

Abstract
The bacteria associated with the infectious claw disease bovine digital dermatitis (DD) are spirochetes of the genus Treponema; however, their environmental reservoir remains unknown. To our knowledge, the current study is the first report of the discovery and phylogenetic characterization of rRNA gene sequences from DD-associated treponemes in the dairy herd environment. Although the spread of DD appears to be facilitated by wet floors covered with slurry, no DD-associated treponemes have been isolated from this environment previously. Consequently, there is a lack of knowledge about the spread of this disease among cows within a herd as well as between herds. To address the issue of DD infection reservoirs, we searched for evidence of DD-associated treponemes in fresh feces, in slurry, and in hoof lesions by deep sequencing of the V3 and V4 hypervariable regions of the 16S rRNA gene coupled with identification at the operational-taxonomic-unit level. Using treponeme-specific primers in this high-throughput approach, we identified small amounts of DNA (on average 0.6% of the total amount of sequence reads) from DD-associated treponemes in 43 of 64 samples from slurry and cow feces collected from six geographically dispersed dairy herds. Species belonging to the Treponema denticola/Treponema pedis-like and Treponema phagedenis-like phylogenetic clusters were among the most prevalent treponemes in both the dairy herd environment and the DD lesions. By the high-throughput approach presented here, we have demonstrated that cow feces and environmental slurry are possible reservoirs of DD-associated treponemes. This method should enable further clarification of the etiopathogenesis of DD.
INTRODUCTION
Bovine digital dermatitis (DD) was first described in 1974 and has since become a growing problem worldwide (1). The disease is characterized by focal proliferative to ulcerative dermatitis that is typically located on the plantar aspect of the hoof (1). The consequences of this disease are decreased animal welfare and serious economic losses for the farmers due to factors such as reduced milk yield and premature culling (2–4). DD is considered a multifactorial disease, and moist, unhygienic conditions, such as the slurry to which the animals are frequently exposed, are considered predisposing factors (5, 6). The bacteria most often associated with DD are spirochetes of the genus Treponema, which predominate in the deepest regions of DD lesions (7, 8). To date, at least 20 different phylotypes have been identified from DD biopsy specimens; among these, Treponema phagedenis-like, Treponema medium/Treponema vincentii-like, Treponema denticola-like, and Treponema pedis phylotypes are highly associated with progression of the disease (9–12). DD also appears to be highly infectious based on the rapid intra- and interherd spread of the disease (6, 13, 14). The spread of DD in dairy cattle is facilitated by wet walking surfaces covered with feces (15), although the causative agents have not yet been found in slurry (16). Because the Treponema phylotypes typically observed in DD biopsy specimens have not yet been identified in the dairy farm environment, the transmission route and the environmental reservoirs of these bacteria remain unclear.
Treponemes are notoriously difficult to cultivate, and there have been relatively few investigations of these bacteria in the farm environment (17). Global bacterial studies have shown that there are several phylotypes of Treponema in the bovine gastrointestinal (GI) tract (18–20), including Treponema bryantii and Treponema saccharophilum, which have been isolated from the rumina of cows (21, 22). These cultivable treponemes are phylogenetically very distinct from those associated with DD (17). So far, no DD-associated bacteria have been isolated from feces, suggesting that DD treponemes are not part of the normal microbiota of the bovine GI tract (16).
PCR-based methods circumvent the difficulties associated with cultivation of the fastidious anaerobic treponemes. In a previous PCR-based survey, no evidence was found for the presence of three major DD-related Treponema phylotypes in environmental slurry or GI tract contents, although these phylotypes were found in the rectal and oral tissues of some animals (16). We hypothesized that DD-related treponemes are present in the dairy herd environment—but in very small amounts that cannot be detected by standard PCR methods. The aim of this study was therefore to identify DD-associated treponemes from cow feces, environmental slurry, and DD lesions by using high-throughput, culture-independent 16S rRNA gene sequence analysis. Before sequencing, the samples were PCR amplified with a primer set specific for members of the genus Treponema that was able to amplify most of the Treponema phylotypes hitherto identified in DD lesions (10). Using this approach, we performed phylogenetic characterization of 82 samples from seven dairy herds with variable prevalences of DD.
