

Abstract
A well-accepted method for identification of microorganisms uses matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS) coupled to analysis software which identifies and classifies the organism according to its ribosomal protein spectral profile. The method, called MALDI biotyping, is widely used in clinical diagnostics and has partly replaced conventional microbiological techniques such as biochemical identification due to its shorter time to result (minutes for MALDI biotyping versus hours or days for classical phenotypic or genotypic identification). Besides its utility for identifying bacteria, MS-based identification has been shown to be applicable also to yeasts and molds. A limitation to this method, however, is that accurate identification is most reliably achieved on the species level on the basis of reference mass spectra, making further phylogenetic classification unreliable. Here, it is shown that combining tryptic digestion of the acid/organic solvent extracted (classical biotyping preparation) and resolubilized proteins, nano-liquid chromatography (nano-LC), and subsequent identification of the peptides by MALDI-tandem TOF (MALDI-TOF/TOF) mass spectrometry increases the discrimination power to the level of subspecies. As a proof of concept, using this targeted proteomics workflow, we have identified subspecies-specific biomarker peptides for three Salmonella subspecies, resulting in an extension of the mass range and type of proteins investigated compared to classical MALDI biotyping. This method therefore offers rapid and cost-effective identification and classification of microorganisms at a deeper taxonomic level.
INTRODUCTION
Matrix-assisted laser desorption ionization (MALDI) biotyping is based on unique ribosomal protein profiles matched to a reference database and identified accordingly (1–5). The resulting fingerprints of these constantly expressed and highly abundant proteins are highly reproducible and mostly independent of the culture medium, incubation temperature, and growth state (6–8). MALDI biotyping has gained much attention in recent years due to its speed, simple handling, cost-effectiveness, and high-throughput capabilities (9). Conventional methods assessing phenotypic traits based on biochemical reactions, antigen-antibody reactions, Gram staining, colony morphology, or growth pattern are often time-consuming and expensive. Molecular biology techniques for assessment of genotypic traits such as PCR, sequencing, pulsed-field gel electrophoresis (PFGE), multilocus sequence typing (MLST), restriction fragment length polymorphism (RFLP), and microarrays need great expertise and are expensive as well. However, MALDI biotyping has limitations, too. The identification rates using clinical isolates are reported to be 79.9% to 93.6% at the species level (10–13) and 94.5% to 97.2% at the genus level (10, 12, 14). These rates are mainly due to high protein identity among bacterial subspecies and serovars. Therefore, there is a need for a more sensitive and accurate method. This is particularly important in food safety, where contamination by food-borne bacteria such as salmonellae can lead to serious illness or even death and can cause economic and reputational losses to agriculture and the food industry. In most cases, contamination is introduced during production, processing, or storage. The rapid bacterial profiling through MALDI-TOF MS has also found application in clinical microbiology, where characterization of certain species proved more accurate with MALDI biotyping than with 16S rRNA gene sequencing (15). Other fields of application are biodefense and environmental microbiology (16), where rapid detection and differentiation of bacteria from surface, air, soil, or water samples below the species level are key to evaluating their commensal, mutualistic, or pathogenic characteristics. It is worth mentioning that the extent and quality of entries as well as the correct taxonomy of the microorganisms deposited in the database are crucial. Here we extend this method to the level of peptides. In this report, we present methods designed to improve ultrafast generation of peptides that could act as Salmonella subspecies biomarkers. We investigate the use of high-intensity focused ultrasound (HIFU) (17) to aid solubilization of the pelleted proteins after acid/organic solvent extraction and study the course of tryptic digestion under HIFU conditions to optimize peptide output and generation of subspecies-specific peptide biomarkers.
MATERIALS AND METHODS
Bacterial culture and protein extraction (classical biotyping).
Protein extraction was performed in accordance with a procedure previously described (18) and lately applied by many research groups (19–26). For classical biotyping, which uses undigested cell extracts, Salmonella enterica subsp. arizonae (German Collection of Microorganisms and Cell Cultures; DSMZ 9386), S. enterica subsp. enterica (isolate from a gastroenteritis outbreak in Scandinavia [27]), and S. enterica subsp. houtenae (DSMZ 9221) were grown overnight on tryptic soy agar (Sigma-Aldrich, St. Louis, MO). An inoculation loop full of cells was then introduced into 1.2 ml of 75% ethanol (Sigma-Aldrich) and suspended by mixing with a vortex device. The sample was centrifuged for 2 min at 16,100 × g, the supernatant was discarded, and the residual ethanol was evaporated for 5 min at room temperature. In order to extract the proteins, 50 μl of 70% formic acid (Sigma-Aldrich) and 50 μl of 100% acetonitrile (Biosolve B.V., Valkenswaard, Netherlands) were added to the pellet and thoroughly mixed using a vortex device. Another centrifugation at 16,100 × g for 2 min resulted in a protein-containing supernatant which was used for spotting onto the MALDI target (MSP 96 polished steel BC target; Bruker Daltonics, Bremen, Germany) and the subsequent MALDI-TOF measurement.
