of Sydney Re-entry Fellowship, J.G. by the Estate of Dr Clem Jones AO, and grants from the Australian Research Council and the National Health and Medical Research Council of Australia.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.jcs124404/-/DC1
References
- Alcedo J., Kenyon C. (2004). Regulation of C. elegans longevity by specific gustatory and olfactory neurons. Neuron 41, 45–55 10.1016/S0896-6273(03)00816-X [DOI] [PubMed] [Google Scholar]
- Apfeld J., Kenyon C. (1999). Regulation of lifespan by sensory perception in Caenorhabditis elegans. Nature 402, 804–809 10.1038/45544 [DOI] [PubMed] [Google Scholar]
- Avila J., Lucas J. J., Perez M., Hernandez F. (2004). Role of tau protein in both physiological and pathological conditions. Physiol. Rev. 84, 361–384 10.1152/physrev.00024.2003 [DOI] [PubMed] [Google Scholar]
- Avila J., Gómez de Barreda E., Engel T., Lucas J. J., Hernández F. (2010). Tau phosphorylation in hippocampus results in toxic gain-of-function. Biochem. Soc. Trans. 38, 977–980 10.1042/BST0380977 [DOI] [PubMed] [Google Scholar]
- Baas P. W., Pienkowski T. P., Kosik K. S. (1991). Processes induced by tau expression in Sf9 cells have an axon-like microtubule organization. J. Cell Biol. 115, 1333–1344 10.1083/jcb.115.5.1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt R., Gergou A., Wacker I., Fath T., Hutter H. (2009). A Caenorhabditis elegans model of tau hyperphosphorylation: induction of developmental defects by transgenic overexpression of Alzheimer's disease-like modified tau. Neurobiol. Aging 30, 22–33 10.1016/j.neurobiolaging.2007.05.011 [DOI] [PubMed] [Google Scholar]
- Chalfie M., Sulston J. (1981). Developmental genetics of the mechanosensory neurons of Caenorhabditis elegans. Dev. Biol. 82, 358–370 10.1016/0012-1606(81)90459-0 [DOI] [PubMed] [Google Scholar]
- Chalfie M., Sulston J. E., White J. G., Southgate E., Thomson J. N., Brenner S. (1985). The neural circuit for touch sensitivity in Caenorhabditis elegans. J. Neurosci. 5, 956–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Kanai Y., Cowan N. J., Hirokawa N. (1992). Projection domains of MAP2 and tau determine spacings between microtubules in dendrites and axons. Nature 360, 674–677 10.1038/360674a0 [DOI] [PubMed] [Google Scholar]
- Clavaguera F., Bolmont T., Crowther R. A., Abramowski D., Frank S., Probst A., Fraser G., Stalder A. K., Beibel M., Staufenbiel M. et al. (2009). Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 11, 909–913 10.1038/ncb1901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleveland D. W., Hwo S. Y., Kirschner M. W. (1977a). Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J. Mol. Biol. 116, 227–247 10.1016/0022-2836(77)90214-5 [DOI] [PubMed] [Google Scholar]
- Cleveland D. W., Hwo S. Y., Kirschner M. W. (1977b). Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J. Mol. Biol. 116, 207–225 10.1016/0022-2836(77)90213-3 [DOI] [PubMed] [Google Scholar]
- Crittenden S. L., Kimble J. (1999). Confocal methods for Caenorhabditis elegans. Methods Mol. Biol. 122, 141–151 [DOI] [PubMed] [Google Scholar]
- Dawson H. N., Ferreira A., Eyster M. V., Ghoshal N., Binder L. I., Vitek M. P. (2001). Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J. Cell Sci. 114, 1179–1187 [DOI] [PubMed] [Google Scholar]