MATERIALS AND METHODS
Sample collection and preparation.
Samples were collected from six Holstein Friesian dairy herds with recurrent cases of DD and one small free-range herd (mixed breeds) with no known history of DD. These free-range animals had access to shelter and were kept in pastures year-round. In the remaining six herds, the animals were kept in loose house systems with slatted or element floors. The details of the farms are listed in Table 1. Except for the free-range herd, the samples were taken from the herds at the time of hoof trimming, which enabled sampling of fresh feces from individual cows as well as sampling from a small number of skin lesions. Fecal samples were obtained with a wooden spatula, and the surfaces of the skin lesions were scraped with a small blade. All the lesion samples—eight from DD-affected areas and one from an exudative udder lesion—were obtained from the Gilleleje herd. Samples were immediately transferred to RNAlater stabilization solution (Ambion, Austin, TX, USA). After 24 h at 5°C, according to the manufacturer's instructions, the samples were kept at −20°C until use.
TABLE 1.
Details of the 7 farms included in the study
| Herd | Herd size | Breed(s) | Floor type | Occurrence of DD | Preventive measures |
|---|---|---|---|---|---|
| Melose | 7 | Hereford/Jersey/Holstein Frisian | Pasture | No | None |
| Soroe | 150 | Holstein Friesian | Slatted | Yes | None |
| Gilleleje | 247 | Holstein Friesian | Slatted | Yes | Hydrated lime |
| Holmegaard | 109 | Holstein Friesian | Slatted | Yes | None |
| Ribe | 301 | Holstein Friesian | Slatted | Yes | None |
| Slagelse | 100 | Holstein Friesian | Element | Yes | None |
| Oroe | 183 | Holstein Friesian | Slatted | Yes | None |
Bacterial DNA was extracted from feces and slurry samples using the QIAamp DNA stool minikit (Qiagen, Hilden, Germany). Portions (200 mg) of feces were first homogenized in 1.4 ml of ASL buffer (included in the kit) and subsequently heated for 10 min at 70°C to lyse the bacteria. Then bacterial DNA from the skin lesions was purified with the DNeasy Blood and Tissue kit (Qiagen). Briefly, samples were resuspended in 180 μl of ATL buffer (Qiagen). A sterile 5-mm steel bead (Qiagen) was added for complete bacterial lysis in a Qiagen TissueLyser (Qiagen), which was run at 20 Hz twice, for 2 min each time. Next, 20 μl of proteinase K was added, and the samples were incubated for 1 h at 56°C. All subsequent steps were performed according to the protocol provided. The concentrations and purity of the samples were evaluated using a Nanodrop 1000 spectrophotometer (Fisher Scientific, Wilmington, MA), and only samples with A260/A280 ratios of >1.5 were used for further analyses.
Preparation of 16S rRNA gene amplicon libraries and sequencing.
DNA was amplified by PCR using oligonucleotide primers designed to target the V3 and V4 hypervariable regions of the 16S rRNA gene sequences of Treponema putidum, T. pedis, T. denticola, T. vincentii, Treponema calligyrum, and Treponema refringens (10). These Treponema-specific primers have been shown to cross-react with the majority of treponemes hitherto identified from DD lesions (10). Each sample was amplified with unique forward and reverse primers that included an added hexamer barcode at their 5′ ends. The target region was amplified in a 50-μl reaction mixture that contained 1× AmpliTaq buffer (Applied Biosystems, Carlsbad, CA), 100 μM each deoxynucleoside triphosphate (Amersham Biosciences, Piscataway, NJ), 0.2 pmol each primer, 2.5 U AmpliTaq DNA polymerase (Applied Biosystems), and 2 μl of template DNA. Thermal cycling was performed using a T3 thermocycler (Biometra, Göttingen, Germany) with the following protocol: denaturation at 94°C for 3.5 min, followed by 30 cycles of denaturation at 94°C for 45 s, annealing at 59°C for 45 s, and extension at 72°C for 1 min, with a final elongation step of 5 min. A positive control and a negative control (water) were included for every PCR. The DNA concentrations and quality of the PCR amplicons from all samples were assessed with an Agilent 2100 bioanalyzer (Agilent Technologies Inc., Santa Clara, CA) prior to high-throughput sequencing (data not shown). Equal amounts of all 40 amplicons were pooled and were purified by using the Qiagen MinElute kit (Qiagen) according to the protocol provided by the manufacturer.