MALDI-TOF measurement.
Protein-containing supernatant (1 μl) was spotted onto the MALDI target, allowed to dry, and covered with 1 μl of saturated matrix solution containing HCCA (α-cyano-4-hydroxycinnamic acid) (Bruker Daltonics) at a concentration of 10 mg/ml in acetonitrile-water-trifluoroacetic acid (TFA) (50:47.5:2.5 [vol/vol/vol]) (Sigma-Aldrich). Direct smearing of bacterial cells was performed in parallel: cell material from the agar plate was transferred onto the MALDI target using a toothpick, and the smear was covered with saturated HCCA matrix. Sample spectra were collected with a microflex LT MALDI-TOF mass spectrometer (Bruker Daltonics) using flexControl software (Version 3.4) (Bruker Daltonics) and applying the measurement parameters suggested by the manufacturer for classical biotyping: laser frequency of 60 Hz in the positive linear mode with acquisition ranging from 2 to 20 kDa. Final spectra consisted of 240 shots per spot (40 shots per raster spot). The laser intensity was chosen so as to obtain spectra with maximal absolute peak intensities ranging from about 5 × 103 to 104 arbitrary units. The spectra were evaluated using the accompanying MALDI BioTyper OC software (Version 3.1) (Bruker Daltonics).
Bacterial culture and protein extraction (biomarker search).
For generation of biomarker peptides, an overnight culture of S. enterica subsp. arizonae, S. enterica subsp. enterica, and S. enterica subsp. houtenae grown in tryptic soy broth (Sigma-Aldrich) was diluted to an optical density at 600 nm (OD600) of 1. A 1-ml volume thereof was spun at 5,000 × g for 1 min, and the resulting pellet was washed with deionized water. After a second spin, the pellet was resuspended in 1.2 ml of 75% ethanol and mixed using a vortex device. The sample was then centrifuged for 2 min at 16,100 × g, the supernatant was discarded, and the residual ethanol was evaporated for 5 min at room temperature. In order to extract the proteins, 100 μl of 70% formic acid and 100 μl of 100% acetonitrile were added to the pellet and thoroughly mixed using a vortex device. After another centrifugation at 16,100 × g for 2 min, the protein-containing supernatant was transferred to a new Eppendorf tube and put into the freezer at −20°C until used for further preparation.
Trypsin digestion.
The supernatant (175 μl) of the samples described in the above section was subjected to complete evaporation using a SpeedVac concentrator (Savant Instruments Inc., New York, NY) at room temperature. The resulting pellet was resuspended in 3 μl of 100% acetonitrile, 3 μl of RapiGest (Waters, Guyancourt, France), 18 μl of 100 mM Tris-HCl (Sigma-Aldrich) (pH 8.2), and 3 μl of 1 M Tris-HCl (Sigma-Aldrich) (pH 8.2). This sample was then subjected to 5 min of HIFU treatment (model UTR200, Hielscher Ultrasonics, Teltow, Germany) in order to resolubilize the pellet. Settings were as following: intensity, 90%; cycles, 0.8. A 3-μl volume of a solution of 0.1 mg/ml trypsin (Roche Diagnostics, Mannheim, Germany)–10 mM HCl (Merck, Darmstadt, Germany) was added subsequently, and the proteins were digested during a 15-min HIFU treatment (intensity, 90%; cycles, 0.8) in an ice water bath.
Nano-LC spotting.
The resulting peptides were separated with an easy-nLC II nano-liquid chromatography (nano-LC) system (Bruker Daltonics) which was coupled to a Proteineer fc II fraction collector (Bruker Daltonics). Mobile phase A consisted of water containing 0.1% TFA (Sigma-Aldrich), and mobile phase B consisted of 90% acetonitrile (Merck) containing 0.1% TFA. Separation was performed on a C18 column (Acclaim PepMap100; Dionex Benelux B.V., Amsterdam, Netherlands) (15 cm by 75 μm, 3 μm pore size, 100 Å) with a linear gradient of 2% to 45% mobile phase B in 64 min and a flow rate of 300 nl/min. A C18 reversed-phase solid-phase extraction trap (BioSphere NS-MP-10; NanoSeparations, Nieuwkoop, Netherlands) (2 cm by 100 μm, 5 μm, 120 Å) was used to prevent the separation column from being jammed. The separated peptides were mixed with the HCCA matrix directly in the Proteineer fc II fraction collector and spotted onto an MTP AnchorChip 1536 TF instrument (Bruker Daltonics) with an interval of six spots per min. The HCCA matrix used for spotting was prepared as follows: 748 μl of TA95 (acetonitrile-water-TFA, 95:4.9:0.1 [vol/vol/vol]), 36 μl of saturated HCCA–TA90 (acetonitrile-water-TFA, 90:9.9:0.1 [vol/vol/vol]), 8 μl of 10% TFA, and 8 μl of 100 mM NH4H2PO4 (Sigma-Aldrich) dissolved in water. One per eight spots was manually spotted with peptide calibration standard II (Bruker Daltonics) diluted 1:200 in HCCA matrix prepared with 748 μl of TA85 (acetonitrile-water-TFA, 85:14.9:0.1 [vol/vol/vol]) instead of TA95.
