

Abstract
The vast majority of microscopic life on earth consists of microbes that do not grow in laboratory culture. To profile the microbial diversity in environmental and clinical samples, we have devised and employed molecular probe technology, which detects and identifies bacteria that do and do not grow in culture. The only requirement is a short sequence of contiguous bases (currently 60 bases) unique to the genome of the organism of interest. The procedure is relatively fast, inexpensive, customizable, robust, and culture independent and uses commercially available reagents and instruments. In this communication, we report improving the specificity of the molecular probes substantially and increasing the complexity of the molecular probe set by over an order of magnitude (>1,200 probes) and introduce a new final readout method based upon Illumina sequencing. In addition, we employed molecular probes to identify the bacteria from vaginal swabs and demonstrate how a deliberate selection of molecular probes can identify less abundant bacteria even in the presence of much more abundant species.
INTRODUCTION
It is now widely appreciated that only a small fraction of the earth's bacteria may be grown in laboratory culture (for example, see reference 1). That statement is particularly true for the bacteria found in and on human beings (2). The Human Microbiome Project employs a metagenomic approach, principally amplifying and sequencing a small portion of the 16S rRNA gene, to identify bacteria. Like any technology, sequencing rRNA genes has strengths and weaknesses. Its principal strengths are that it overcomes the serious limitations of bacterial unculturability and genomic diversity. Its principal weaknesses are that different bacteria have different copy numbers of rRNA genes (3–5), that the application of rRNA gene “universal” primers has specificity performance limitations for some bacterial species (6–8), that chimeras are formed at a relatively high frequency when rRNA genes are amplified and cloned (for example, see reference 9), and that the presence of a highly abundant bacterium can mask the detection of less abundant species. Microarrays containing probes that hybridize to rRNA genes sequences are also effective metagenomic tools for detecting unculturable bacteria (10), but, as mentioned above, they can be limited in their ability to discern between some species.
To overcome these weaknesses, we developed a genome-based strategy that targets single-copy bacterial genome sequences with high specificity and sensitivity. Originally referred to as molecular inversion probes (11), the technology was simplified for high-throughput analysis, and thus, the name was revised simply to “molecular probes.” This approach is rapid, scalable, robust, and culture independent (12, 13). This assay requires only a short stretch of contiguous DNA sequence unique to the bacterial genome of interest. That means that probe technology cannot detect novel microbes, i.e., microbes for which genome sequence information is not yet available. In addition, molecular probe technology is highly dependent on probe design and hybridization conditions and, in its current form, is challenged by quantitation. Nevertheless, molecular probes are a powerful technology that can be used alone or to complement other metagenomic discovery approaches, including those based on rRNA gene sequencing or hybridization.
We have made four important improvements to the molecular probe designs and assay. (i) To improve the specificity of the molecular probes, the length of the homology sequence between molecular probe and target DNA has been increased from 40 to 60 bases. (ii) The complexity of the assay has been increased by over an order of magnitude to include over 1,200 molecular probes targeting 61 bacteria. We demonstrate that an effective molecular probe set can be constructed from individually synthesized oligonucleotides or from a less-expensive array-based synthesis platform. (iii) We have employed Illumina sequencing to quantitate the oligonucleotide barcodes following the molecular probe reaction. Because of recent, dramatic decreases in DNA sequencing cost and the ability to sequence multiple samples in a single experiment, there is now a significant cost advantage of sequencing over array-based barcode quantitation. (iv) Lastly, we demonstrate how a deliberate selection of molecular probes can identify less abundant bacteria even in the presence of much more abundant species.
MATERIALS AND METHODS
DNAs.
Bacterial genomic DNAs for the control experiments were purchased from the American Type Culture Collection. Four genomic DNAs were mixed to produce a simulated clinical sample (SCS), which was employed in control experiments. SCS2 contains, per μl, 3,510,000 molecules (36.0%) of Escherichia coli DNA, 570,000 molecules (5.9%) of Lactobacillus acidophilus DNA, 2,886,000 molecules (29.6%) of Neisseria gonorrhoeae DNA, and 2,772,000 molecules (28.5%) of Streptococcus agalactiae DNA. Vaginal swab DNAs were published previously (14). All of these DNAs were sheared to an average double-strand size of ∼500 bp by employing a Covaris instrument, as described by the manufacturer. Following shearing, the DNAs were concentrated and dialyzed into TE buffer (0.01 Tris, 0.001 EDTA [pH 8.0]) by use of Amicon Ultra centrifugal filter units (Millipore) and stored at –20°C until use.
Molecular probe designs.
Molecular probes are 5′-phosphorylated, long oligonucleotides (12, 13). Our goal was to design and synthesize ∼20 molecular probes for each genome sequence. We reasoned that some would work and some would not work but that, with 20 probes, a sufficient number would work to identify bacteria reliably. The molecular probes are 116-mers. As diagrammed in Fig. 1A, these oligonucleotides are composed of three domains: a 60-base homology sequence known as a “Homer,” identical in sequence to the target bacterial genome and divided into two 30-base end sections; a common 36-base PCR primer section (15); and a 20-base barcode compatible with the Affymetrix GenFlex Tag16K array (referred to here as the Tag4 array) (16). The Homers were derived from the public bacterial genome sequences in GenBank by applying our custom script, blaster.rb, which is freely available at http://med.stanford.edu/sgtc/research/blaster.html.
FIG 1.

Molecular probe design and assays. (A) Design of the molecular probes synthesized at the SGTC. The purple color represents the 60-base sequence homology domain (the Homer), which is divided into two 30-base segments. Blue represents the 20-base oligonucleotide barcode from the Tag4 array. Green represents the 36-base domain for the two 20-base PCR primers. The two 20-base primers overlap by 4 bases at their respective 5′ ends. The total is 116 bases. The 5′ end is phosphorylated. (B) Assays. (a) The molecular probe mixture is incubated with the denatured target DNA (wiggly lines) under annealing conditions. (b) Where sufficient sequence similarity exists between the molecular probe and the target single-stranded DNA (in purple), 60 bp of duplex DNA form. The 5′-phosphorylated end of the molecular probe is adjacent to the 3′-hydroxyl end of the probe with no bases missing. (c) Ligation creates a phosphodiester bond between the 5′ and 3′ ends. (d) Exonucleases digest the remaining single-stranded linear DNA, leaving only the single-stranded circular DNA. (e) PCR amplifies the single-stranded circular DNA. The 5′ ends of the PCR primers are biotinylated for the Tag4 readout but not for the Illumina readout. (f) The final readout is by either fluorescence on the Tag4 array (top) or sequencing by the Illumina MiSeq instrument (bottom).