The DNA was separated into 2 batches (pool 1, 45 samples; pool 2, 41 samples) and was submitted to the National High-Throughput DNA Sequencing Centre at the University of Copenhagen, Copenhagen, Denmark, for sequencing on an Illumina MiSeq platform. The 251-bp reads obtained were analyzed using BION-meta software. Demultiplexing was performed according to the primer and barcode sequences. Forward and reverse sequences were joined, allowing no gaps, a maximum mismatch percentage of 80%, and a minimum overlap length of 20 bp. Next, the sequences were cleaned at both ends by the removal of bases with a quality of <98%, which is equivalent to a Phred score of 17. Identical sequences were dereplicated into consensus sequences of 320 to 321 bp. Consensus sequences of at least 190 nucleotides were mapped into a table according to the individual barcodes. Finally, the consensus sequences were taxonomically classified against the Ribosomal Database Project database II (RDP II) (http://rdp.cme.msu.edu/index.jsp) using a word length of 8 and a minimum match of 90%. To allow comparison of the relative abundances in samples, the number of reads for each barcode was normalized. The resulting microbial profiles were further analyzed using Excel and GraphPad Prism, version 5 (GraphPad Software, Inc., La Jolla, CA). Spearman's rho test was used to measure the size and statistical significance of the association between the DD-associated phylotype distributions in the environmental samples and the samples from the skin lesions.
BioProject accession number.
The data discussed in this publication have been deposited in the NCBI database and are accessible through accession number PRJNA240271.
RESULTS
A total of 10,993,304 (pool 1) and 6,729,168 (pool 2) reads were obtained from the sequencing center. After demultiplexing according to the sequences of barcodes and primers, 6,979,106 and 4,597,162 sequences remained in pools 1 and 2, respectively. The 3′ and 5′ ends of these sequences were further trimmed according to quality. Sequences with a quality below 98% were discarded. In total, 2,798,092 (pool 1) and 2,151,644 (pool 2) sequences, equivalent to 62,180 and 52,479 average reads per sample, respectively, were used for taxonomic classification. Of these, 34.4% of pool 1 and 25.9% of pool 2 were taxonomically classifiable according to the RDP database.
In total, 86 samples were sequenced. Four samples with input reads below 20,000 sequences were discarded from the data set. The consensus sequences were taxonomically assigned using the RDP II database. The sequences that could not be classified to the species level were combined into the groups Bacteria spp., Spirochaeta spp., and Treponema spp. The remaining sequences showed high sequence homology with previously identified Treponema spp. or clones that have not yet been cultivated.
No significant differences in species composition could be observed between the slurry and cow manure samples (referred to below as “environmental samples”). In most cases, more than 50% of the reads from the environmental samples could not be classified further than the genus level. Most of these reads corresponded to the genus Treponema, while on average 4% corresponded to the genus Spirochaeta. A relatively large number of sequences were highly similar to a not-yet-cultivated variant, Treponema KO1_aai43a12 (GenBank accession no. EU776449), which was isolated from red kangaroo (Macropus rufus) feces at the Saint Louis Zoological Park, St. Louis, MO, USA (19) (Fig. 1). On average, this phylotype constituted 93% and 28.5% of the sequences amplified from the free-range herd and environmental samples, respectively, while it was present in very low numbers in the skin lesions (0.5%) (Fig. 1). Another phylotype, Treponema SP2_g07_2 (GenBank accession no. EU469011), which resembled a clone identified from Speke's gazelle (Gazella spekei) feces (19), was likewise present in most of the samples; however, its prevalence was much lower than that of KO1_aai43a12 (Fig. 1).