MALDI-TOF/TOF measurement.
MALDI measurements of trypsin-digested samples were performed on an ultrafleXtreme MALDI-tandem TOF (MALDI-TOF/TOF) MS equipped with a SmartBeam laser (Bruker Daltonics). WARP-LC (Version 1.3) was used to set up data acquisition methods, define values for tandem MS (MS/MS) precursor selection (signal-to-noise [S/N] ratio threshold, 10), and merge compounds that were separated by less than six fractions (mass tolerance, ±50 ppm). The measurement parameters were again programmed in flexControl (Version 3.4) as follows: a laser frequency of 1,000 Hz in the positive reflectron mode was used, with acquisition ranging from 700 to 4,000 Da. Final spectra consisted of 3,000 shots per spot (100 shots per raster spot). The laser intensity and detector sensitivity were adjusted such that the highest peak of the spectrum was in the range of 104 to 105 arbitrary units.
MS/MS data acquired through WARP-LC were searched on MASCOT (Version 2.4.1) using ProteinScape (Version 3.0; Bruker Daltonics) with the search restricted to Salmonella on the NCBI database (status as of June 2013). The search was limited to tryptic peptides with variable methionine, histidine, and tryptophan oxidation and an MS/MS tolerance of ±0.7 Da. The peptide tolerance was set to ±50 ppm. The peptide charge was defined as +1, and one miscleavage was allowed. A peptide decoy database was used as well as the MASCOT Percolator algorithm to improve the significance of the search results. A second search was conducted on the UniProtKB/Swiss-Prot database, the results of which are shown in Table S1 in the supplemental material.
Data analysis.
Data evaluation and a biomarker search were performed using a Microsoft Excel 2010 macro developed in house. Briefly, the flexAnalysis (Version 3.4) peak lists or WARP-LC compound lists from classical biotyping or protein digests, respectively, were exported and merged in one Excel sheet. In a first step, an S/N ratio and a ppm value were defined. The values chosen for evaluation of the MS data were an S/N ratio of 3 and a ppm value of ±200, whereas for the MS/MS data the S/N ratio was set to 10 and the effect of various ppm values (±25, ±50, and ±100 ppm) was investigated. The macro then selected data sets with an S/N ratio above the defined value and created a pivot table with the corresponding m/z values in the row fields and the name of each of the different measured sets in the column fields. Each m/z value was then taken as the center and m/z values within the surrounding ppm window were counted. The counts of the names of the different measured sets within the defined ppm window were the data items. This pivot table gives an overview of the number of m/z signals within the ppm window per set of subspecies. In a second step, m/z values of biomarkers were selected and pasted into a new Excel sheet. An m/z value was determined to be a biomarker for a subspecies when it was present in all sets of the corresponding subspecies but in none of the other subspecies.
RESULTS
MALDI-TOF (classical biotyping).
MALDI-TOF MS mass spectra were obtained for three different subspecies of S. enterica (S. enterica subsp. arizonae, S. enterica subsp. enterica, and S. enterica subsp. houtenae) by either directly smearing bacterial cells from the agar plate onto the MALDI target or spotting cell extracts. For both direct smearing and extraction, the experiment was repeated three times, each time using the same overnight culture for both approaches. Direct smearing yielded identification scores (data not shown) that were slightly lower overall than those seen with the cell extracts. Therefore, only the results obtained from the cell extracts are presented.
By classical biotyping according to the manufacturer's protocol, all three S. enterica samples could be correctly identified on the species level (Fig. 1A to C). However, identification on the subspecies level could not be achieved consistently.
FIG 1.

MALDI-TOF MS mass spectra of cell extracts of S. enterica subsp. arizonae (A), S. enterica subsp. enterica (B), and S. enterica subsp. houtenae (C). Each experiment was performed in triplicate. The absolute intensities of the ions are shown on the y axis. The mass-to-charge (m/z) ratios of the ions are shown on the x axis, which correspond to the masses of the single positively charged ions.
MALDI-TOF/TOF (biomarker search).
MALDI-TOF/TOF MS experiments were performed in triplicate using trypsin-digested cell extracts of the same three Salmonella samples. The optimized workflow established in the current work is depicted in Fig. 2. For that figure, tryptic digestion of resolubilized proteins and nano-LC separation of the resulting peptides were combined with identification of those peptides by MALDI-TOF/TOF MS. The analysis of the acquired data by searching the NCBI database for each sample yielded a number of proteins, ranging from 465 to 627 proteins.
FIG 2.