At the time of our molecular probe design, there were two Atopobium vaginae genome sequences available in GenBank: A. vaginae DSM 15829 (35.37× coverage) and A. vaginae PB189-T1-4 (57× coverage). These two sequences are somewhat different. Therefore, because the presence or absence of the bacterium A. vaginae is very important to the health of the human vagina, we designed 20 molecular probes for each genome sequence. Similarly, the genome sequences of Pseudomonas fluorescens in GenBank have significant variation. Therefore, we chose two different P. fluorescens genome sequences and designed 20 molecular probes for each. For 61 bacterial species, there were a total of 1,096 molecular probes, with an average of 18.6 molecular probes per genome. For 10 species' genomes, blaster.rb could not define 20 unique Homers. In those cases, we allowed blaster.rb to define additional Homers that identified the species of interest plus one or more other species within the same genus. There were 108 such genus-level molecular probes. Thus, overall, there were 1,204 molecular probes.
Four molecular probes were positive in over half of the samples, including those where no target DNA was present. Since the same four molecular probes were promiscuous in both the Stanford Genome Technology Center (SGTC) and Agilent Technologies molecular probe sets (see below) and with both detection methods (see below), we assumed that the problem was inherent in the probes' designs. To determine if secondary structure could account for these probes' promiscuity, the sequence of each oligonucleotide was entered into the m-fold software (17) in the public UNAFold Web Server at the RNA Institute at SUNY Albany (http://mfold.rit.albany.edu/). There was no significant predicted secondary structure for any of the four promiscuous probes. To determine if any promiscuous probe could form a dimer with itself or with another promiscuous probe, we entered the sequences into the Integrated DNA Technologies OligoAnalyzer 3.1 website (http://www.idtdna.com/analyzer/applications/oligoanalyzer/). No dimerization of any type could be predicted. The sequences of the four promiscuous probes were searched with BLAST against GenBank. Only the appropriate Homer homologies were detected. While we have no explanation as to why these four probes were promiscuous, data derived from them were removed informatically from the data sets. Therefore, overall, the molecular probe set was composed effectively of 1,200 (i.e., 1,204 − 4) probes.
Syntheses of the molecular probe sets.
There were two sources of molecular probes. We synthesized one set of oligonucleotides by employing our 96-well automated multiplex oligonucleotide synthesizer. We refer to these oligonucleotides as the SGTC probe set (for details, see below) (18). The second set was purchased from Agilent Technologies under a collaborative technology access program. These oligonucleotides were provided in a pooled format. Since the two sets yielded comparable data, all consideration of the Agilent Technologies probe set can be found in the supplemental material.
SGTC oligonucleotide synthesis (50-nmol scale) was accomplished with our custom 96-well plate DNA synthesizer using standard 1,000-Å controlled-pore glass columns (Biosearch Technologies). Cycle conditions were similar to the AB3900 (Applied Biosystems) manufacturer's recommended protocol, which included the following reagents: trichloroacetic acid as a 3% solution in dichloromethane (American International Chemicals [AIC]), acetonitrile (AIC), 0.02 M oxidizing solution (Sigma), cap A/B (Glen Research), 0.1 M solutions of dA, dC, dG, and dT (Sigma), and 0.25 M 5-benzylthio-1H-tetrazole (AIC). Postsynthesis steps included strand cleavage from the support followed by base deprotection overnight (15 h) at 55°C with ammonium hydroxide (28 to 30% in water) (J.T. Baker). After lyophilization, oligonucleotides were resuspended in 30 μl 0.1× TE buffer, and the absorbance of each was measured at 260 nm employing a Spectramax 384 Plus 96-well plate reader (Molecular Devices). All samples were then normalized to 100 μM in 0.1× TE buffer using a Biomek FX robot and then analyzed for purity using reverse-phase high-performance liquid chromatography (Transgenomic WAVE system). After mixing the individual molecular probes together in equimolar amounts, the 5′ ends were phosphorylated. A deliberately reduced molecular probe set was also constructed. All Lactobacillus probes and the four promiscuous probes were omitted, yielding the Lactobacillus-negative probe set. After being mixed in equimolar amounts, the molecular probes were 5′ phosphorylated.
Two readouts for the molecular probe assay.
A description of the assay itself has been published previously (12, 13). Briefly, the procedure is as follows, as outlined in Fig. 1B. (i) The probe set is incubated with the denatured target DNA under hybridization conditions. (ii) Where sufficient DNA base sequence homology exists between the Homer of the probe and the target DNA, a 60-bp duplex is formed, with the 5′-phosphate of the probe immediately adjacent to the 3′-hydroxyl. (iii) Ligase creates a 5′-to-3′ phosphodiester bond, closing the circle. (iv) Exonuclease digests single-stranded DNA, leaving only the circular DNA. (v) PCR is performed to amplify the circular DNA. (vi) Two barcode detection methods were employed in our molecular probe assay. The final readout was either fluorescence on a Tag4 array or (new here) Illumina sequencing of the barcode plus the homology region (20 bases + 60 bases = 80 bases). Because we have published the Tag4 array readout previously (12, 13) and since the two methods yielded comparable data, all further consideration of the Tag4 readout is presented in the supplemental material.
Illumina sequencing.
The PCR fragments generated after amplification of the ligated molecular probes were sequenced in an Illumina MiSeq instrument, as described by the manufacturer. The PCR fragments were end repaired with a quick blunting kit purchased from New England BioLabs (NEB, Beverly, MA). Then, the fragments were tailed with dATP by incubation with 25 U Klenow fragment (NEB), Klenow buffer, and 0.2 mM dATP for 30 min at 37°C. Duplex indexed adapters with a 7-base unique barcode sequence (19) were ligated onto the PCR products by using 1 μl of quick DNA ligase (NEB) and ligase buffer with 90 min incubation at 12°C. The ligated products were pooled and enriched for 180- to 220-bp fragments using a Pippin Prep electrophoresis system (Sage Sciences, Beverly, MA). The pool of size-selected fragments was amplified with the Illumina Pair end primers 5′-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGA-3′ (forward) and 5′-CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAAC-3′ (reverse), and the library was sequenced on an Illumina MiSeq using a 300-cycle MiSeq reagent V2 kit.