FIG 1.

Color-coded bar plot showing the distribution of bacterial groups across the 82 samples from 7 individual herds. The bacterial sequences that could not be determined to the species/phylotype level were designated Bacteria spp. Spirochaeta/Treponema spp., Treponema KO1_aai43a12, and Treponema SP2_g07_2 were treated as separate groups, since they were among the most prevalent of the non-DD-related amplicons. Sequences homologous to the 14 DD-associated phylotypes were included in the DD Treponema group.
Overall, 14 different phylotypes of known DD-associated treponemes could be mapped from the sequenced fragments (Table 2), and they belonged to 4 phylogenetic groups: T. medium/T. vincentii-like, T. phagedenis-like, T. denticola/T. pedis-like, and T. refringens-like. While no DD-related treponemes were found in the samples from the free-range herd, we identified 12 different DD Treponema phylotypes distributed in 43 of the remaining 64 environmental samples: 16 cow manure samples and 27 slurry samples. With a few exceptions, the number of DD-related sequences identified from the environmental samples was very low. Figure 2 displays the prevalence of DD-associated treponemes in the 43 samples from six different herds. In 33 of the 46 samples, the prevalence of these treponemes was below 0.5%. In only two of the samples (both from slurry) did the sequences of DD-related treponemes exceed 5% of the total bacterial sequence population. On average, 0.22% and 0.75% of the cow manure and slurry PCR-amplified sequences, respectively, were homologous to DD-related treponeme sequences.
TABLE 2.
Treponema groups and phylotypes identified from sequenced 16S rRNA gene fragments
| Group | Phylotype/species mapped |
|---|---|
| T. medium/T. vincentii-like | T. medium |
| T. phagedenis-like | T. phagedenis |
| PT13 | |
| T. denticola/T. pedis-like | T. denticola |
| T. pedis | |
| PT8 | |
| 9T-42 | |
| T. refringens-like | T. refringens |
| PT1 | |
| PT2 | |
| PT3 | |
| PT4 | |
| PN-20 (PT12) | |
| PT15 |
FIG 2.
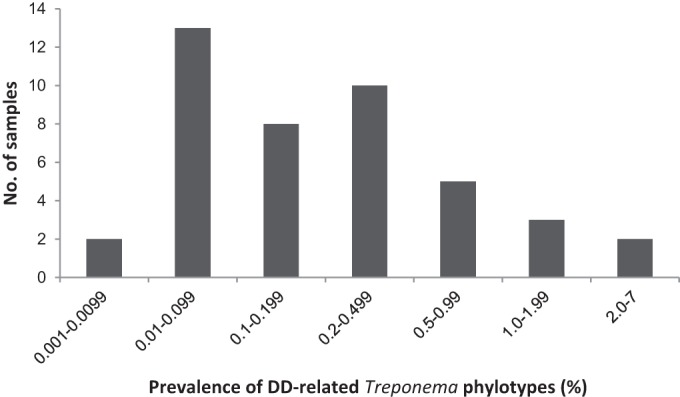
Prevalence of DD-related Treponema phylotypes in 43 positive environmental samples from the 6 positive herds.
In contrast, the samples obtained from the 8 DD lesions and the single udder lesion were entirely dominated by DD-related species (Fig. 1); DD-related sequences constituted an average of 86.6% of the total sequence population. All 14 phylotypes of known DD-associated treponemes were present in these 9 samples, and the average number of DD-related species identified in a single sample was 12 (Fig. 3a). Figure 3b shows the prevalence distribution of the T. medium/T. vincentii-like, T. phagedenis-like, T. denticola/T. pedis-like, and T. refringens-like phylogenetic clusters in the 9 skin lesions. From this figure, it can be observed that treponemes from the T. denticola/T. pedis-like group were the most prevalent representatives of the DD-associated Treponema species in these lesion samples. The distribution of the total number of reads for the DD-associated treponemes in the environmental samples was significantly correlated with that in the samples from DD lesions (Fig. 4) (Spearman's rho, 0.83; P, 0.0002).