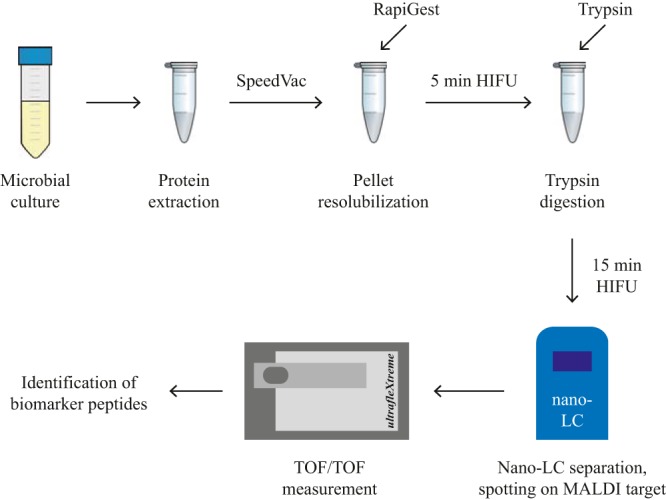
Workflow for the preparation of tryptic digests of cell extracts from S. enterica subsp. arizonae, S. enterica subsp. enterica, and S. enterica subsp. houtenae and subsequent MALDI-TOF/TOF MS analysis. The cell extract used for classical biotyping is dried on a SpeedVac. After resolubilizing the pellet through the addition of RapiGest detergent under 5 min of high-intensity focused ultrasound (HIFU) treatment, trypsin is added and the proteins are digested for 15 min under the assistance of HIFU (in an ice water bath). The peptides are separated by nano-LC, mixed with HCCA matrix directly in the fraction collector, and spotted onto the MALDI target. MALDI-TOF/TOF analysis is performed, and the resulting data are screened for biomarker peptide candidates using an Excel macro developed in house.
Further data analysis with the Excel macro developed in house yielded a list of potential biomarker peptide masses for the three subspecies. The following searches were conducted. The S/N ratio was set to 10 according to the value defined for MS/MS precursor selection, and specific ppm values (±25, ±50, and ±100 ppm) in the range of the mass tolerance defined for MS/MS spectra acquisition were tested. An m/z value was classified as being a biomarker candidate when it was present in all three sets of the corresponding subspecies but in none of the other subspecies. Table 1 shows the number of potential biomarker peptide masses found for each subspecies as well as the number of masses assigned to actual tryptic peptides of Salmonella proteins found in the MASCOT search and belonging to the correct subspecies (±200 ppm; S/N ratio = 3). The analysis exhibited 51, 48, and 26 potential biomarker peptide masses for S. enterica subsp. arizonae, S. enterica subsp. enterica, and S. enterica subsp. houtenae, respectively. Of those masses, about 50% were assigned to actual Salmonella peptides (Table 1).
TABLE 1.
Numbers of potential biomarker peptide masses found for S. enterica subsp. arizonae, S. enterica subsp. enterica, and S. enterica subsp. houtenae and numbers of peptides assigned to actual peptides in Salmonella proteins of the correct subspecies found by the MASCOT search using the NCBI databasea
| Salmonella subspecies | No. of potential biomarker peptides | No. of assigned biomarker peptides |
|---|---|---|
| S. enterica subsp. arizonae | 51 | 29 |
| S. enterica subsp. enterica | 48 | 22 |
| S. enterica subsp. houtenae | 26 | 17 |
Data represent ±200 ppm at an S/N ratio of 3.
In a second approach, the acquired data were analyzed by searching the UniProtKB/Swiss-Prot database. Details on the procedure are given in the supplemental material. All biomarker peptides identified for the three subspecies which were assigned to Salmonella proteins in this second MASCOT search are listed in Table S1 in the supplemental material.
Subspecies biomarker sequence comparison.
For each subspecies, a sequence comparison of the assigned peptides to the analogous peptide sequences of the other two subspecies was conducted, revealing the amino acid exchanges responsible for the subspecies specificity of each peptide. Table 2 displays some selected examples from the NCBI search for biomarker peptides, all of which are distinctive for each of the three investigated subspecies (i.e., all three peptide sequences differ from one another). For example, a biomarker peptide in the isoprenoid biosynthesis protein of S. enterica subsp. houtenae (range, 73 to 99) shows amino acid exchanges from serine to alanine in S. enterica subsp. arizonae and from glutamic acid to aspartic acid in S. enterica subsp. enterica. Of note, all biomarker peptides listed in Table 2 were found at ±25 ppm, except for the flagellar synthesis protein FlgN, which was detected only when the ppm value was set to ±50 or ±100. The ferric uptake regulator was additionally found in the ±50-ppm search, and another nine biomarker peptide masses were found in the ±50- and ±100-ppm searches (marked with an italic superscript “b” in Table 2). A complete overview of all potential biomarker peptides which were annotated to Salmonella proteins in the NCBI database search, including the respective ppm values, can be found in the supplemental material (see Table S2 in the supplemental material).
TABLE 2.
