

Abstract
Magnetotactic bacteria have emerged as excellent model systems to study bacterial cell biology, biomineralization, vesicle formation, and protein targeting because of their ability to synthesize single-domain magnetite crystals within unique organelles (magnetosomes). However, only few species are amenable to genetic manipulation, and the limited methods for site-specific mutagenesis are tedious and time-consuming. Here, we report the adaptation and application of a fast and convenient technique for markerless chromosomal manipulation of Magnetospirillum gryphiswaldense using a single antibiotic resistance cassette and galK-based counterselection for marker recycling. We demonstrate the potential of this technique by genomic excision of the phbCAB operon, encoding enzymes for polyhydroxyalkanoate (PHA) synthesis, followed by chromosomal fusion of magnetosome-associated proteins to fluorescent proteins. Because of the absence of interfering PHA particles, these engineered strains are particularly suitable for microscopic analyses of cell biology and magnetosome biosynthesis.
INTRODUCTION
Magnetotactic bacteria (MTB) are exceptional in their ability to synthesize unique organelles (magnetosomes) that consist of membrane-enveloped, nanometer-sized, single-domain magnetite crystals. Magnetosomes are associated with a specific set of proteins (1) and are attached to a filamentous cytoskeletal structure (2, 3), which enables them to assemble into a cohesive chain positioned at midcell (4). Therefore, MTB have emerged as excellent model organisms to study the biogenesis of bacterial organelles, biomineralization, protein targeting, and bacterial cell biology. In addition, magnetosomes have been genetically engineered with respect to both their magnetite core as well their enveloping membrane, and numerous applications of functionalized magnetosomes have been demonstrated (5–8). However, the progress in exploring the biology of MTB and in engineering magnetosomes has been impeded by the limited genetic tools available for these fastidious bacteria.
To date, genetic systems are available for only two Magnetospirillum species from the alphaproteobacteria, but their genetic manipulation has remained rather inefficient, laborious, and time-consuming (2, 9). In Magnetospirillum gryphiswaldense (MSR-1), for example, the genome has been routinely mutated by Cre-lox recombination (10–12), which relies on the integration of 34-bp loxP sequences directly up- and downstream of the genomic target by one double homologous (thereby replacing the target DNA by a resistance marker) or two single homologous recombination events. Because of the low frequency of double recombination in MSR-1, two distinct integrating vectors have been used, each one providing a specific resistance marker for clonal selection. After integration of both loxP-carrying vectors, Cre recombinase becomes expressed from a third, nonintegrating plasmid. The recombinase specifically recognizes the integrated loxP sites, and when their sequences are parallel, enclosed nucleotides become excised. Since one loxP sequence remains after excision, the system had to be advanced for repeated deletions in the same host (13). This advanced system has proven useful to delete single genes, entire operons, and even larger genomic loci in MSR-1 (14).
However, the Cre-lox technology exhibits several practical disadvantages. First, two different vectors for genome integration need to be constructed. To positively select for double integration, two antibiotics have to be applied, which impedes cell growth. Second, three consecutive cycles of transformation, each accompanied by clonal selection and screening, are necessary. These procedures are particularly time-consuming for slow-growing magnetospirilla. Third, lox nucleotides remain in the genomic target region and complicate the design of in-frame deletion vectors. More importantly, these scar sequences render the introduction of targeted single-base exchanges nearly impossible.
An alternative technique to manipulate bacterial genomes relies on RecA-mediated chromosomal integration and excision of a nonreplicating vector that carries the mutated allele, an antibiotic resistance cassette for positive selection, and a conditionally lethal gene as essential components for counterselection.
In magnetospirilla, counterselection can be mediated by SacB, which confers sensitivity to sucrose (15, 16). This selection marker is commonly used to mutate Magnetospirillum magneticum AMB-1 (17–20), but it has been applied to MSR-1 in only a few cases (21). The reason for this is that in our hands, sacB counterselection has proven not to be reliable, likely because of rapid spontaneous gene inactivation upon selective pressure leading to numerous false-positive colonies on counterselective plates (11; E. Katzmann, unpublished data), requiring laborious and cumbersome replica platings.
Thus, to enhance targeted mutagenesis techniques for MSR-1 and potentially other magnetospirilla, we tested alternative conditional marker genes, and as one promising candidate, we analyzed the galactokinase-encoding gene galK. Galactokinase confers sensitivity to galactose or 2-deoxygalactose in the absence of a galactose-metabolizing pathway (22, 23) and is utilized for counterselection in several Gram-positive and Gram-negative bacteria (24–26).
In our study, we found that GalK represents a reliable and robust marker for counterselection in MSR-1. Using galK, we constructed a universal vector for efficient and markerless genome manipulation. To prove its practical use, we abolished synthesis of intracellular polyhydroxyalkanoate (PHA) inclusions by deletion of the putative phbCAB operon in MSR-1, which resulted in cells with reduced autofluorescence and diminished distortion of magnetosome chains. We used this technique further for native-site genomic in-frame fluorescent tagging of magnetosome key proteins in the wild type (wt) and in the phbCAB mutant, and we found the fusion proteins to be functional. In summary, we developed an efficient and powerful tool for genome manipulation of MSR-1 and generated strains particularly suitable for analysis of subcellular structures by light and electron microscopy.
MATERIALS AND METHODS
Bacterial strains, vectors, and culture conditions.