Sequence analysis.
After demultiplexing and sample index removal, sequence reads were aligned to 80-bp amplicon sequences using the Burrows-Wheeler Aligner (BWA) and allowing a maximum of a 2-base difference (20). There are two domains within our alignment targets. The first domain is a 20-base barcode compatible with the Tag4 array (16, 21). The second domain is a 60-base Homer unique to the microbial genome of interest. Since the 60-base sequence is formed by a ligation of the two 30-base ends of the molecular probe (Fig. 1), the requirement of a proper alignment to the 80-base (i.e., 20 + 60 bases) amplicon indicates that a successful ligation has taken place and generates high-confidence mapping by enforcing both the sequence homology and the uniqueness of the barcode.
Statistical analysis.
For Illumina sequence data, the P values for presence were computed using a negative binomial model (22). Each probe was assigned a P value for presence, measuring whether the number of observed reads was significantly larger than random assignment of reads to all probes in the probe library. The P value was computed as
where p is the probability that a random probe receives a sequencing read (1/1,200), k is the number of reads observed, n is the total number of reads mapped, and r is an iterator from 0 to n − k. A probe with a P value of less than 0.05 was considered positive. Further, a microorganism was designated “present” if at least three molecular probes directed against that organism were positive, since the P value of such an event is less than 0.05 even after the Bonferroni multiple test correction (see Fig. S2 in the supplemental material) (23). All statistical analysis was carried out in R 2.15.2. The heat maps showing the presence of the microorganisms were generated using the ggplot2 packages (24).
RESULTS
Figure 1A is a diagram of the molecular probes. There are three domains: the 60-base Homer, divided into two 30-base segments at the 5′ and 3′ ends of the probe; the 36-base region homologous to the PCR primers; and the 20-base barcode compatible with the Tag4 array. Thus, the molecular probes are 116 bases and are 5′ phosphorylated. Figure 1B presents a diagram of the procedure (see Materials and Methods), starting with the molecular probe set and its single-stranded target DNA and ending with the two alternate readouts, Tag4 array and Illumina MiSeq.
As a test case, we constructed a simulated clinical sample (SCS) by mixing known amounts of four bacterial DNAs, from E. coli, L. acidophilus, N. gonorrhoeae, and S. agalactiae DNAs. To determine the optimum molecular probe concentration to employ in the molecular probe reaction, we first titrated the SGTC molecular probe set against the SCS (see Materials and Methods). The goal was to achieve optimum bacterial detection, that is, to define the probe concentration that minimized the number of false positives while also minimizing the number of false negatives. That molecular probe concentration was determined to be 100 zeptomoles (zmol)/μl (6 × 104 molecules/μl) of each probe.
As an additional control, we titrated the SGTC molecular probe set against 10-fold dilutions of the SCS. Three 10-fold dilutions of the SCS were interrogated with our 1,200 molecular probes, and the barcodes of probes that successfully hybridized to their target were subsequently quantitated by Illumina sequencing. The resulting sequence reads were mapped to amplicon sequences corresponding to the molecular probes, and positive probes were determined as described in Materials and Methods. Figure 2 presents the data for each of the 80 individual probes targeting the four bacterial genomic DNAs (20 probes per bacterium) present in the sample. (The numerical results are found in Table S1 in the supplemental material.)
FIG 2.
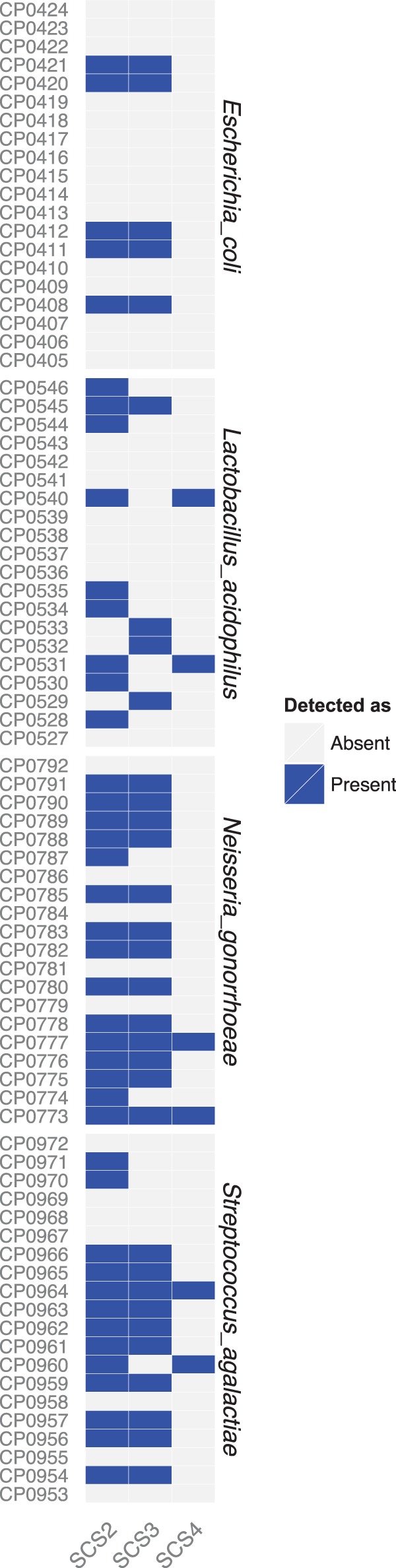
Titration of SCS with the SGTC molecular probe set. SCS2 was sequentially diluted 10-fold to produce SCS3 and SCS4. Each was reacted separately with the SGTC probe set. The final readout was by Illumina MiSeq sequencing of the barcodes plus the Homer (20 + 60 bases = 80 bases). The vertical axis lists the 20 individual probes for each of the four bacteria. Blue signifies that that individual probe was detected. The absence of color signifies that that individual probe was not detected.
Each bacterial DNA was detected by multiple probes. However, there were many falsely negative molecular probes (defined as negative by the analysis described in Materials and Methods). For example, only five of the molecular probes directed against E. coli DNA were positive, thus illustrating the importance of employing a multiple-probe strategy for each bacterial DNA to ensure reliable detection of the microbe. In general, the probes observed to be falsely negative were found to be falsely negative regardless of their source (see Fig. S1 in the supplemental material). This observation suggests that the probe designs, rather than the syntheses, were at fault.