Selected examples for biomarker peptides which are discriminative for all three investigated subspeciesa
| Salmonella subspecies | Assigned protein | Protein mass (kDa) | Biomarker peptide mass (Da) | Amino acid exchange(s) (subspecies)c |
|---|---|---|---|---|
| S. arizonae | 6-Phosphogluconolactonase | 36.4 | 3,004.52 ± 0.03b | A→C, Y→H, A→V (S. enterica) |
| A→C, Y→H, A→V, E→A (S. houtenae) | ||||
| S. arizonae | Arsenate reductase | 13.4 | 1,420.81 ± 0.00 | E→D (S. enterica*) |
| E→D, G→S (S. houtenae*) | ||||
| S. arizonae | Ferric uptake regulator | 17.0 | 3,291.42 ± 0.01 | Q→H (S. enterica) |
| E→D, Q→H (S. houtenae) | ||||
| S. arizonae | Membrane protein | 15.7 | 3,051.48 ± 0.02b | L→I, T→A (S. enterica*) |
| L→I, M→V, T→A (S. houtenae*) | ||||
| S. arizonae | N-Ethylmaleimide reductase | 39.5 | 2,590.17 ± 0.02 | K→E, E→A (S. enterica*) |
| E→A (S. houtenae*) | ||||
| S. arizonae | NADH dehydrogenase subunit G | 100.0 | 3,009.44 ± 0.01b | H→Q (S. enterica) |
| A→T, D→G, H→Q (S. houtenae) | ||||
| S. enterica | Cell division protein DamX | 45.5 | 3,153.62 ± 0.03b | TT→AP, TA→KV (S. arizonae•) |
| A→T, T→K, A→T (S. houtenae) | ||||
| S. enterica | Fumarate hydratase | 51.8 | 2,460.30 ± 0.01b | P→S (S. arizonae) |
| L→M (S. houtenae*) | ||||
| S. enterica | H-NS histone family | 15.5 | 1,979.88 ± 0.01 | MD→IN (S. arizonae*) |
| D→G (S. houtenae*) | ||||
| S. enterica | Peptidyl-prolyl cis-trans-isomerase PpiD | 58.2 | 2,187.04 ± 0.00 | V→A, V→T (S. arizonae*) |
| V→T (S. houtenae*) | ||||
| S. enterica | Probable thiol peroxidase | 18.0 | 2,379.23 ± 0.01 | S→N (S. arizonae*) |
| T→A (S. houtenae*) | ||||
| S. enterica | Putative outer membrane protein | 12.8 | 3,224.54 ± 0.02b | L→F, S→R, S→I, N→Y (S. arizonae•) |
| S→T (S. houtenae*) | ||||
| S. enterica | RNase E | 119.3 | 2,706.37 ± 0.00b | S→T (S. arizonae*) |
| P→T, S→T (S. houtenae*) | ||||
| 3,046.42 ± 0.02b | T→A, S→T (S. arizonae*) | |||
| T→A, H→Y (S. houtenae*) | ||||
| S. houtenae | Flagellar synthesis protein FlgN | 16.0 | 3,102.61 ± 0.05 | S→A (S. arizonae) |
| N→D (S. enterica*) | ||||
| S. houtenae | Isoprenoid biosynthesis protein | 22.9 | 2,763.46 ± 0.02 | S→A (S. arizonae*) |
| E→D (S. enterica*) | ||||
| S. houtenae | NlpB/DapX lipoprotein | 36.9 | 2,504.25 ± 0.01 | D→E, V→P (S. arizonae) |
| V→A (S. enterica*) | ||||
| S. houtenae | Outer membrane protein A | 38.4 | 3,521.70 ± 0.02b | A→T, Y→GA, T→Y (S. arizonae*) |
| A→V, Y→GP (S. enterica*) |
The measured peptide masses are given as the arithmetic mean calculated from the values of the three replicates of the subspecies, and the corresponding standard deviations are presented. Specific amino acids in the biomarker peptides of a subspecies and their exchanges in peptides of the other subspecies are given. All numbers were rounded to the second digit after the comma. The NCBI database was used.
Biomarker peptide found in all investigated ppm windows (±25, ±50, and ±100 ppm).
*, a sequence analogous to the biomarker peptide was observed; •, a shorter or longer peptide sequence containing at least one relevant amino acid exchange was observed.
DISCUSSION
Identification of Salmonella on the genus level by classical MALDI biotyping was reported earlier by Sparbier and colleagues (26). In the present study, the identification was correct on the species level; however, the results were inconsistent on the subspecies level, the spectra being too similar to consistently distinguish the three S. enterica subspecies (Fig. 1). The impression from the visual spectra comparison was confirmed by a biomarker search conducted with an Excel macro, analyzing peak lists generated from spectra with the flexAnalysis software. With an S/N ratio of 3 and a ppm value of ±200, no characteristic biomarker peaks could be found for any of the three subspecies. The S/N ratio of 3 was chosen since the same value is used by the MALDI BioTyper OC software for spectra evaluation. The selected ppm value comprises the approximate resolution range of the MALDI-TOF device according to its general specifications.