Bacterial strains and vectors are listed in Table 1. Escherichia coli strains were cultivated in lysogeny broth (LB) medium as described previously (27). Kanamycin was added to 25 μg/ml, and 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) was added to 40 μg/ml when necessary. E. coli BW29427 cultures (K. A. Datsenko and B. L. Wanner, unpublished data) were supplemented with 1 mM dl-α,ε-diaminopimelic acid (DAP). M. gryphiswaldense cultures were grown microaerobically in modified flask standard medium (FSM) at 30°C (28) with agitation at 120 rpm, unless otherwise stated. When appropriate, kanamycin was added to 5 μg/ml; galactose was added to 0.5, 1, 2.5, or 5% (wt/vol); and anhydrotetracycline was added to 100 ng/ml after autoclaving. Media were solidified by the addition of 1.5% (wt/vol) agar. The optical density and magnetic response (Cmag) of exponentially growing MSR-1 cultures were measured photometrically at 565 nm, as reported previously (29).
TABLE 1.
Bacterial strains and vectors
| Strain or vector | Application and/or characteristic(s) | Reference and/or source |
|---|---|---|
| Strains | ||
| E. coli | ||
| DH5α | Host for cloning; fhuA2 Δ(argF-lacZ)U169 phoA glnV44 ϕ80 Δ(lacZ)M15 gyrA96 recA1 relA1 endA1 thi-1 hsdR17 | 47 |
| BW29427 | Donor for conjugation; thrB1004 pro thi rpsL hsdSlacZ ΔM15 RP4-1360 Δ(araBAD)567 ΔdapA1341::[erm pir]tra | K. A. Datsenko and B. L. Wanner (Purdue University), unpublished |
| M. gryphiswaldense | ||
| MSR-1 R/S | wt | 21 |
| FM019 | mamC-mCherry | This work |
| FM021 | mamC-egfp | This work |
| FM022 | mCherry-mamK | This work |
| FM025 | ΔphbCAB | This work |
| FM046 | ΔphbCAB mamC-egfp | This work |
| FM047 | ΔphbCAB mamC-mCherry | This work |
| FM048 | ΔphbCAB mCherry-mamK | This work |
| Vectors | ||
| pK19 mob GII | Backbone for pORFM suicide vectors; npt mobRK2 pMB-1 replicon | 36; GenBank accession no. AF012346 |
| pJET 1.2/blunt | Cloning vector; bla | Thermo Scientific |
| pAP160 | Source of tetR | A. Pollithy, unpublished |
| pAP173 | Source of Ptet and terminator sequences | A. Pollithy, unpublished |
| pOR014 | Construction vector for pORFM GalK; npt terminator mobRK2 | This work |
| pOR025a | Intermediate for pORFM GalK construction; ter npt mobRK2 tetR | This work |
| pORFM GalK | General backbone vector for GalK counterselection; npt galK tetR mobRK2 | This work |
| pORFM blu | General backbone vector for GalK counterselection, blue-white screening; lacZα npt galK tetR mobRK2 | This work |
| pFM234 | phbCAB deletion; npt galK tetR mobRK2 | This work |
| pFM236 | mamC-egfp chromosomal fusion; npt galK tetR mobRK2 | This work |
| pFM237 | mamC-mCherry chromosomal fusion; npt galK tetR mobRK2 | This work |
| pFM245 | mCherry-mamK chromosomal fusion; npt gal, tet, mobRK2 | This work |
Correlation of optical density and cell counts.
To test whether the correlation of optical density (measured photometrically at 565 nm) and cell numbers per ml between the wt and the phbCAB mutant was identical, cultures of the wt and three mutant strains grown overnight were diluted to an optical density at 565 nm (OD565) of 0.1 and fixed with formaldehyde (1% final concentration). Samples of each strain were applied to a hemocytometer, and cells per chamber (n = 11) were enumerated. Mean values were calculated from the cell counts, and the mean value of wt cells was set to 100%.
Molecular and genetic techniques.
Plasmids (Table 1) were constructed by standard recombinant techniques. Oligonucleotides that were used as primers for PCRs are listed in Table S1 in the supplemental material. PCR-amplified DNA fragments for cloning were routinely sequenced with BigDye Terminator v3.1 chemistry on an ABI 3700 capillary sequencer (Applied Biosystems).
Construction of integrative vectors for markerless gene deletion and chromosomal fluorescent fusion.
To avoid transcriptional readthrough from upstream regions in pK19mobGII, the bacteriophage lambda T0 and the E. coli rrnB T1 transcription terminators were amplified from plasmid pAP173 (A. Pollithy, unpublished data) by using primer pair oOR059/oOR060 and cloned upstream of the pK19mobGII multiple-cloning site (MCS) after HindIII and PstI restriction, yielding pOR014. To ensure galK transcription in MSR-1, a tet promoter-galK fusion was generated. Therefore, the galK gene was amplified from E. coli K-12 genomic DNA by using primer pair oOR063/oOR077 and ligated downstream of the tet promoter in pAP173 after NdeI and BamHI restriction. This Ptet-galK cassette was intended to be cloned into pOR014. However, since no colonies grew after transformation in E. coli, the Ptet-galK fusion was amplified from the ligation reaction by using primer pair oOR082/oOR083 and cloned into pJET 1.2/blunt (Fermentas), but again, no colonies grew after transformation. Thus, the tet repressor gene (tetR) under the control of the neomycin promoter (Pneo-tetR) was amplified from pAP160 (A. Pollithy, unpublished) by using primer pair tetRfwSacI/tetRrevSacI and cloned into pOR014, yielding pOR025a. Subsequently, to construct pORFM GalK (see Fig. S1A in the supplemental material), the Ptet-galK PCR product was cloned into pOR025a by using MunI and Bsp119I restriction sites. To facilitate the design and cloning of homologous up- and downstream regions into pORFM GalK, the MCS of pORFM GalK was amended by a lacZα gene fragment containing EcoRV and SmaI/XmaI restriction sites, both suitable for blunt-end cloning, yielding pORFM blu (Fig. 1; see also Fig. S1B in the supplemental material).