As the target DNA concentration was diluted, some molecular probes did not detect their cognate DNAs. As examples, S. agalactiae probes CP0970 and CP0971 were positive only at the highest DNA concentration, and N. gonorrhoeae probes CP0790 and CP0791 were positive only at the two highest DNA concentrations. The most sensitive molecular probes detected their cognate DNAs at the lowest concentration tested: e.g., L. acidophilus probes CP0531 and CP0540 each detected 5,700 molecules of L. acidophilus DNA. Since the target DNAs were not tested at even lower concentrations, the minimum detection limits of CP0531 and CP0540 have not been determined. In general, the minimum detection limit for each molecular probe is a function of the complexity of the sample and the total number of Illumina sequence reads achieved and thus should not be generalized.
While the qualitative results for SCS were excellent (Fig. 2), the quantitative results were not. The number of Illumina sequence reads per bacterium did not follow the known relative amounts of the four bacterial DNAs in the sample (see Table S1 in the supplemental material). In an attempt at “normalization” of the data to take into account the different numbers of positive probes per bacterium, we calculated the average number of reads per positive probe per bacterium. This process did not improve the quantitation. This problem with quantitation has been reported previously for targeted RNA sequencing (25). Presumably, biases were introduced by unequal hybridization and amplification efficiencies across different molecular probes. Potentially, quantitation could be achieved by the addition of molecular indexing (25).
Since we knew which four bacterial DNAs comprised the SCS, we examined molecular probe specificity in terms of each probe's ability to discriminate between species belonging to the same genus. There were 200 Lactobacillus probes in our probe set. Only the nine probes for L. acidophilus were positive in SCS2. There were 20 molecular probes for Neisseria meningitidis in our probe set. All were negative in SCS2; i.e., none of the 20 N. meningitidis probes reacted with N. gonorrhoeae DNA. Despite the presence of S. agalactiae DNA in SCS2, none of the 20 S. mutans probes, none of the 20 S. pneumoniae probes, and none of the 20 S. pyogenes probes were positive.
To determine how many positive molecular probes, on average, are needed to confidently identify any given bacterium, we evaluated the specificity of the SCS titration series. First, we observed an empirical false-positive rate of 0.004 for those 1,120 (i.e., 1,200 − 80) probes that have no target DNA in these samples. Second, we computed the probability that a bacterium was observed by random chance by summing the binomial probability of having equal or more positive probes than observed. Third, the Bonferroni method was applied to correct the P values for multiple testing (23). As shown in Fig. S2 in the supplemental material, requiring multiple positive probes improved the significance of detection of a bacterium. If three molecular probes are required to be positive, the probability of its being a false positive is less than 0.05. Accordingly, in the experiments described below, we required three probes to be positive to designate a bacterium as being present. Analogously, utilizing 20 probes per target reduces the potential for false negatives. For the 80 probes that had target DNA in the three SCS of the titration series, we observed that on average, 67% of them appeared to be negative according to our criteria (Fig. 2). However, when the data for presence or absence were aggregated across 20 probes, the chance of a bacterium's being called negative when it was actually present fell below 0.05; i.e., the sum of binomial probabilities of having 0, 1, or 2 positive probes was less than 0.05. Therefore, we estimate the probability of false negatives to be <0.05 for other bacterial targets present at concentrations similar to those in the SCS titration series.
In a previous study (14), we identified the bacteria in vaginal swabs by amplifying and Sanger sequencing (nearly) the entire rRNA genes. For samples where we had sufficient vaginal swab DNA remaining, we employed our current molecular probe sets to identify the bacterial DNAs and then compared these data to our previous results. The comparative data from Sanger sequencing the amplified rRNA genes (16S/Sanger) and the molecular probe technology with the SGTC probe set with the Illumina readout (MP/Illumina) are presented in Fig. 3. (Results of the equivalent experiment employing the SGTC probe set with the Tag4 method are shown in Fig. S3 in the supplemental material. The number of supporting Illumina sequence reads is shown in Table S2 in the supplemental material.) The microbiomes of 13 of 24 vaginal swabs were dominated by Lactobacillus species, as determined by both assays. As one example, swab 038-2 was dominated by Lactobacillus helveticus, an unusual Lactobacillus species to be found in the vagina: 98% of the sequence reads in the 16S/Sanger assay and 91% of the reads in the MP/Illumina assay supported the presence of L. helveticus. However, the two technologies did not always detect the same Lactobacillus species. For example, for sample 049-1, the 16S/Sanger assay detected only L. acidophilus, whereas the MP/Illumina assay detected L. acidophilus and Lactobacillus crispatus. The additional detection of L. crispatus may be explained by the vast difference in sequencing depth (369 total reads versus 237,276 total reads). Both technologies also detected Enterococcus faecalis in sample 049-1.
FIG 3.
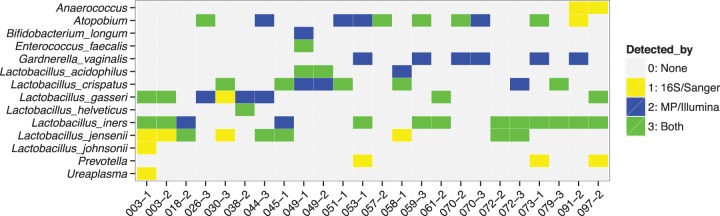
For 24 vaginal swabs, a comparison of the bacteria detected by Sanger sequencing rRNA genes (16S/Sanger) and the SGTC molecular probe set with the Illumina readout was made. The 16S/Sanger data have been published previously (14). The ordinate lists only those bacteria found by either assay. The abscissa lists the 24 individual vaginal swabs. Yellow signifies a bacterium detected only by 16S/Sanger. Blue signifies a bacterium detected only by the molecular probes. Green signifies a bacterium detected by both assays. The absence of color signifies that that bacterium was not detected by either assay.