- Dawson H. N., Cantillana V., Jansen M., Wang H., Vitek M. P., Wilcock D. M., Lynch J. R., Laskowitz D. T. (2010). Loss of tau elicits axonal degeneration in a mouse model of Alzheimer's disease. Neuroscience 169, 516–531 10.1016/j.neuroscience.2010.04.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehmelt L., Halpain S. (2005). The MAP2/Tau family of microtubule-associated proteins. Genome Biol. 6, 204 10.1186/gb-2004-6-1-204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drubin D. G., Kirschner M. W. (1986). Tau protein function in living cells. J. Cell Biol. 103, 2739–2746 10.1083/jcb.103.6.2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushige T., Siddiqui Z. K., Chou M., Culotti J. G., Gogonea C. B., Siddiqui S. S., Hamelin M. (1999). MEC-12, an alpha-tubulin required for touch sensitivity in C. elegans. J. Cell Sci. 112, 395–403 [DOI] [PubMed] [Google Scholar]
- Goedert M., Baur C. P., Ahringer J., Jakes R., Hasegawa M., Spillantini M. G., Smith M. J., Hill F. (1996). PTL-1, a microtubule-associated protein with tau-like repeats from the nematode Caenorhabditis elegans. J. Cell Sci. 109, 2661–2672 [DOI] [PubMed] [Google Scholar]
- Gómez-Isla T., Hollister R., West H., Mui S., Growdon J. H., Petersen R. C., Parisi J. E., Hyman B. T. (1997). Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann. Neurol. 41, 17–24 10.1002/ana.410410106 [DOI] [PubMed] [Google Scholar]
- Gómez-Ramos A., Díaz-Hernández M., Cuadros R., Hernández F., Avila J. (2006). Extracellular tau is toxic to neuronal cells. FEBS Lett. 580, 4842–4850 10.1016/j.febslet.2006.07.078 [DOI] [PubMed] [Google Scholar]
- Gordon P., Hingula L., Krasny M. L., Swienckowski J. L., Pokrywka N. J., Raley-Susman K. M. (2008). The invertebrate microtubule-associated protein PTL-1 functions in mechanosensation and development in Caenorhabditis elegans. Dev. Genes Evol. 218, 541–551 10.1007/s00427-008-0250-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götz J., Ittner L. M. (2008). Animal models of Alzheimer's disease and frontotemporal dementia. Nat. Rev. Neurosci. 9, 532–544 10.1038/nrn2420 [DOI] [PubMed] [Google Scholar]
- Götz J., Gladbach A., Pennanen L., van Eersel J., Schild A., David D., Ittner L. M. (2010). Animal models reveal role for tau phosphorylation in human disease. Biochim. Biophys. Acta 1802, 860–871 10.1016/j.bbadis.2009.09.008 [DOI] [PubMed] [Google Scholar]
- Harada A., Oguchi K., Okabe S., Kuno J., Terada S., Ohshima T., Sato-Yoshitake R., Takei Y., Noda T., Hirokawa N. (1994). Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature 369, 488–491 10.1038/369488a0 [DOI] [PubMed] [Google Scholar]
- Hirokawa N., Funakoshi T., Sato-Harada R., Kanai Y. (1996). Selective stabilization of tau in axons and microtubule-associated protein 2C in cell bodies and dendrites contributes to polarized localization of cytoskeletal proteins in mature neurons. J. Cell Biol. 132, 667–679 10.1083/jcb.132.4.667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hueston J. L., Herren G. P., Cueva J. G., Buechner M., Lundquist E. A., Goodman M. B., Suprenant K. A. (2008). The C. elegans EMAP-like protein, ELP-1 is required for touch sensation and associates with microtubules and adhesion complexes. BMC Dev. Biol. 8, 110 10.1186/1471-213X-8-110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal K., Liu F., Gong C. X., Grundke-Iqbal I. (2010). Tau in Alzheimer disease and related tauopathies. Curr. Alzheimer Res. 7, 656–664 10.2174/156720510793611592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ittner L. M., Fath T., Ke Y. D., Bi M., van Eersel J., Li K. M., Gunning P., Götz J. (2008). Parkinsonism and impaired axonal transport in a mouse model of frontotemporal dementia. Proc. Natl. Acad. Sci. USA 105, 15997–16002 10.1073/pnas.0808084105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ittner L. M., Ke Y. D., Delerue F., Bi M., Gladbach A., van Eersel J., Wölfing H., Chieng B. C., Christie M. J., Napier I. A. et al. (2010). Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell 142, 387–397 10.1016/j.cell.2010.06.036 [DOI] [PubMed] [Google Scholar]