FIG 1.
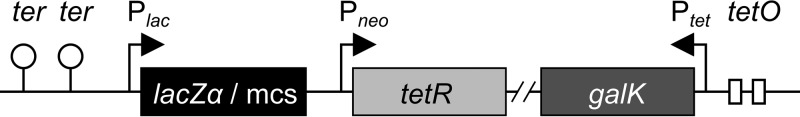
Vector design for galK-based counterselection in MSR-1 by pORFM blu. To repress galK expression under nonselective conditions, the gene is placed under the control of the tet promoter/operator (Ptet-tetO). The tet repressor (tetR) is constitutively expressed from a neomycin promoter (Pneo). Upon induction with anhydrotetracycline, the tet promoter becomes active, and galK expression increases. To enable blue-white screening in E. coli, the multiple-cloning site (mcs) was combined with a lacZα gene fragment.
For pFM234 construction, the ∼1.6-kb regions up- and downstream of the putative phbCAB operon were PCR amplified by using a proofreading DNA polymerase and primer pairs oFM341/oFM342 and oFM343/oFM344. PCR products were fused in a second PCR (30) using primer pair oFM341/oFM344, thereby generating the mutated allele where the phbCAB operon is replaced by a truncated open reading frame (ORF) consisting of 5′ phbC and 3′ phbB codons. This DNA fragment was blunt-end ligated into the EcoRV-digested pORFM blu vector and transformed into E. coli DH5α, and transformed cells were plated onto LB medium supplemented with kanamycin and X-Gal. White colonies were selected from the plates, and the presence of the cloned fragment was confirmed by colony PCR using primer pair oFM280b/oFM281b.
To generate fluorescent chromosomal fusions to MamC and MamK, monomeric DsRed in its variant mCherry and enhanced green fluorescent protein (EGFP; Clontech) were used. MamC was C-terminally (31) fused to mCherry and EGFP, and MamK was N-terminally (32) fused to mCherry.
pFM236 (for the mamC-egfp fusion) was generated essentially as described above but with pORFM GalK as the vector backbone and without blue-white screening of E. coli colonies. Briefly, the ∼1.6-kb regions up- and downstream of the mamC 3′ end were PCR amplified by using primer pairs oFM270/oFM271 and oFM274/oFM275, respectively. egfp was amplified with primer pair oFM272/oFM273. The upstream fragment was cloned into pORFM GalK after digestion with SalI and KpnI, followed by egfp after digestion with KpnI and EcoRI. The downstream fragment was cloned into the resulting vector after digestion with EcoRI and NheI.
For pFM237 (mamC-mCherry) construction, mCherry was PCR amplified with primer pair oFM276/oFM277. egfp was then cut out from pFM236 by KpnI and EcoRI digestion and replaced by mCherry.
To construct pFM245 (mCherry-mamK fusion), the ∼1.4-kb regions up- and downstream of the mamK start codon were PCR amplified by using primer pairs oFM369/oFM370 and oFM373/oFM374, and mCherry was amplified with primer pair oFM371/oFM372, including a spacer sequence. The upstream fragment and mCherry were fused by a second PCR using primer pair oFM369/oFM372. The fused fragment was cloned into pORFM GalK after digestion with SalI and BamHI. The downstream fragment was cloned behind this insert after restriction with BamHI and SpeI.
Conjugation experiments.
Plasmid transfer by biparental conjugation was performed with E. coli BW29427 as the donor strain and M. gryphiswaldense MSR-1 as the acceptor strain. The conjugation procedure was performed as described previously (9, 12).
Screening of MSR-1 insertion mutants.
Kanamycin-resistant colonies were transferred into 100 μl FSM in 96-well plates and grown microaerobically overnight. The cultures were screened for up- or downstream integration of the vector by PCR (see Fig. S3 in the supplemental material) using a vector-specific oligonucleotide primer (oFM280a or -281 for pORFM GalK derivatives and oFM280b or -281b for pORFM blu derivatives) and one primer specific for a sequence adjacent to one homologous region (verification primer) (see Table S1 in the supplemental material). If possible, at least one insertion mutant strain with either up- or downstream integration was used for counterselection.
Galactose counterselection of insertion mutants.
PCR-verified insertion mutants were transferred into 1 ml FSM in 24-well plates and grown overnight. Two hundred microliters of the culture grown overnight was plated onto FSM containing 0.5% (wt/vol) galactose and 100 ng/ml anhydrotetracycline. Plates were incubated at 30°C under microaerobic conditions for 5 days, as described previously (33).
Screen for in-frame deletion and fusion.
To discriminate between reconstituted wt and mutated genotypes, colonies were transferred from counterselective plates into 100 μl FSM and incubated microaerobically overnight in 96-well plates. The genotype was determined by PCR using oligonucleotide primers specific to sequences adjacent to the cloned homologous regions (verification primers) (see Table S1 and Fig. S4 in the supplemental material). Loss of the vector was further confirmed by reinoculating mutant strains into FSM with kanamycin, where no growth was observed, and in medium with galactose, where growth occurred.
Fluorescence microscopy.