All but two of the remaining swabs contained Atopobium and/or Gardnerella vaginalis. Atopobium was found in a total of 10 vaginal swabs. Both assays detected Atopobium in five swabs. The 16S/Sanger assay alone detected Atopobium in one additional swab (091-2). The molecular probes with the MP/Illumina readout alone detected Atopobium in four additional swabs (044-3, 051-1, 053-1, and 073-1). As discussed in Materials and Methods, we suspect that there is a large amount of sequence variation within Atopobium genomes. That suspicion will be confirmed or negated as more Atopobium genome sequences become available.
The situation is particularly interesting for Gardnerella vaginalis, a bacterium that is very important to the health (or lack thereof) of the human vagina. The 16S/Sanger assay did not find G. vaginalis in any of these vaginal swabs. The molecular probes with the MP/Illumina readout found G. vaginalis in six vaginal swabs (Fig. 3). We return to these important data in the Discussion.
The 16S/Sanger assay detected Prevotella in three swabs, whereas the molecular probes did not detect Prevotella in any swab. It is possible that the Prevotella genome sequence that was used to define the Homers had insufficient base sequence homology to the particular Prevotella species in the swabs. That situation can be corrected easily as more Prevotella genome sequences become available. Conversely, the probes with the MP/Illumina readout detected Bifidobacterium longum as supported by the substantial majority of the Illumina sequence reads for sample 049-1, whereas the 16S/Sanger assay just barely detected B. longum. An analogous explanation may apply.
We also interrogated these clinical samples with the probe set derived from array-based synthesis (see Fig. S4 and S5 in the supplemental material). The declining costs of array-based DNA synthesis make it an attractive tool for a variety of biological applications, particularly those where the resulting oligonucleotides are to be used as a pool. The results produced by the SGTC probe set and the Agilent Technologies probe set were in very good agreement (see Fig. S6 in the supplemental material). These results underscore the utility of array-based synthesis of molecular probes for future metagenomic applications.
One of the unique advantages of the molecular probe technology is that molecular probe sets may be customized. For the vaginal swabs, we wanted to identify as many bacteria as possible, including those at low concentrations whose presence may be masked by the abundant Lactobacillus species. To that end, we constructed a deliberately reduced molecular probe set which lacked probes directed at Lactobacillus, i.e., the Lactobacillus-negative molecular probe set. This reduced probe set was reacted with eight vaginal-swab DNAs (those for which we had sufficient sample DNA remaining), and the results were compared to those obtained by employing the complete probe set. These comparisons are presented in Fig. 4. (The numbers of Illumina reads are presented in Table S3 in the supplemental material.) As expected, the Lactobacillus-negative probe set did not identify any of the Lactobacillus species that were detected by the complete probe set (Fig. 3 and 4). This observation further underscores the specificity of the molecular probe assay. For three swabs (030-3, 049-1, and 073-1), no additional bacteria were detected, although, of course, the relative numbers of sequence reads supporting each bacterium were different. As an example, for 030-3, E. coli went from 3.2% of the sequence reads (full probe set) to 100% of the sequence reads (Lactobacillus-negative probe set) (Fig. 4; also, see Table S3 in the supplemental material). For the other five vaginal swabs (049-2, 053-1, 079-3, 091-2, and 097-2), the Lactobacillus-negative probe set detected bacteria not found with the full probe set. As one example, for swab 049-2, B. longum and G. vaginalis were not detected by the full molecular probe set but were supported by 95.5% and 4.5% of the sequence reads, respectively, by the Lactobacillus-negative probe set (Fig. 4; also, see Table S3 in the supplemental material). As a second example, for swab 091-2, Atopobium was found by the Lactobacillus-negative molecular probe set (15.4% of the sequence reads) but not by the full probe set (Fig. 4; also, see Table S3 in the supplemental material). These examples emphasize the ability and usefulness of the molecular probe technology to target subsets of bacteria.
FIG 4.
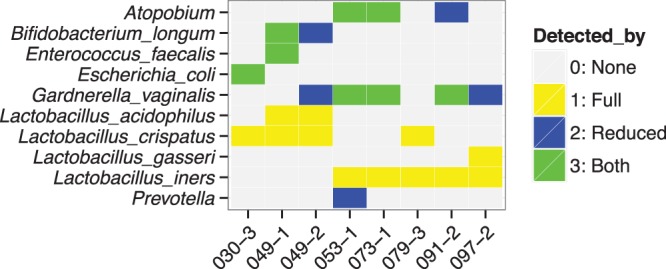
Comparison of the bacteria detected by the full SGTC molecular probe set with the bacteria detected by the Lactobacillus-negative probe set. The Lactobacillus-negative probe set contained none of the Lactobacillus probes. The ordinate lists only those bacteria found by either probe set. We had sufficient remaining DNA for eight comparisons. The abscissa lists the eight individual vaginal swabs. The final readout was by the Illumina MiSeq. Yellow signifies a bacterium detected only by the full probe set. Blue signifies a bacterium detected only by the Lactobacillus-negative probe set. Green signifies a bacterium detected by both probe sets. The absence of color signifies that that bacterium was not detected by either assay.
DISCUSSION
The nature of the molecular probe assay is to use probes directed against many microbial genomes, of which only a fraction are expected to be present (and thus detected) in a typical sample. Even though the Tag4 array-based readout has proven robust (12, 13), only a small percentage of the array features are positive in a typical sample. Thus, the molecular probe assay does not make efficient use of the array. In contrast, Illumina sequencing reads reflect only the bacteria present in the sample. This fact, coupled with the ability to multiplex many samples in one sequencing run, makes next-generation sequencing an extremely cost-effective readout method for the molecular probe assay.
In the current study, we interrogated the 24 clinical samples in Fig. 3 in a single MiSeq experiment. To determine the feasibility of additional multiplexing, we combined the data for all of the clinical samples as a test set and repeated the determination of bacterial presence or absence as a function of the depth of sequencing, from 100% down to 0.01% of the original. As presented in Fig. 5, increasing the level of multiplexing 100-fold caused only an ∼15% drop in the number of times bacteria were detected. This observation translates into detecting the most abundant bacteria in a multiplex of 2,400 samples in one MiSeq experiment. In practical terms, a 3-fold multiplex increases the total number of samples to 72, while sacrificing the detection of only one bacterium (Fig. 5, inset). Of course, the number of samples that can be multiplexed depends strongly on the composition of each sample to be included and thus should be considered carefully when an experiment is being designed.
FIG 5.