The approach to develop a workflow beyond the classical MALDI biotyping (Fig. 2) in order to discriminate on the subspecies level revealed that about 10 times more proteins can be detected than with classical MALDI-TOF MS. The number of proteins found by classical MALDI biotyping ranged from 51 to 59 in a mass range of 3 to 14 kDa, including many ribosomal proteins (7). Applying the HIFU-assisted proteomics workflow clearly extended the mass range and type of possible biomarker target peptides, e.g., the S. enterica subsp. houtenae-biomarker peptide detected in the 170-kDa cell division MukB protein (see Table S2 in the supplemental material), and thus gave rise to more taxonomic discrimination power. The potential of a broader mass range to increase discrimination power was shown for protein biomarkers of Gram-positive as well as Gram-negative bacteria from 15 to 75 kDa by Madonna and coworkers (28).
The MALDI-TOF/TOF biomarker peptide search was conducted using an S/N ratio of 10 and ppm values of ±25, ±50, and ±100. Most biomarker peptides were detected with more than one ppm value. Of special interest are the masses which were found with all three ppm values (entries marked with an italic superscript “b” in Table 2). Having been found at ±25 ppm, the masses of the three replicates lie relatively close to one another, and the fact that they still appear as a biomarker in the ±100-ppm search means that there are no relevant interfering peaks within a relatively broad mass window around them. They can therefore be considered more robust candidates than other biomarker peptides. More examples can be found in Tables S1 and S2 in the supplemental material.
For some proteins which have been identified via the MALDI-TOF/TOF workflow (Fig. 2), more than one tryptic biomarker peptide could be found. These proteins are the DamX cell division protein, 60-kDa chaperonin, NIpB/DapX lipoprotein, RNase E, and outer membrane protein A (see Table S2 in the supplemental material). Also, a biomarker peptide was measured in some cases and annotated in more than one subspecies, making it an especially robust candidate for subspecies distinction (marked with an asterisk in Table 2). Thus, the biomarker peptide in the isoprenoid biosynthesis protein, which is distinctive for all three subspecies, was measured and annotated for each of the three, with average masses of 2,747.45 ± 0.02, 2,749.45 ± 0.01, and 2,763.46 ± 0.02 for S. enterica subsp. arizonae, S. enterica subsp. enterica, and S. enterica subsp. houtenae, respectively. Other examples are the flagellar synthesis protein FlgN, RNase E, N-ethylmaleimide reductase, and outer membrane protein A (Table 2).
It is noteworthy that other working groups reported the identification of Salmonella subspecies- or serovar-specific protein biomarkers as well. Dieckmann and coworkers (29) analyzed 126 Salmonella strains by whole-cell MALDI-TOF MS for variations in protein mass spectral profiles revealing protein biomarker peaks useful for discrimination of S. enterica subspecies. We could confirm two of the detected protein biomarker peaks, those corresponding to ribosomal protein L17 and glutaredoxin-1, to be specific for S. enterica subsp. enterica. The responsible amino acid exchanges are listed in Table S1 of the supplemental material.
In a later study, Dieckmann and Malorny (30) screened 913 S. enterica subsp. enterica strains by whole-cell MALDI-TOF MS in order to find serovar-specific biomarker proteins. They found the L17 ribosomal protein to be a major subspecies-specific biomarker for S. enterica subsp. enterica, being present in almost all 89 S. enterica subsp. enterica serovars tested and absent in all other S. enterica subspecies as well as S. bongori. The biomarker peptide responsible for this subspecies specificity could be identified in the present study in the L17 50S ribosomal protein (see Table S1 in the supplemental material), containing an additional serine residue and a threonine-to-alanine mutation for S. enterica subsp. arizonae and S. enterica subsp. houtenae, as stated by Dieckmann and Malorny and confirmed in the present work.
It is noteworthy that the abundance of existing serovars within a subspecies makes it difficult to ascertain that a peptide sequence is identical in all serovars, especially when considering S. enterica subsp. enterica, for which above 2,500 serovars are known. Thus, Dieckmann and colleagues (29) found DNA-binding protein H-NS (theoretical mass, 15,412.5 Da) to be a genus-identifying biomarker since it was present in all subspecies of S. enterica and even in S. bongori. In a later study (30), however, the same protein was found to have a glycine-to-aspartic acid mutation yielding a predicted mass of 15,470.5 Da in S. enterica subsp. enterica serovar Paratyphi A. In our studies, the H-NS histone family was found to contain a biomarker peptide specific for S. enterica subspecies, with an exchange of methionine-aspartic acid for isoleucine-asparagine in S. enterica subsp. arizonae and an aspartic acid-to-glycine mutation in S. enterica subsp. houtenae.
A very extensive review has been published recently on the application of MALDI-TOF MS profiling of bacteria at the strain level (16). Studies conducted so far cover a variety of microorganisms and have used different procedures for data evaluation, namely, library-based approaches and bioinformatics-enabled approaches. Of special interest to the present study are the bioinformatics-enabled approaches. For example, Fagerquist et al. (31) employed bottom-up proteomics to identify five species of Campylobacter from protein extracts previously separated by high-performance LC (HPLC) and one-dimensional (1D) sodium dodecyl sulfate polyacrylamide gel electrophoresis. They could identify one protein biomarker, a DNA-binding protein, whose amino acid sequence displayed intra- and interspecies variations, thereby facilitating strain differentiation. In a follow-up study, the same researchers investigated three strains of Campylobacter jejuni (32) and found several protein biomarkers displaying mass shifts in MALDI-TOF MS profiles due to amino acid exchanges rather than posttranslational modifications. As in the present work, MASCOT analysis was used to confirm protein identities.