M. gryphiswaldense strains were grown in 15-ml polypropylene tubes with sealed screw caps and a culture volume of 11 ml to early log phase. To image fluorescent proteins, 10-μl samples were directly immobilized on 1% (wt/vol) agarose pads and covered with a coverslip. For Nile red staining, 1-ml samples were withdrawn, and 1 μl 0.5 mg/ml Nile red (in dimethyl sulfoxide [DMSO]) was added. Cells were incubated for 5 min, harvested by centrifugation, and washed with phosphate-buffered saline (PBS) before immobilization on agarose. The samples were imaged with an Olympus BX81 microscope equipped with a 100× UPLSAPO100XO objective and an Orca-ER camera (Hamamatsu).
Transmission electron microscopy.
For transmission electron microscopy (TEM) analysis, cells were grown at 25°C under microaerobic conditions to an OD565 of 0.1, fixed in formaldehyde (1%), concentrated, adsorbed onto carbon-coated copper mesh grids, and washed three times with particle-free water. Samples were viewed and recorded with a Morgagni 268 microscope (FEI, Eindhoven, the Netherlands) at an 80-kV accelerating tension.
Cryo-electron tomography.
Cryo-electron tomography (CET) was performed on logarithmic MSR-1 cultures embedded in vitreous ice by plunge freezing into liquid ethane, as described previously (34).
Image acquisition and processing.
Fluorescence images were recorded and processed (brightness and contrast adjustments) by using Olympus Xcellence software, TEM images were acquired with the iTEM software program (5.0), and CET tilt series were recorded with Serial EM and FEI software. Three-dimensional (3D) reconstructions of the tomograms were performed with the weighted back-projection method using TOMtoolbox (35) and visualized with Amira 3D image processing software. Images were assembled with the GNU Image Manipulation Program (GIMP 2.8), and graphics were drawn by using Inkscape (0.48) software.
RESULTS AND DISCUSSION
Generation of a universal GalK-based counterselection vector for MSR-1.
To investigate whether GalK may be a suitable counterselection marker, we first verified the absence of a potential galactose utilization pathway from the MSR-1 genome. To preclude adverse effects of increased galactose concentrations on MSR-1, we next tested growth on medium supplemented with 0, 0.5, 1, 2.5, and 5.0% (wt/vol) galactose. Similar numbers of colonies emerged under all conditions, although colonies on plates with 5.0% galactose were somewhat smaller, indicating a slight growth impairment at this high concentration (data not shown).
Based on these results, we pursued construction of a galK-containing suicide plasmid, and we selected the mobilizable broad-host-range vector pK19mobGII (conferring kanamycin resistance) (36) as the backbone. To introduce galK, we first amplified the gene from E. coli K-12 and cloned it under the control of the tet promoter (Ptet), which is constitutively active in MSR-1 and of intermediate strength (37). However, we failed to obtain E. coli colonies, suggesting that constitutive (over)expression of galK in E. coli was lethal. To prevent this effect, we first cloned the tetracycline repressor gene (tetR) into pK19mobGII and repeated the insertion of the Ptet-galK construct (see Materials and Methods for details). This yielded the counterselective vector with tetracycline-inducible galK expression, designated pORFM GalK.
In addition, to facilitate direct blunt-end cloning of PCR-amplified genomic sections for homologous recombination, we reintroduced a multiple-cloning site into the plasmid and combined it with a lacZα gene fragment as a chromogenic marker for blue-white screening in E. coli, as outlined in Fig. 1. We designated this vector pORFM blu.
Deletion of the phbCAB operon eliminates PHA granules.
In MSR-1 growing on standard FSM, large parts of the intracellular volume are frequently occupied by PHA granules. These inclusions tend to distort magnetosome chains (Fig. 2A and B) and interfere with fluorescence microscopy by autofluorescence or adsorption of lipophilic membrane stains (Fig. 2A and C, insets). To prove the function of pORFM blu and to generate a strain with enhanced properties for light and electron microscopy, we intended to abolish PHA granule formation by the deletion of genes essential for PHA synthesis. Inspection of the MSR-1 genome revealed a set of three genes (Mgr_4240 to Mgr_4242), encoding a putative PHA polymerase, an acetyl coenzyme A (acetyl-CoA) acetyltransferase, and an acetyl-CoA reductase (organized in a presumed phbCAB operon), as the most promising target for deletion. We constructed the deletion vector as diagrammed in Fig. S2 in the supplemental material and transferred it into MSR-1 by biparental conjugation. Eight of the kanamycin-resistant colonies were screened for vector insertion up- or downstream of the phbCAB operon by PCR. All strains contained a downstream insertion, suggesting that vector integration upstream of the phbCAB genes was lethal. Three of the downstream insertion mutants were processed further and transferred onto FSM plates supplemented with 0.5, 1, or 2.5% (wt/vol) galactose and 100 ng/ml anhydrotetracycline for counterselection. The numbers of colonies on all plates were similar, suggesting that the lowest galactose concentration of 0.5% was entirely sufficient to suppress growth of cells which did not recombine. We therefore set 0.5% (wt/vol) galactose as the default concentration and obtained 29 colonies from these counterselective plates. A PCR screen suggested that 15 of them converted back to the wt, whereas 13 contained the desired deletion and 1 was inconclusive (see Fig. S4 in the supplemental material). This result indicated an approximate 1:1 ratio between deletion and reconstitution, as expected for an unbiased loop-out of the plasmid.
FIG 2.