Percentage of targets detected at various sequencing depths. The figure shows the percentage of microbes and molecular probes detected in all clinical samples combined when sequencing reads are down-sampled to 0.01 to 100% of the original. The inset shows the same information with a denser sampling, to 10 to 100% of the original.
The presence or absence of G. vaginalis is an important determinant of the state of health of the human vagina. Thus, it is necessary that the method selected to identify the bacteria of the vaginal microbiome be able to detect G. vaginalis. Unfortunately, the “universal” amplification primers commonly employed to carry out PCR on the entire 16S rRNA gene have mismatches to G. vaginalis rRNA genes (6, 7, 26). Since amplification primers work in pairs, we employed the NCBI Primer-BLAST software to query the nonredundant GenBank database (which includes rRNA gene sequences). The software did not identify G. vaginalis sequences. We also did a BLAST search for each primer against three complete G. vaginalis genome sequences in GenBank (GI:311113888, GI:283782520, and GI:385800986). While the two reverse primers (conventionally called 1492R) had a perfect match, the two forward primers (conventionally called 8F) had no perfect match to any of the three G. vaginalis genome sequences. Ahmed et al. (27) compared the genome sequences of 17 G. vaginalis clinical isolates, most of which they sequenced themselves. They found that there were four nonrecombining groups of G. vaginalis genomes and proposed that it “may be appropriate to treat these four groups as separate species.” Ahmed et al. (27) determined that there are 746 genes in the core G. vaginalis genome and that these core genes make up only ∼52% of the genome sequences, on average. Therefore, going forward, we would derive Homers for each of the four groups separately based upon sequence differences. In addition, since we do not include rRNA gene sequences in our Homer search, we could easily distinguish N. meningitidis from N. gonorrhoeae even when the N. meningitidis isolate expressed N. gonorrhoeae rRNA genes (28).
Our molecular probe technology may be compared to other contemporary or proposed methods for identifying bacteria. Importantly, probe technology does not require growth of the bacteria. Therefore, probe technology can detect and identify bacteria that do not grow in culture. However useful, phenotypic growth methods of bacterial identification, such as the Verigene Gram-positive blood culture assay (for example, see references 29 and 30), are not comparable because they require growth in culture, as does the ingenious intrinsic fluorescence method (31).
A nonexhaustive comparison to methods that may not require growth in culture follows. (i) Matrix-assisted laser desorption ionization mass spectrometry (MALDI MS) and matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS) could, in principle, be employed to identify bacteria (for example, see reference 32). To identify the bacterium, the mass-to-charge pattern achieved with either instrument requires comparison to the pattern of known bacteria, for which an extensive database is currently nonexistent. In addition, it is unclear how many different bacteria in a sample at how many different concentrations MALDI MS or MALDI-TOF MS could detect (for example, see reference 33). Further evaluation must await a more complete database. (ii) A quaternized magnetic nanoparticle-fluorescent polymer system has been proposed for identification of bacteria (34). At present, it is also unclear how many different bacteria in a sample at how many different concentrations this method can detect. As with MALDI-TOF MS, further evaluation must await a more complete database. (iii) The rapid drop in cost is making the whole-genome shotgun sequencing approach more and more feasible for metagenomic analyses. Nevertheless, it still costs much more than a targeted approach for microbial profiling. Furthermore, the usefulness of this method depends, in large part, upon the public availability of new, more robust bioinformatic tools to sort genome sequences when many similar bacteria are present and at very different concentrations (for example, see reference 35). (iv) In theory, real-time PCR (RT-PCR) is promising as a fast and inexpensive technology to diagnose infectious agents. However, successful applications of RT-PCR have concentrated on only three bacteria (36), five bacteria (37), or six pathogens (38) and, recently, a 10-species biofilm model (39). It seems unlikely that fluorescence resonance energy transfer coupled with the melting temperatures of the amplicons will succeed in highly multiplexed identification and detection assays. (v) Another technology to identify bacteria is the PhyloChip (for example, see references 10, 40, 41, and 42). The PhyloChip is a microarray upon which there are oligonucleotides whose sequences are from the sequences of rRNA genes. However useful, the PhyloChip suffers from two serious problems. The first problem is that bacterial rRNA genes have very little sequence diversity. In some cases, the PhyloChip cannot distinguish genera, let alone species. The second problem is that multiple primer pairs need to be employed. That introduces the issue of differing PCR efficiencies.
Our goal has been to devise a technology that detects and identifies bacteria that do and do not grow in culture and where many different bacteria may be present at very different concentrations. Molecular probe technology meets that goal. In addition, the molecular probes need not target only bacterial DNAs. Presumably, probes could be designed to detect any given genome or even individual genes, such as toxin genes. The molecular probe technology has several advantages over rRNA gene sequencing, such as detection of organisms with poor homology to the “universal” sequencing primers, better discrimination between species, and the ability to omit from the analysis highly abundant organisms that may mask signal. Indeed, the dominance of certain DNAs greatly complicates rRNA gene-based characterization of microbial diversity in soil (43). Dramatic increases in publically available genome sequences, coupled with dramatic decreases in the costs of oligonucleotide synthesis and DNA sequencing, now make molecular probe technology an attractive option for metagenomic analyses of complex samples.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a grant from the National Human Genome Research Institute (HG000205) to R.W.D.
R.W.H. and R.P.S. conceived the experiments. C.P. adapted the Homer-calling software to expand the Homers from 40 to 60 bases. M.F. called the Homers. E.F. put the molecular probe sequences together. M.A.J. synthesized the 116-mers at the SGTC. R.W.H. carried out the molecular probe reactions. M.M. and R.P.S. performed and analyzed the Tag4 assays. M.M. and S.K. undertook the Illumina sequencing. W.X. undertook the statistical analyses of the data. R.W.H., R.P.S., S.K., and W.X. wrote the manuscript. R.W.D. provided the financial, physical, and intellectual milieu for these experiments.
R.W.D. is a coholder of the patent on molecular inversion probes. We report no conflicts of interest.
Footnotes
Published ahead of print 2 May 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00666-14.