FIG 3.

Bar plots comparing the DD-associated phylotype compositions of the samples isolated from 8 DD lesions and 1 udder lesion (DD3) (a) and the distributions of the 4 major phylogenetic clusters of DD-related Treponema species in the same 9 skin lesions (b).
FIG 4.
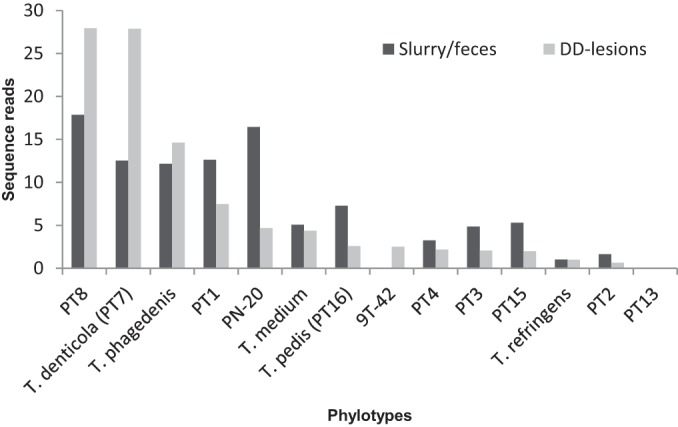
Distribution of the 14 phylotypes of known DD-associated treponemes, expressed as percentages of the total number of reads for DD-associated treponemes, in the DD lesion samples compared with that in the slurry/cow manure samples. Note that DD-associated treponeme sequence reads account for 86% of total sequence reads in lesion samples compared with <1% of total sequence reads in environmental samples.
However, it should be noted that although the proportions of DD-associated treponeme phylotypes in the environmental samples and the DD lesions appear similar (Fig. 4), they are percentages of DD-associated treponeme sequence reads, not of all sequence reads, in the samples, and their magnitudes are in fact very different. DD-related Treponema sequences are highly abundant in the lesion samples, while they only make up a very small percentage of the environmental samples.
DISCUSSION
Despite the apparent contagiousness of DD, DD-associated Treponema has not been identified in the dairy herd environment or the contents of the bovine GI tract (16, 17). Thus, it seemed reasonable to hypothesize that any DD treponemes present in this environment would be sporadic. We hypothesized that high-throughput sequencing of the 16S rRNA gene might be sensitive enough to detect low levels of spirochetes in a complex environment. However, the use of universal bacterial primers may underestimate spirochete populations, especially the less abundant species/phylotypes for which we were searching (23). Therefore, to obtain high levels of coverage of any DD-associated Treponema species present in the dairy herd environment, we targeted our sequencing analysis specifically against members of this genus, using primers designed to amplify the Treponema spp. that had been identified in DD lesions previously (9, 11, 12, 24, 25).
The present study involved 7 samples from a free-range herd with no history of DD, 64 environmental samples, consisting of slurry and fresh cow feces from individual animals in six herds, 8 samples from DD lesions, and 1 sample from an udder lesion, possibly due to bovine ulcerative mammary dermatitis (UMD). Most of the amplicons from the environmental sequences could be taxonomically assigned only to the genus level. There are several phylotypes of Treponema in the bovine GI tract (17, 26, 27); however, because of the difficulties associated with the isolation, cultivation, and purification of treponemes, many species of this genus have yet to be characterized. This may explain why a large fraction of the amplified sequences could not be classified further than the genus level.