The phbCAB mutant is devoid of PHA granules but forms wt-like crystals, magnetosome chains, and polyphosphate inclusions. (A) TEM image of typical wt cells containing multiple PHA granules (indicated by white arrows) and attached polyphosphate inclusions (black arrows) beside a magnetosome chain (series of crystals). (Top insets) Differential interference contrast (DIC) and fluorescence images of Nile red-stained wt cells. PHA granules appear as three-dimensional globules by differential interference contrast and as brightly stained dots under fluorescence illumination. (Bottom right inset) Close-up view of a magnetosome chain entrapped by a PHA granule (white arrow, with the boundary marked by a dotted line) and a polyphosphate inclusion (black arrow). (Bottom left inset) Dividing wt cell with incipient division septum and buckling magnetosome chain, which is displaced by PHA granules. (B) Section of a segmented cryo-electron tomogram from a wt cell. PHA and polyphosphate inclusions are marked with white and black arrows, respectively. Magnetite crystals are depicted in red, magnetosome membranes are yellow, magnetosome filaments are in green, and the cell membrane is shown in blue. (C) TEM image of phbCAB mutant cells. Note the absence of PHA granules and the preserved regular spacing of polyphosphate inclusions (black arrows). (Top insets) Differential interference contrast and fluorescence images of Nile red-stained ΔphbCAB cells. The cells appear smooth by differential interference contrast. The fluorescence image suggests membrane-specific staining in the absence of PHA granules. (Bottom right inset) wt-like magnetite crystals of the phbCAB mutant. (Bottom left inset) Dividing cell with a characteristically buckling magnetosome chain opposite the asymmetrically inward growing division septum. (D) Segmented tomogram of a phbCAB mutant cell. The absence of PHA inclusions facilitates reconstruction of intracellular structures.
Nile red staining (38) and fluorescence microscopy revealed the absence of PHA inclusions in all 13 putative deletion mutants, suggesting that the cells had become deficient in PHA granule formation. Transmission electron microscopy (TEM) and cryo-electron tomography (CET) corroborated this observation and further revealed wt-like magnetosome chains and crystals (Fig. 2). Consistently, the phbCAB mutant strains exhibited a wt-like magnetic response, although cultivation experiments suggested a slight growth impairment of the mutants (see Fig. S5 in the supplemental material). To distinguish whether this in fact relies on reduced cell density or may be caused by different light scattering properties of the PHA granule-free cells, we determined absolute cell numbers by counting. The results indeed suggested a difference in the correlation of optical density and cell counts between wt and mutant strains (about 126% of wt cells). However, this difference did not completely compensate for the lower optical density (see Fig. S5 in the supplemental material), which might indicate that deletion of the phbCAB genes interferes with other metabolic pathways. Diminished growth (depending on the carbon source) upon deletion of PHA polymerase genes has also been reported, for example, for Rhodospirillum rubrum (39).
The only further MSR-1 mutant for which perturbed PHA synthesis has been reported to date accumulated 71% less PHA but hydrolyzed more ATP and consumed more oxygen than the wt. In contrast to our targeted deletion, this strain originated from aberrant recombination of a suicide vector next to an ATPase gene, which likely caused increased transcription of the gene (40). Thus, the reduced PHA synthesis in this mutant was presumably due to higher energy consumption and, hence, a secondary effect.
Since there is growing evidence that the distribution and segregation of intracellular macromolecules, organelles, and storage inclusions in bacteria are nonrandom (4, 41–43), we compared the positionings of polyphosphate inclusions and magnetosome chains in the phbCAB mutant to those in the wt. We found that the formation and cellular distribution of polyphosphate were not affected (Fig. 2, black arrows) and that the formation and positioning of magnetosome chains in the phbCAB mutant were indistinguishable from those of the wt (Fig. 2C).
Construction of unmarked and functional MamC and MamK fluorescent fusions.
Fluorescent fusions to magnetosome-associated proteins have been described previously and have proven useful for analyses of subcellular protein localization patterns and dynamics (2, 19, 31, 32, 44–46). However, these genes were expressed either from replicating vectors that cause cell-to-cell heterogeneity and overexpression of the fusion protein due to plasmid copy number variation or as additional variants from ectopic positions in the chromosome with the native, untagged gene present. However, we wished to demonstrate the functionality of fused key magnetosome marker proteins when expressed solely from their native chromosomal position. Therefore, we constructed markerless fluorescent fusions of mamC (carboxy terminal) or mamK (amino terminal) to mCherry or egfp within the mamGFDC or mamAB operon, respectively, in both the wt and the phbCAB mutant using the newly established GalK counterselection technique. We selected MamC because of its abundance and specificity for the magnetosome membrane and the actin-like MamK for its function as a cytoskeletal element and its central role in magnetosome chain assembly and segregation (2, 4).
Fluorescence microscopy revealed filamentous fluorescence signals for both proteins, similar to previous reports (2, 4, 31, 44). MamC-EGFP and MamC-mCherry fluorescence was confined to intracellular spots, which concatenated into a nonresolvable string-like structure at midcell, corresponding to the magnetosome chain (Fig. 3B, C, E, and F). However, mCherry-MamK formed a filamentous structure of constant intensity reaching from pole to pole (Fig. 3A and B). Interestingly, the fluorescence signals were of uniform strength throughout the population (see Fig. S6 and S7 in the supplemental material), which is not observed when fluorescent protein fusions are expressed from plasmids. Electron microscopy showed wt-like magnetosome crystals and chains in all strains, indicating that the fusion proteins were functional as a sole copy in the chromosome and that no polar effects on downstream genes occurred (Fig. 3, insets). Markedly, in the mCherry-mamK strain, magnetosomes were organized into single or double chains at midcell, which is not observed in mutants with nonfunctional or missing MamK (4, 44). These characteristics of the mutant strains suggest that they are most favorable for microscopic and ultrastructural analyses as well as live-cell imaging.