- Ittner A., Ke Y. D., van Eersel J., Gladbach A., Götz J., Ittner L. M. (2011). Brief update on different roles of tau in neurodegeneration. IUBMB Life 63, 495–502 10.1002/iub.467 [DOI] [PubMed] [Google Scholar]
- Ke Y. D., Suchowerska A. K., van der Hoven J., De Silva D. M., Wu C. W., van Eersel J., Ittner A., Ittner L. M. (2012). Lessons from tau-deficient mice. Int. J. Alzheimers Dis. 2012, 873270 10.1155/2012/873270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon C., Chang J., Gensch E., Rudner A., Tabtiang R. (1993). A C. elegans mutant that lives twice as long as wild type. Nature 366, 461–464 10.1038/366461a0 [DOI] [PubMed] [Google Scholar]
- Kosik K. S., Finch E. A. (1987). MAP2 and tau segregate into dendritic and axonal domains after the elaboration of morphologically distinct neurites: an immunocytochemical study of cultured rat cerebrum. J. Neurosci. 7, 3142–3153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraemer B. C., Zhang B., Leverenz J. B., Thomas J. H., Trojanowski J. Q., Schellenberg G. D. (2003). Neurodegeneration and defective neurotransmission in a Caenorhabditis elegans model of tauopathy. Proc. Natl. Acad. Sci. USA 100, 9980–9985 10.1073/pnas.1533448100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. J., Kenyon C. (2009). Regulation of the longevity response to temperature by thermosensory neurons in Caenorhabditis elegans. Current biology 19, 715–722 10.1016/j.cub.2009.03.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G., Leugers C. J. (2012). Tau and tauopathies. Prog. Mol. Biol. Transl. Sci. 107, 263–293 10.1016/B978-0-12-385883-2.00004-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G., Newman S. T., Gard D. L., Band H., Panchamoorthy G. (1998). Tau interacts with src-family non-receptor tyrosine kinases. J. Cell Sci. 111, 3167–3177 [DOI] [PubMed] [Google Scholar]
- Lee V. M., Goedert M., Trojanowski J. Q. (2001). Neurodegenerative tauopathies. Annu. Rev. Neurosci. 24, 1121–1159 10.1146/annurev.neuro.24.1.1121 [DOI] [PubMed] [Google Scholar]
- Lewis J. A., Wu C. H., Levine J. H., Berg H. (1980). Levamisole-resistant mutants of the nematode Caenorhabditis elegans appear to lack pharmacological acetylcholine receptors. Neuroscience 5, 967–989 10.1016/0306-4522(80)90180-3 [DOI] [PubMed] [Google Scholar]
- Liao H., Li Y., Brautigan D. L., Gundersen G. G. (1998). Protein phosphatase 1 is targeted to microtubules by the microtubule-associated protein Tau. J. Biol. Chem. 273, 21901–21908 10.1074/jbc.273.34.21901 [DOI] [PubMed] [Google Scholar]
- McDermott J. B., Aamodt S., Aamodt E. (1996). ptl-1, a Caenorhabditis elegans gene whose products are homologous to the tau microtubule-associated proteins. Biochemistry 35, 9415–9423 10.1021/bi952646n [DOI] [PubMed] [Google Scholar]
- McIntire S. L., Reimer R. J., Schuske K., Edwards R. H., Jorgensen E. M. (1997). Identification and characterization of the vesicular GABA transporter. Nature 389, 870–876 10.1038/39908 [DOI] [PubMed] [Google Scholar]
- McKay S. J., Johnsen R., Khattra J., Asano J., Baillie D. L., Chan S., Dube N., Fang L., Goszczynski B., Ha E. et al. (2003). Gene expression profiling of cells, tissues, and developmental stages of the nematode C. elegans. Cold Spring Harb. Symp. Quant. Biol. 68, 159–169 10.1101/sqb.2003.68.159 [DOI] [PubMed] [Google Scholar]
- Mitani S., Du H., Hall D. H., Driscoll M., Chalfie M. (1993). Combinatorial control of touch receptor neuron expression in Caenorhabditis elegans. Development 119, 773–783 [DOI] [PubMed] [Google Scholar]