Because the primers applied in this study target only a relatively narrow range of the Treponema genus, we did not observe the species T. bryantii and T. saccharophilum, which have been isolated from the rumina of cows previously (21, 22). A relatively large number of amplicons were homologous to two not-yet-cultivated ruminant clones, Treponema KO1_aai43a12 and Treponema SP2_g07_2, which were isolated from red kangaroo and Speke's gazelle feces, respectively. KO1_aai43a12 was highly prevalent, especially in the samples from the Melose free-range herd (Fig. 1). The red kangaroo is also a foregut fermenter, and its gut microbial community clusters with that of the Holstein cow in a phylogenetic analysis of gut microbe relatedness (19). The closest match to this phylotype in GenBank was an uncultured cattle rumen bacterium (93% sequence identity) and a cultivable species, Treponema brennaborense (91% sequence homology). The two clones most likely represent commensal GI tract treponemes.
Using a deep-sequencing approach, we were also able to identify treponeme phylotypes usually identified only in bovine DD lesions in 43 of the 64 environmental samples, including both cow manure and slurry samples. In a previous study, all environmental slurry samples and GI tract contents were negative for DD-associated Treponema DNA from the three most prevalent phylotypes (16). However, DD treponemes have previously been identified in the oral cavity and rectum in approximately 14% of the cows tested (16). This raises the question of whether our observations in cow manure are due to rectal contamination of the feces or whether the DD treponemes are in fact also present in the GI tract contents, where their DNA can be detected if a sensitive enough method is applied. Future studies employing highly sensitive detection methods will be required to clarify this issue.
Not surprisingly, we detected sequences that were homologous to 14 different DD-related phylotype variants in the bovine lesion samples (Fig. 3a). On average, 12 different variants were identified. All variants had been identified in DD lesions previously (9–12). As in previous investigations, species from the T. denticola/T. pedis-like, T. phagedenis-like, T. refringens-like, and T. medium/T. vincentii-like phylogenetic clusters were represented in the lesions, including the putative UMD lesion (Fig. 3b) (9, 10, 28, 29).
With the caveats that all the DD skin samples were collected in 1 herd (Gilleleje) and that the proportions of phylotypes in the two sample types represented very different magnitudes, the overall sequence distribution of the DD-related phylotypes in the environmental samples (n = 64) was highly correlated with that observed in the DD skin samples (n = 9). Considering that the distribution of the phylogenetic clusters of the DD treponemes in the Gilleleje samples resembled the distributions observed in previous investigations (9, 12), we assumed that the Gilleleje herd was suitable as a representative herd. One of the main aims of this study was to identify a sensitive method capable of detecting treponeme phylotypes associated with DD in the dairy herd environment. Here we demonstrated that it was possible to detect and characterize DD-related treponeme sequences in this habitat by using a targeted PCR approach in combination with deep sequencing methods, even if the sequences were present at a very low frequency. Additionally, the frequency distribution of phylotypes in the dairy herd environment appeared to mirror the average distribution of treponemes found in the DD lesions. However, the question of how bacteria with such a low prevalence can be so successful in infecting a herd remains to be clarified. The targeted sequencing approach presented here can now be used to investigate both environmental and whole-animal samples in order to determine potential transmission routes and possible infection reservoirs of DD. We hope that this approach will allow us to better understand the etiopathogenesis of this disease.
ACKNOWLEDGMENTS
This research was funded by the Danish Mælkeafgiftsfond.
We thank the participating farmers and claw trimmers for collaboration with regard to the herds, and we thank the technical staff members at DTU-VET for excellent assistance.