FIG 3.

Differential interference contrast, fluorescence, and TEM images of MSR-1 wt (left column) and phbCAB mutant (right column) cells with markerless chromosomal fusions of mamC and mamK to mCherry (red) and egfp (green). The filamentous fluorescence signals are of even intensity throughout the cell populations (see also Fig. S5 and S6 in the supplemental material). All strains display wt-like magnetosome chains and crystals, indicating that the fusion proteins are functional and that there are no polar effects on downstream genes. (A and D) mCherry-mamK; (B and E) mamC-egfp; (C and F) mamC-mCherry. Membranes in panels B and E were stained with FM4-64 (red), and those in panels A, C, D, and F were stained with Cellbrite Blue cytoplasmic membrane stain.
Conclusions.
In summary, we were able to enhance the toolbox for genetic manipulation of MSR-1 and potentially of other magnetospirilla by a quick, efficient, and reliable technique. Effort and time to generate mutants in MSR-1 could be reduced to less than one-third compared to the Cre-lox technology. These savings result mainly because only one deletion vector is constructed, only one conjugation procedure is necessary, and insertion mutants grow in the presence of only one antibiotic. In contrast to sacB-mediated counterselection, in our hands, the use of galK resulted in strict selection for recombination events and obviated the need for replica platings, reducing the time typically required for an unmarked mutation from several months to 3 to 5 weeks. Since no scar sequence is left in the chromosome, the introduction of tailored in-frame deletions, in-frame fusions, and site-specific point mutations is feasible. Recently, this technique was used to reliably introduce a number of single nucleotide exchanges into the MSR-1 chromosome and to precisely delete genome fragments of >19 kb (data not shown), illustrating that the adapted counterselection technique presented here is currently the most powerful tool for chromosomal manipulation of MSR-1.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Isabelle Mai for technical assistance and to Günter Pfeifer for continuously supporting CET. We also thank A. Pollithy for providing vectors pAP160 and pAP173.
This research was supported by Deutsche Forschungsgemeinschaft grants Schu1080/9-1 and Schu1080/15-1 and by HFSP grant RGP0052/2012 to D.S.
Footnotes
Published ahead of print 9 May 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00588-14.
REFERENCES
- 1.Grünberg K, Müller EC, Otto A, Reszka R, Linder D, Kube M, Reinhardt R, Schüler D. 2004. Biochemical and proteomic analysis of the magnetosome membrane in Magnetospirillum gryphiswaldense. Appl. Environ. Microbiol. 70:1040–1050. 10.1128/AEM.70.2.1040-1050.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Komeili A, Li Z, Newman DK, Jensen GJ. 2006. Magnetosomes are cell membrane invaginations organized by the actin-like protein MamK. Science 311:242–245. 10.1126/science.1123231 [DOI] [PubMed] [Google Scholar]
- 3.Scheffel A, Gruska M, Faivre D, Linaroudis A, Plitzko JM, Schüler D. 2006. An acidic protein aligns magnetosomes along a filamentous structure in magnetotactic bacteria. Nature 440:110–114. 10.1038/nature04382 [DOI] [PubMed] [Google Scholar]
- 4.Katzmann E, Müller FD, Lang C, Messerer M, Winklhofer M, Plitzko JM, Schüler D. 2011. Magnetosome chains are recruited to cellular division sites and split by asymmetric septation. Mol. Microbiol. 82:1316–1329. 10.1111/j.1365-2958.2011.07874.x [DOI] [PubMed] [Google Scholar]
- 5.Lang C, Schüler D, Faivre D. 2007. Synthesis of magnetite nanoparticles for bio- and nanotechnology: genetic engineering and biomimetics of bacterial magnetosomes. Macromol. Biosci. 7:144–151. 10.1002/mabi.200600235 [DOI] [PubMed] [Google Scholar]
- 6.Ohuchi S, Schüler D. 2009. In vivo display of a multisubunit enzyme complex on biogenic magnetic nanoparticles. Appl. Environ. Microbiol. 75:7734–7738. 10.1128/AEM.01640-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pollithy A, Romer T, Lang C, Müller FD, Helma J, Leonhardt H, Rothbauer U, Schüler D. 2011. Magnetosome expression of functional camelid antibody fragments (nanobodies) in Magnetospirillum gryphiswaldense. Appl. Environ. Microbiol. 77:6165–6171. 10.1128/AEM.05282-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sugamata Y, Tanaka T, Matsunaga T, Yoshino T. 2014. Functional expression of an scFv on bacterial magnetic particles by in vitro docking. Biochem. Biophys. Res. Commun. 445:1–5. 10.1016/j.bbrc.2013.12.102 [DOI] [PubMed] [Google Scholar]
- 9.Schultheiss D, Schüler D. 2003. Development of a genetic system for Magnetospirillum gryphiswaldense. Arch. Microbiol. 179:89–94 http://magneticliquid.narod.ru/autority/573.pdf [DOI] [PubMed] [Google Scholar]
- 10.Marx C, Lidstrom M. 2002. Broad-host-range cre-lox system for antibiotic marker recycling in Gram-negative bacteria. Biotechniques 33:1062–1067 http://www.biotechniques.com/multimedia/archive/00010/02335rr01_10092a.pdf [DOI] [PubMed] [Google Scholar]