- Miyasaka T., Ding Z., Gengyo-Ando K., Oue M., Yamaguchi H., Mitani S., Ihara Y. (2005). Progressive neurodegeneration in C. elegans model of tauopathy. Neurobiol. Dis. 20, 372–383 10.1016/j.nbd.2005.03.017 [DOI] [PubMed] [Google Scholar]
- Morris M., Maeda S., Vossel K., Mucke L. (2011). The many faces of tau. Neuron 70, 410–426 10.1016/j.neuron.2011.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan C. L., Peng C. Y., Chen C. H., McIntire S. (2011). Genetic analysis of age-dependent defects of the Caenorhabditis elegans touch receptor neurons. Proc. Natl. Acad. Sci. USA 108, 9274–9279 10.1073/pnas.1011711108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praitis V., Casey E., Collar D., Austin J. (2001). Creation of low-copy integrated transgenic lines in Caenorhabditis elegans. Genetics 157, 1217–1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds C. H., Garwood C. J., Wray S., Price C., Kellie S., Perera T., Zvelebil M., Yang A., Sheppard P. W., Varndell I. M. et al. (2008). Phosphorylation regulates tau interactions with Src homology 3 domains of phosphatidylinositol 3-kinase, phospholipase Cgamma1, Grb2, and Src family kinases. J. Biol. Chem. 283, 18177–18186 10.1074/jbc.M709715200 [DOI] [PubMed] [Google Scholar]
- Santacruz K., Lewis J., Spires T., Paulson J., Kotilinek L., Ingelsson M., Guimaraes A., DeTure M., Ramsden M., McGowan E. et al. (2005). Tau suppression in a neurodegenerative mouse model improves memory function. Science 309, 476–481 10.1126/science.1113694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage C., Xue Y., Mitani S., Hall D., Zakhary R., Chalfie M. (1994). Mutations in the Caenorhabditis elegans beta-tubulin gene mec-7: effects on microtubule assembly and stability and on tubulin autoregulation. J. Cell Sci. 107, 2165–2175 [DOI] [PubMed] [Google Scholar]
- Semple J. I., Biondini L., Lehner B. (2012). Generating transgenic nematodes by bombardment and antibiotic selection. Nat. Methods 9, 118–119 10.1038/nmeth.1864 [DOI] [PubMed] [Google Scholar]
- Sontag J. M., Nunbhakdi-Craig V., White C. L., 3rd, Halpain S., Sontag E. (2012). The protein phosphatase PP2A/Bα binds to the microtubule-associated proteins Tau and MAP2 at a motif also recognized by the kinase Fyn: implications for tauopathies. J. Biol. Chem. 287, 14984–14993 10.1074/jbc.M111.338681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tank E. M., Rodgers K. E., Kenyon C. (2011). Spontaneous age-related neurite branching in Caenorhabditis elegans. J. Neurosci. 31, 9279–9288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tien N. W., Wu G. H., Hsu C. C., Chang C. Y., Wagner O. I. (2011). Tau/PTL-1 associates with kinesin-3 KIF1A/UNC-104 and affects the motor's motility characteristics in C. elegans neurons. Neurobiol. Dis. 43, 495–506 10.1016/j.nbd.2011.04.023 [DOI] [PubMed] [Google Scholar]
- Toth M. L., Melentijevic I., Shah L., Bhatia A., Lu K., Talwar A., Naji H., Ibanez-Ventoso C., Ghose P., Jevince A. et al. (2012). Neurite sprouting and synapse deterioration in the aging Caenorhabditis elegans nervous system. J. Neurosci. 32, 8778–8790 10.1523/JNEUROSCI.1494-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker K. L., Meyer M., Barde Y. A. (2001). Neurotrophins are required for nerve growth during development. Nat. Neurosci. 4, 29–37 10.1038/82868 [DOI] [PubMed] [Google Scholar]
- Wade-Martins R. (2012). Genetics: The MAPT locus-a genetic paradigm in disease susceptibility. Neurology 8, 477–478 10.1038/nrneurol.2012.169 [DOI] [PubMed] [Google Scholar]
- Weingarten M. D., Lockwood A. H., Hwo S. Y., Kirschner M. W. (1975). A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 72, 1858–1862 10.1073/pnas.72.5.1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.