Footnotes
Published ahead of print 9 May 2014
REFERENCES
- 1.Cheli R, Mortellaro C. 1974. La dermatite digitale del bovino, p 208–213 In Proceedings of the 8th International Conference on Diseases of Cattle, Piacenza, Milan, Italy [Google Scholar]
- 2.Amory JR, Barker ZE, Wright JL, Mason SA, Blowey RW, Green LE. 2008. Associations between sole ulcer, white line disease and digital dermatitis and the milk yield of 1824 dairy cows on 30 dairy cow farms in England and Wales from February 2003-November 2004. Prev. Vet. Med. 83:381–391. 10.1016/j.prevetmed.2007.09.007 [DOI] [PubMed] [Google Scholar]
- 3.Willshire JA, Bell NJ. 2009. An economic review of cattle lameness. Cattle Practice 17:136–141 [Google Scholar]
- 4.Whay HR, Waterman AE, Webster AJ. 1997. Associations between locomotion, claw lesions and nociceptive threshold in dairy heifers during the peri-partum period. Vet. J. 154:155–161. 10.1016/S1090-0233(97)80053-6 [DOI] [PubMed] [Google Scholar]
- 5.Palmer MA, Donnelly RF, Garland MJ, Majithiya R, O'Connell NE. 2013. The effect of slurry on skin permeability to methylene blue dye in dairy cows with and without a history of digital dermatitis. Animal 7:1731–1737. 10.1017/S1751731113001274 [DOI] [PubMed] [Google Scholar]
- 6.Wells SJ, Garber LP, Wagner BA. 1999. Papillomatous digital dermatitis and associated risk factors in US dairy herds. Prev. Vet. Med. 38:11–24. 10.1016/S0167-5877(98)00132-9 [DOI] [PubMed] [Google Scholar]
- 7.Moter A, Leist G, Rudolph R, Schrank K, Choi BK, Wagner M, Göbel UB. 1998. Fluorescence in situ hybridization shows spatial distribution of as yet uncultured treponemes in biopsies from digital dermatitis lesions. Microbiology 144:2459–2467. 10.1099/00221287-144-9-2459 [DOI] [PubMed] [Google Scholar]
- 8.Rasmussen N, Capion N, Klitgaard K, Rogdo T, Fjeldaas T, Boye M, Jensen TK. 2012. Bovine digital dermatitis: possible pathogenic consortium consisting of Dichelobacter nodosus and multiple Treponema species. Vet. Microbiol. 160:151–161. 10.1016/j.vetmic.2012.05.018 [DOI] [PubMed] [Google Scholar]
- 9.Evans NJ, Brown JM, Demirkan I, Singh P, Getty B, Timofte D, Vink WD, Murray RD, Blowey RW, Birtles RJ, Hart CA, Carter SD. 2009. Association of unique, isolated treponemes with bovine digital dermatitis lesions. J. Clin. Microbiol. 47:689–696. 10.1128/JCM.01914-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klitgaard K, Foix BA, Boye M, Jensen TK. 2013. Targeting the treponemal microbiome of digital dermatitis infections by high-resolution phylogenetic analyses and comparison with fluorescent in situ hybridization. J. Clin. Microbiol. 51:2212–2219. 10.1128/JCM.00320-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nordhoff M, Moter A, Schrank K, Wieler LH. 2008. High prevalence of treponemes in bovine digital dermatitis—a molecular epidemiology. Vet. Microbiol. 131:293–300. 10.1016/j.vetmic.2008.04.019 [DOI] [PubMed] [Google Scholar]
- 12.Yano T, Moe KK, Yamazaki K, Ooka T, Hayashi T, Misawa N. 2010. Identification of candidate pathogens of papillomatous digital dermatitis in dairy cattle from quantitative 16S rRNA clonal analysis. Vet. Microbiol. 143:352–362. 10.1016/j.vetmic.2009.12.009 [DOI] [PubMed] [Google Scholar]
- 13.Blowey RW, Sharp MW, Done SH. 1992. Digital dermatitis. Vet. Rec. 131:39. 10.1136/vr.131.2.39-a [DOI] [PubMed] [Google Scholar]
- 14.Read DH, Walker RL. 1998. Papillomatous digital dermatitis (footwarts) in California dairy cattle: clinical and gross pathologic findings. J. Vet. Diagn. Invest. 10:67–76. 10.1177/104063879801000112 [DOI] [PubMed] [Google Scholar]