In 2010, the same group could distinguish two pathogenic O157:H7 strains from one non-O157:H7 strain of Escherichia coli through MALDI-TOF/TOF MS in a top-down approach based on six proteins (33). For one of the six, the YahO protein of unknown function, the exchange of a single amino acid was sufficient to enable a distinction of the pathogenic E. coli strains from the nonpathogenic strain.
An alternative to the identification of proteins is the analysis of nucleic acids by MALDI-TOF MS. In the so-called Sequenom approach, similarly to the trypsin digestion step of the improved proteomics workflow described in the present work, a base cleavage step is integrated to generate more products for identification through MALDI-TOF. For example, Syrmis et al. (34) detected single nucleotide polymorphisms (SNPs) useful for differentiation of various methicillin-resistant Staphylococcus aureus strains. Such a targeted approach could be useful for the detection of serovar differences in, e.g., Salmonella strains for which no sequence data are available. However, one should keep in mind that viable or dead contaminating bacteria might be detected by the required PCR amplification. Thus, the proteomics-based workflow offers an advantage by focusing on viable bacteria.
In conclusion, a fast and efficient proteomics workflow was developed in the present work which could be used for identification of subspecies-specific biomarker peptides of a multitude of bacterial species. In future studies, unknown samples will be screened for biomarker peptides by application of selected reaction monitoring (SRM) experiments to rapidly identify the subspecies to which they belong. The most suitable candidates would be those biomarker peptides with a high peptide score, meaning that the MS/MS peptide sequencing yielded satisfactory sequence coverage. Such biomarker peptides displaying sequence coverage of about 70% or higher were found, e.g., in fructose-bisphosphate aldolase, cell division protein DamX, and DNA-binding transcriptional regulator PhoP for S. enterica subsp. arizonae, in a putative outer membrane protein and a probable thiol peroxidase for S. enterica subsp. enterica, or in 60-kDa chaperonin, NAD(P)H:quinone oxidoreductase, and flagellar synthesis protein FlgN for S. enterica subsp. houtenae (see Table S2 in the supplemental material). Also, smaller peptides should be preferred since they are usually better suited from a chromatographic point of view in performing LC-ESI-MS/MS. The SRM approach would allow a rapid and cost-effective screening and classification of unknown bacterial samples. Finally, prior studies (30) have documented Salmonella serovar-level differences in protein profiles and have traced the responsible amino acid exchanges. Thus, the procedure presented here could be further applied to identify biomarker peptides discriminatory of different serovars within a given subspecies.
Supplementary Material
ACKNOWLEDGMENTS
We are indebted to the protein analysis group of the Functional Genomics Center Zurich for valuable discussions, Brion Duffy and Kerstin Brankatschk for supplying Salmonella enterica subsp. enterica, and Daniel Baumgartner for his valuable help in developing the Excel macro.
Footnotes
Published ahead of print 2 May 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00740-14.
REFERENCES
- 1.Claydon MA, Davey SN, Edwards-Jones V, Gordon DB. 1996. The rapid identification of intact microorganisms using mass spectrometry. Nat. Biotechnol. 14:1584–1586. 10.1038/nbt1196-1584 [DOI] [PubMed] [Google Scholar]
- 2.Dieckmann R, Strauch E, Alter T. 2010. Rapid identification and characterization of Vibrio species using whole-cell MALDI-TOF mass spectrometry. J. Appl. Microbiol. 109:199–211. 10.1111/j.1365-2672.2009.04647.x [DOI] [PubMed] [Google Scholar]
- 3.Fenselau C, Demirev PA. 2001. Characterization of intact microorganisms by MALDI mass spectrometry. Mass Spectrom. Rev. 20:157–171. 10.1002/mas.10004 [DOI] [PubMed] [Google Scholar]
- 4.Holland RD, Wilkes JG, Rafii F, Sutherland JB, Persons CC, Voorhees KJ, Lay JO., Jr 1996. Rapid identification of intact whole bacteria based on spectral patterns using matrix-assisted laser desorption/ionization with time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 10:1227–1232. [DOI] [PubMed] [Google Scholar]
- 5.Krishnamurthy T, Ross PL, Rajamani U. 1996. Detection of pathogenic and non-pathogenic bacteria by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 10:883–888. [DOI] [PubMed] [Google Scholar]