REFERENCES
- 1.Rappé MS, Giovannoni SJ. 2003. The uncultured microbial majority. Annu. Rev. Microbiol. 57:369–394. 10.1146/annurev.micro.57.030502.090759 [DOI] [PubMed] [Google Scholar]
- 2.Human Microbiome Project Consortium. 2012. Structure, function and diversity of the healthy human microbiome. Nature 486:207–214. 10.1038/nature11234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Farrelly V, Rainey FA, Stackebrandt E. 1995. Effect of genome size and rrn gene copy number on PCR amplification of 16S rRNA genes from a mixture of bacterial species. Appl. Environ. Microbiol. 61:2798–2801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klappenbach JA, Dunbar JM, Schmidt TM. 2000. rRNA operon copy number reflects ecological strategies of bacteria. Appl. Environ. Microbiol. 66:1328–1333. 10.1128/AEM.66.4.1328-1333.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Acinas SG, Marcelino LA, Klepac-Ceraj V, Polz MF. 2004. Divergence and redundancy of 16S rRNA sequences in genomes with multiple rrn operons. J. Bacteriol. 186:2629–2635. 10.1128/JB.186.9.2629-2635.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baker GC, Smith JJ, Cowan DA. 2003. Review and re-analysis of domain specific 16S primers. J. Microbiol. Methods 55:541–555. 10.1016/j.mimet.2003.08.009 [DOI] [PubMed] [Google Scholar]
- 7.Frank JA, Reich CI, Sharma S, Weisbaum JS, Wilson BA, Olsen GJ. 2008. Critical evaluation of two primers commonly used for amplification of bacterial 16S rRNA genes. Appl. Environ. Microbiol. 74:2461–2470. 10.1128/AEM.02272-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jumpstart Consortium Human Microbiome Project Data Generation Working Group. 2012. Evaluation of 16S rDNA-based community profiling for human microbiome research. PLoS One 7:e39315. 10.1371/journal.pone.0039315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haas BJ, Gevers D, Earl AM, Feldgarden M, Ward DV, Giannoukos G, Ciulla D, Tabbaa D, Highlander SK, Sodergren E, Methé B, DeSantis TZ, Human Microbiome Consortium. Petrosino JF, Knight R, Birren BW. 2011. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 21:494–504. 10.1101/gr.112730.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brodie EL, Desantis TZ, Joyner DC, Baek SM, Larsen JT, Andersen GL, Hazen TC, Richardson PM, Herman DJ, Tokunaga TK, Wan JM, Firestone MK. 2006. Application of a high-density oligonucleotide microarray approach to study bacterial population dynamics during uranium reduction and reoxidation. Appl. Environ. Microbiol. 72:6288–6298. 10.1128/AEM.00246-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hardenbol P, Yu F, Belmont J, Mackenzie J, Bruckner C, Brundage T, Boudreau A, Chow S, Eberle J, Erbilgin A, Falkowski M, Fitzgerald R, Ghose S, Iartchouk O, Jain M, Karlin-Neumann G, Lu X, Miao X, Moore B, Moorhead M, Namsaraev E, Pasternak S, Prakash E, Tran K, Wang Z, Jones HB, Davis RW, Willis TD, Gibbs RA. 2005. Highly multiplexed molecular inversion probe genotyping: over 10,000 targeted SNPs genotyped in a single tube assay. Genome Res. 15:269–275. 10.1101/gr.3185605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hyman RW, St. Onge RP, Allen EA, Miranda M, Aparicio AM, Fukushima M, Davis RW. 2010. Multiplex identification of microbes. Appl. Environ. Microbiol. 76:3904–3910. 10.1128/AEM.02785-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hyman RW, St. Onge RP, Kim H, Tamaresis JS, Miranda M, Aparicio AM, Fukushima M, Pourmand N, Giudice LC, Davis RW. 2012. Molecular probe technology detects bacteria without culture. BMC Microbiol. 12:29. 10.1186/1471-2180-12-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hyman RW, Fukushima M, Jiang H, Fung E, Rand L, Johnson B, Vo KC, Caughey AB, Hilton JF, Davis RW, Giudice LC. 2014. Diversity of the vaginal microbiome correlates with preterm birth. Reprod. Sci. 21:32–40. 10.1177/1933719113488838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baner J, Marits P, Nilsson M, Winqvist O, Landegren U. 2005. Analysis of T-cell receptor V beta gene repertoires after immune stimulation and in malignancy by use of padlock probes and microarrays. Clin. Chem. 51:768–775. 10.1373/clinchem.2004.047266 [DOI] [PubMed] [Google Scholar]
- 16.Pierce SE, Fung EL, Jaramillo DF, Chu AM, Davis RW, Nislow C, Giaever G. 2006. A unique and universal molecular barcode array. Nat. Methods 3:601–603. 10.1038/nmeth905 [DOI] [PubMed] [Google Scholar]
- 17.Zuker M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31:3406–3415. 10.1093/nar/gkg595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jensen MA, Akhras MS, Fukushima M, Pourmand N, Davis RW. 2013. Direct oligonucleotide synthesis onto super-paramagnetic beads. J. Biotechnol. 167:448–453. 10.1016/j.jbiotec.2013.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang C, Krishnakumar S, Wilhelmy J, Babrzadeh F, Stepanyan L, Su LF, Levinson D, Fernandez-Viña MA, Davis RW, Davis MM, Mindrinos M. 2012. High-throughput, high-fidelity HLA genotyping with deep sequencing. Proc. Natl. Acad. Sci. U. S. A. 109:8676–8681. 10.1073/pnas.1206614109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Affymetrix, Inc. 2005. Exon array background correction. Affymetrix, Inc., Santa Clara, CA [Google Scholar]
- 22.Robinson MD, Smyth GK. 2008. Small-sample estimation of negative binomial dispersion, with applications to SAGE data. Biostatistics 9:321–332. 10.1093/biostatistics/kxm030 [DOI] [PubMed] [Google Scholar]
- 23.Holm S. 1979. A simple sequentially rejective multiple test procedure. Scand. J. Stat. 6:65–70 [Google Scholar]
- 24.Wickham H. 2009. Ggplot2: elegant graphics for data analysis. Springer, New York, NY [Google Scholar]