- 11.Scheffel A, Gardes A, Grünberg K, Wanner G, Schüler D. 2008. The major magnetosome proteins MamGFDC are not essential for magnetite biomineralization in Magnetospirillum gryphiswaldense but regulate the size of magnetosome crystals. J. Bacteriol. 190:377–386. 10.1128/JB.01371-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ullrich S, Schüler D. 2010. Cre-lox-based method for generation of large deletions within the genomic magnetosome island of Magnetospirillum gryphiswaldense. Appl. Environ. Microbiol. 76:2439–2444. 10.1128/AEM.02805-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suzuki N, Nonaka H, Tsuge Y, Inui M, Yukawa H. 2005. New multiple-deletion method for the Corynebacterium glutamicum genome, using a mutant lox sequence. Appl. Environ. Microbiol. 71:8472–8480. 10.1128/AEM.71.12.8472-8480.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lohße A, Ullrich S, Katzmann E, Borg S, Wanner G, Richter M, Voigt B, Schweder T, Schüler D. 2011. Functional analysis of the magnetosome island in Magnetospirillum gryphiswaldense: the mamAB operon is sufficient for magnetite biomineralization. PLoS One 6:e25561. 10.1371/journal.pone.0025561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gay P, Le Coq D, Steinmetz M, Berkelman T, Kado CI. 1985. Positive selection procedure for entrapment of insertion sequence elements in gram-negative bacteria. J. Bacteriol. 164:918–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ried JL, Collmer A. 1987. An nptI-sacB-sacR cartridge for constructing directed, unmarked mutations in Gram-negative bacteria by marker exchange-eviction mutagenesis. Gene 57:239–246. 10.1016/0378-1119(87)90127-2 [DOI] [PubMed] [Google Scholar]
- 17.Kanetsuki Y, Tanaka M, Tanaka T, Matsunaga T, Yoshino T. 2012. Effective expression of human proteins on bacterial magnetic particles in an anchor gene deletion mutant of Magnetospirillum magneticum AMB-1. Biochem. Biophys. Res. Commun. 426:7–11. 10.1016/j.bbrc.2012.07.116 [DOI] [PubMed] [Google Scholar]
- 18.Komeili A, Vali H, Beveridge TJ, Newman DK. 2004. Magnetosome vesicles are present before magnetite formation, and MamA is required for their activation. Proc. Natl. Acad. Sci. U. S. A. 101:3839–3844. 10.1073/pnas.0400391101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murat D, Quinlan A, Vali H, Komeili A. 2010. Comprehensive genetic dissection of the magnetosome gene island reveals the step-wise assembly of a prokaryotic organelle. Proc. Natl. Acad. Sci. U. S. A. 107:5593–5598. 10.1073/pnas.0914439107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tanaka M, Mazuyama E, Arakaki A, Matsunaga T. 2011. MMS6 protein regulates crystal morphology during nano-sized magnetite biomineralization in vivo. J. Biol. Chem. 286:6386–6392. 10.1074/jbc.M110.183434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schultheiss D, Kube M, Schüler D. 2004. Inactivation of the flagellin gene flaA in Magnetospirillum gryphiswaldense results in nonmagnetotactic mutants lacking flagellar filaments. Appl. Environ. Microbiol. 70:3624–3631. 10.1128/AEM.70.6.3624-3631.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McKenney K, Shimatake H, Court D, Schmeissner U, Brady C, Rosenberg M. 1981. A system to study promoter and terminator signals recognized by Escherichia coli RNA polymerase. Gene Amplif. Anal. 2:383–415 [PubMed] [Google Scholar]
- 23.Ueki T, Inouye S, Inouye M. 1996. Positive-negative KG cassettes for construction of multi-gene deletions using a single drug marker. Gene 183:153–157. 10.1016/S0378-1119(96)00546-X [DOI] [PubMed] [Google Scholar]
- 24.Barkan D, Stallings CL, Glickman MS. 2011. An improved counterselectable marker system for mycobacterial recombination using galK and 2-deoxy-galactose. Gene 470:31–36. 10.1016/j.gene.2010.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Merritt J, Tsang P, Zheng L, Shi W, Qi F. 2007. Construction of a counterselection-based in-frame deletion system for genetic studies of Streptococcus mutans. Oral Microbiol. Immunol. 22:95–102. 10.1111/j.1399-302X.2007.00329.x [DOI] [PubMed] [Google Scholar]
- 26.Müller FD, Schink CW, Hoiczyk E, Cserti E, Higgs PI. 2012. Spore formation in Myxococcus xanthus is tied to cytoskeleton functions and polysaccharide spore coat deposition. Mol. Microbiol. 83:486–505. 10.1111/j.1365-2958.2011.07944.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 28.Heyen U, Schüler D. 2003. Growth and magnetosome formation by microaerophilic Magnetospirillum strains in an oxygen-controlled fermentor. App Microbiol. Biotechnol. 61:536–544. 10.1007/s00253-002-1219-x [DOI] [PubMed] [Google Scholar]
- 29.Schüler D, Uhl R, Bäuerlein E. 1995. A simple light scattering method to assay magnetism in Magnetospirillum gryphiswaldense. FEMS Microbiol. Lett. 132:139–145. 10.1111/j.1574-6968.1995.tb07823.x [DOI] [Google Scholar]
- 30.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77:51–59. 10.1016/0378-1119(89)90358-2 [DOI] [PubMed] [Google Scholar]