- 15.Laven RA. 2001. Control of digital dermatitis in cattle. In Practice 23:336–341. 10.1136/inpract.23.6.336 [DOI] [Google Scholar]
- 16.Evans NJ, Timofte D, Isherwood DR, Brown JM, Williams JM, Sherlock K, Lehane MJ, Murray RD, Birtles RJ, Hart CA, Carter SD. 2012. Host and environmental reservoirs of infection for bovine digital dermatitis treponemes. Vet. Microbiol. 156:102–109. 10.1016/j.vetmic.2011.09.029 [DOI] [PubMed] [Google Scholar]
- 17.Evans NJ, Brown JM, Murray RD, Getty B, Birtles RJ, Hart CA, Carter SD. 2011. Characterization of novel bovine gastrointestinal tract Treponema isolates and comparison with bovine digital dermatitis treponemes. Appl. Environ. Microbiol. 77:138–147. 10.1128/AEM.00993-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jami E, Israel A, Kotser A, Mizrahi I. 2013. Exploring the bovine rumen bacterial community from birth to adulthood. ISME J. 7:1069–1079. 10.1038/ismej.2013.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, Schlegel ML, Tucker TA, Schrenzel MD, Knight R, Gordon JI. 2008. Evolution of mammals and their gut microbes. Science 320:1647–1651. 10.1126/science.1155725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang S, Ma S, Chen J, Mao H, He Y, Xi D, Yang L, He T, Deng W. 2010. Bacterial diversity in the rumen of Gayals (Bos frontalis), Swamp buffaloes (Bubalus bubalis) and Holstein cow as revealed by cloned 16S rRNA gene sequences. Mol. Biol. Rep. 37:2063–2073. 10.1007/s11033-009-9664-6 [DOI] [PubMed] [Google Scholar]
- 21.Paster BJ, Canale-Parola E. 1985. Treponema saccharophilum sp. nov., a large pectinolytic spirochete from the bovine rumen. Appl. Environ. Microbiol. 50:212–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stanton TB, Canale-Parola E. 1980. Treponema bryantii sp. nov., a rumen spirochete that interacts with cellulolytic bacteria. Arch. Microbiol. 127:145–156. 10.1007/BF00428018 [DOI] [PubMed] [Google Scholar]
- 23.Sakamoto M, Siqueira JF, Jr, Rocas IN, Benno Y. 2009. Diversity of spirochetes in endodontic infections. J. Clin. Microbiol. 47:1352–1357. 10.1128/JCM.02016-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi BK, Nattermann H, Grund S, Haider W, Göbel UB. 1997. Spirochetes from digital dermatitis lesions in cattle are closely related to treponemes associated with human periodontitis. Int. J. Syst. Bacteriol. 47:175–181. 10.1099/00207713-47-1-175 [DOI] [PubMed] [Google Scholar]
- 25.Demirkan I, Carter SD, Hart CA, Woodward MJ. 1999. Isolation and cultivation of a spirochaete from bovine digital dermatitis. Vet. Rec. 145:497–498. 10.1136/vr.145.17.497 [DOI] [PubMed] [Google Scholar]
- 26.Patton TG, Scupham AJ, Bearson SM, Carlson SA. 2009. Characterization of fecal microbiota from a Salmonella endemic cattle herd as determined by oligonucleotide fingerprinting of rDNA genes. Vet. Microbiol. 136:285–292. 10.1016/j.vetmic.2008.10.032 [DOI] [PubMed] [Google Scholar]
- 27.Tajima K, Aminov RI, Nagamine T, Ogata K, Nakamura M, Matsui H, Benno Y. 1999. Rumen bacterial diversity as determined by sequence analysis of 16S rDNA libraries. FEMS Microbiol. Ecol. 29:159–169. 10.1111/j.1574-6941.1999.tb00607.x [DOI] [Google Scholar]
- 28.Stamm LV, Walker RL, Read DH. 2009. Genetic diversity of bovine ulcerative mammary dermatitis-associated Treponema. Vet. Microbiol. 136:192–196. 10.1016/j.vetmic.2008.10.022 [DOI] [PubMed] [Google Scholar]
- 29.Moe KK, Yano T, Kuwano A, Sasaki S, Misawa N. 2010. Detection of treponemes in canker lesions of horses by 16S rRNA clonal sequencing analysis. J. Vet. Med. Sci. 72:235–239. 10.1292/jvms.09-0404 [DOI] [PubMed] [Google Scholar]