- 25.Fu GK, Xu W, Wilhelmy J, Mindrinos MN, Davis RW, Xiao W, Fodor SP. 2014. Molecular indexing enables quantitative targeted RNA sequencing and reveals poor efficiencies in standard library preparations. Proc. Natl. Acad. Sci. U. S. A. 111:1891–1896. 10.1073/pnas.1323732111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Verhelst R, Verstraelen H, Claeys G, Verschraegen G, Delanghe J, Van Simaey L, De Ganck C, Temmerman M, Vaneechoutte M. 2004. Cloning of 16S rRNA genes amplified from normal and disturbed vaginal microflora suggests a strong association between Atopobium vaginae, Gardnerella vaginalis and bacterial vaginosis. BMC Microbiol. 4:16. 10.1186/1471-2180-4-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ahmed A, Earl J, Retchless A, Hillier SL, Rabe LK, Cherpes TL, Powell E, Janto B, Eutsey R, Hiller NL, Boissy R, Dahlgren ME, Hall BG, Costerton JW, Post JC, Hu FZ, Ehrlich GD. 2012. Comparative genomic analyses of 17 clinical isolates of Gardnerella vaginalis provide evidence of multiple genetically isolated clades consistent with subspeciation into genovars. J. Bacteriol. 194:3922–3937. 10.1128/JB.00056-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walcher M, Skvoretz R, Montgomery-Fullerton M, Jonas V, Brentano S. 2013. Description of an unusual Neisseria meningitidis isolate containing and expressing Neisseria gonorrhoeae-specific 16S rRNA gene sequences. J. Clin. Microbiol. 51:3199–3206. 10.1128/JCM.00309-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sullivan KV, Turner NN, Roundtree SS, Young S, Brock-Haag CA, Lacey D, Abuzaid S, Blecker-Shelly DL, Doern CD. 2013. Rapid detection of Gram-positive organisms by use of the Verigene gram-positive blood culture nucleic acid test and the BacT/Alert pediatric FAN system in a multicenter pediatric evaluation. J. Clin. Microbiol. 51:3579–3584. 10.1128/JCM.01224-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mestas J, Polanco CM, Felsenstein S, Dien Bard J. 2014. Performance of the Verigene Gram-positive blood culture assay for direct detection of Gram-positive organisms and resistance markers in a pediatric hospital. J. Clin. Microbiol. 52:283–287. 10.1128/JCM.02322-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walsh JD, Hyman JM, Borzhemskaya L, Bowen A, McKellar C, Ullery M, Mathias E, Ronsick C, Link J, Wilson M, Clay B, Robinson R, Thorpe T, van Belkum A, Dunne WM., Jr 2013. Rapid intrinsic fluorescence method for direct identification of pathogens in blood cultures. mBio 4:e00865–13. 10.1128/mBio.00865-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sandrin TR, Demirev PA. 2014. Using mass spectrometry to identify and characterize bacteria. Microbe 9:23–29 [Google Scholar]
- 33.Xiao D, Zhang H, He L, Peng X, Wang Y, Xue G, Su P, Zhang J. 2013. High natural variability bacteria identification and typing: Helicobacter pylori analysis based on peptide mass fingerprinting. J. Proteomics 98C:112–122. 10.1016/j.jprot.2013.11.021 [DOI] [PubMed] [Google Scholar]
- 34.Wan Y, Sun Y, Qi P, Wang P, Zhang D. 2013. Quaternized magnetic nanoparticles-fluorescent polymer system for detection and identification of bacteria. Biosens. Bioelectron. 55C:289–293. 10.1016/j.bios.2013.11.080 [DOI] [PubMed] [Google Scholar]
- 35.Hasman H, Saputra D, Sicheritz-Ponten T, Lund O, Svendsen CA, Frimodt-Møller N, Aarestrup FM. 2014. Rapid whole-genome sequencing for detection and characterization of microorganisms directly from clinical samples. J. Clin. Microbiol. 52:139–146. 10.1128/JCM.02452-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barletta F, Mercado EH, Lluque A, Ruiz J, Cleary TG, Ochoa TJ. 2013. Multiplex real-time PCR for detection of Campylobacter, Salmonella, and Shigella. J. Clin. Microbiol. 51:2822–2829. 10.1128/JCM.01397-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Buchan BW, Olson WJ, Pezewski M, Marcon MJ, Novicki T, Uphoff TS, Chandramohan L, Revell P, Ledeboer NA. 2013. Clinical evaluation of a real-time PCR assay for identification of Salmonella, Shigella, Campylobacter (Campylobacter jejuni and C. coli), and Shiga toxin-producing Escherichia coli isolates in stool specimens. J. Clin. Microbiol. 51:4001–4007. 10.1128/JCM.02056-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koo C, Malapi-Wight M, Kim HS, Cifci OS, Vaughn-Diaz VL, Ma B, Kim S, Abdel-Raziq H, Ong K, Jo YK, Gross DC, Shim WB, Han A. 2013. Development of a real-time microchip PCR system for portable plant disease diagnosis. PLoS One 8:e82704. 10.1371/journal.pone.0082704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ammann TW, Bostanci N, Belibasakis GN, Thurnheer T. 2013. Validation of a quantitative real-time PCR assay and comparison with fluorescence microscopy and selective agar plate counting for species-specific quantification of an in vitro subgingival biofilm model. J. Periodontal Res. 48:517–526. 10.1111/jre.12034 [DOI] [PubMed] [Google Scholar]
- 40.Liu WT, Mirzabekov AD, Stahl DA. 2001. Optimization of an oligonucleotide microchip for microbial identification studies: a non-equilibrium dissociation approach. Appl. Environ. Microbiol. 3:619–629. 10.1046/j.1462-2920.2001.00233.x [DOI] [PubMed] [Google Scholar]
- 41.Small J, Call DR, Brockman FJ, Straub TM, Chandler DP. 2001. Direct detection of 16S rRNA in soil extracts by using oligonucleotide microarrays. Appl. Environ. Microbiol. 67:4708–4716. 10.1128/AEM.67.10.4708-4716.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Loy A, Lehner A, Lee N, Adamczyk J, Meier H, Ernst J, Schleifer K-H, Wagner M. 2002. Oligonucleotide microarray for 16S rRNA gene-based detection of all recognized lineages of sulfate-reducing prokaryotes in the environment. Appl. Environ. Microbiol. 68:5064–5081. 10.1128/AEM.68.10.5064-5081.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Delmont TO, Robe T, Cecillon S, Clark IM, Constancias F, Simonet P, Hirsch PR, Vogel TM. 2011. Accessing the soil metagenome for studies of microbial diversity. Appl. Environ. Microbiol. 77:1315–1324. 10.1128/AEM.01526-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.