- 31.Lang C, Schüler D. 2008. Expression of green fluorescent protein fused to magnetosome proteins in microaerophilic magnetotactic bacteria. Appl. Environ. Microbiol. 74:4944–4953. 10.1128/AEM.00231-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scheffel A, Schüler D. 2007. The acidic repetitive domain of the Magnetospirillum gryphiswaldense MamJ protein displays hypervariability but is not required for magnetosome chain assembly. J. Bacteriol. 189:6437–6446. 10.1128/JB.00421-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Uebe R, Voigt B, Schweder T, Albrecht D, Katzmann E, Lang C, Bottger L, Matzanke B, Schüler D. 2010. Deletion of a fur-like gene affects iron homeostasis and magnetosome formation in Magnetospirillum gryphiswaldense. J. Bacteriol. 192:4192–4204. 10.1128/JB.00319-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raschdorf O, Müller FD, Posfai M, Plitzko JM, Schüler D. 2013. The magnetosome proteins MamX, MamZ and MamH are involved in redox control of magnetite biomineralization in Magnetospirillum gryphiswaldense. Mol. Microbiol. 89:872–886. 10.1111/mmi.12317 [DOI] [PubMed] [Google Scholar]
- 35.Nickell S, Forster F, Linaroudis A, Net WD, Beck F, Hegerl R, Baumeister W, Plitzko JM. 2005. TOM software toolbox: acquisition and analysis for electron tomography. J. Struct. Biol. 149:227–234. 10.1016/j.jsb.2004.10.006 [DOI] [PubMed] [Google Scholar]
- 36.Katzen F, Becker A, Ielmini MV, Oddo CG, Ielpi L. 1999. New mobilizable vectors suitable for gene replacement in Gram-negative bacteria and their use in mapping of the 3′ end of the Xanthomonas campestris pv. campestris gum operon. Appl. Environ. Microbiol. 65:278–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Borg S, Hofmann J, Pollithy A, Lang C, Schüler D. 2014. New vectors for chromosomal integration enable high-level constitutive or inducible magnetosome expression of fusion proteins in Magnetospirillum gryphiswaldense. Appl. Environ. Microbiol. 80:2609–2616. 10.1128/AEM.00192-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spiekermann P, Rehm BHA, Kalscheuer R, Baumeister D, Steinbüchel A. 1999. A sensitive, viable-colony staining method using Nile red for direct screening of bacteria that accumulate polyhydroxyalkanoic acids and other lipid storage compounds. Arch. Microbiol. 171:73–80. 10.1007/s002030050681 [DOI] [PubMed] [Google Scholar]
- 39.Jin H, Nikolau BJ. 2012. Role of genetic redundancy in polyhydroxyalkanoate (PHA) polymerases in PHA biosynthesis in Rhodospirillum rubrum. J. Bacteriol. 194:5522–5529. 10.1128/JB.01111-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu J, Ding Y, Jiang W, Tian J, Li Y, Li J. 2008. A mutation upstream of an ATPase gene significantly increases magnetosome production in Magnetospirillum gryphiswaldense. Appl. Microbiol. Biotechnol. 81:551–558. 10.1007/s00253-008-1665-1 [DOI] [PubMed] [Google Scholar]
- 41.Galán B, Dinjaski N, Maestro B, de Eugenio LI, Escapa IF, Sanz JM, Garcia JL, Prieto MA. 2011. Nucleoid-associated PhaF phasin drives intracellular location and segregation of polyhydroxyalkanoate granules in Pseudomonas putida KT2442. Mol. Microbiol. 79:402–418. 10.1111/j.1365-2958.2010.07450.x [DOI] [PubMed] [Google Scholar]
- 42.Jendrossek D, Pfeiffer D. 21 January 2014. New insights in the formation of polyhydroxyalkanoate granules (carbonosomes) and novel functions of poly(3-hydroxybutyrate). Environ. Microbiol. 10.1111/1462-2920.12356 [DOI] [PubMed] [Google Scholar]
- 43.Pfeiffer D, Wahl A, Jendrossek D. 2011. Identification of a multifunctional protein, PhaM, that determines number, surface to volume ratio, subcellular localization and distribution to daughter cells of poly(3-hydroxybutyrate), PHB, granules in Ralstonia eutropha H16. Mol. Microbiol. 82:936–951. 10.1111/j.1365-2958.2011.07869.x [DOI] [PubMed] [Google Scholar]
- 44.Draper O, Byrne ME, Li Z, Keyhani S, Barrozo JC, Jensen G, Komeili A. 2011. MamK, a bacterial actin, forms dynamic filaments in vivo that are regulated by the acidic proteins MamJ and LimJ. Mol. Microbiol. 82:342–354. 10.1111/j.1365-2958.2011.07815.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schultheiss D, Handrick R, Jendrossek D, Hanzlik M, Schüler D. 2005. The presumptive magnetosome protein Mms16 is a poly(3-hydroxybutyrate) granule-bound protein (phasin) in Magnetospirillum gryphiswaldense. J. Bacteriol. 187:2416–2425. 10.1128/JB.187.7.2416-2425.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zeytuni N, Ozyamak E, Ben-Harush K, Davidov G, Levin M, Gat Y, Moyal T, Brik A, Komeili A, Zarivach R. 2011. Self-recognition mechanism of MamA, a magnetosome-associated TPR-containing protein, promotes complex assembly. Proc. Natl. Acad. Sci. U. S. A. 108:E480–E487. 10.1073/pnas.1103367108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hanahan D. 1983. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166:557–580. 10.1016/S0022-2836(83)80284-8 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.