- 6.Valentine N, Wunschel S, Wunschel D, Petersen C, Wahl K. 2005. Effect of culture conditions on microorganism identification by matrix-assisted laser desorption ionization mass spectrometry. Appl. Environ. Microbiol. 71:58–64. 10.1128/AEM.71.1.58-64.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wunschel DS, Hill EA, McLean JS, Jarman K, Gorby YA, Valentine N, Wahl K. 2005. Effects of varied pH, growth rate and temperature using controlled fermentation and batch culture on matrix assisted laser desorption/ionization whole cell protein fingerprints. J. Microbiol. Methods 62:259–271. 10.1016/j.mimet.2005.04.033 [DOI] [PubMed] [Google Scholar]
- 8.Wunschel SC, Jarman KH, Petersen CE, Valentine NB, Wahl KL, Schauki D, Jackman J, Nelson CP, White EV. 2005. Bacterial analysis by MALDI-TOF mass spectrometry: an inter-laboratory comparison. J. Am. Soc. Mass Spectrom. 16:456–462. 10.1016/j.jasms.2004.12.004 [DOI] [PubMed] [Google Scholar]
- 9.Kliem M, Sauer S. 2012. The essence on mass spectrometry based microbial diagnostics. Curr. Opin. Microbiol. 15:397–402. 10.1016/j.mib.2012.02.006 [DOI] [PubMed] [Google Scholar]
- 10.van Veen SQ, Claas ECJ, Kuijper EJ. 2010. High-throughput identification of bacteria and yeast by matrix-assisted laser desorption ionization-time of flight mass spectrometry in conventional medical microbiology laboratories. J. Clin. Microbiol. 48:900–907. 10.1128/JCM.02071-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seng P, Drancourt M, Gouriet F, La Scola B, Fournier PE, Rolain JM, Raoult D. 2009. Ongoing revolution in bacteriology: routine identification of bacteria by matrix-assisted laser desorption ionization time-of-flight mass spectrometry. Clin. Infect. Dis. 49:543–551. 10.1086/600885 [DOI] [PubMed] [Google Scholar]
- 12.Sogawa K, Watanabe M, Sato K, Segawa S, Ishii C, Miyabe A, Murata S, Saito T, Nomura F. 2011. Use of the MALDI BioTyper system with MALDI-TOF mass spectrometry for rapid identification of microorganisms. Anal. Bioanal. Chem. 400:1905–1911. 10.1007/s00216-011-4877-7 [DOI] [PubMed] [Google Scholar]
- 13.Cherkaoui A, Hibbs J, Emonet S, Tangomo M, Girard M, Francois P, Schrenzel J. 2010. Comparison of two matrix-assisted laser desorption ionization-time of flight mass spectrometry methods with conventional phenotypic identification for routine identification of bacteria to the species level. J. Clin. Microbiol. 48:1169–1175. 10.1128/JCM.01881-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bizzini A, Durussel C, Bille J, Greub G, Prod'hom G. 2010. Performance of matrix-assisted laser desorption ionization-time of flight mass spectrometry for identification of bacterial strains routinely isolated in a clinical microbiology laboratory. J. Clin. Microbiol. 48:1549–1554. 10.1128/JCM.01794-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Croxatto A, Prod'hom G, Greub G. 2012. Applications of MALDI-TOF mass spectrometry in clinical diagnostic microbiology. FEMS Microbiol. Rev. 36:380–407. 10.1111/j.1574-6976.2011.00298.x [DOI] [PubMed] [Google Scholar]
- 16.Sandrin TR, Goldstein JE, Schumaker S. 2013. MALDI TOF MS profiling of bacteria at the strain level: a review. Mass Spectrom. Rev. 32:188–217. 10.1002/mas.21359 [DOI] [PubMed] [Google Scholar]
- 17.López-Ferrer D, Capelo JL, Vázquez J. 2005. Ultra fast trypsin digestion of proteins by high intensity focused ultrasound. J. Proteome Res. 4:1569–1574. 10.1021/pr050112v [DOI] [PubMed] [Google Scholar]
- 18.Sauer S, Freiwald A, Maier T, Kube M, Reinhardt R, Kostrzewa M, Geider K. 2008. Classification and identification of bacteria by mass spectrometry and computational analysis. PLoS One 3:e2843. 10.1371/journal.pone.0002843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barbuddhe SB, Maier T, Schwarz G, Kostrzewa M, Hof H, Domann E, Chakraborty T, Hain T. 2008. Rapid identification and typing of Listeria species by matrix-assisted laser desorption ionization-time of flight mass spectrometry. Appl. Environ. Microbiol. 74:5402–5407. 10.1128/AEM.02689-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dubois D, Leyssene D, Chacornac JP, Kostrzewa M, Schmit PO, Talon R, Bonnet R, Delmas J. 2010. Identification of a variety of Staphylococcus species by matrix-assisted laser desorption ionization-time of flight mass spectrometry. J. Clin. Microbiol. 48:941–945. 10.1128/JCM.00413-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mellmann A, Cloud J, Maier T, Keckevoet U, Ramminger I, Iwen P, Dunn J, Hall G, Wilson D, LaSala P, Kostrzewa M, Harmsen D. 2008. Evaluation of matrix-assisted laser desorption ionization-time-of-flight mass spectrometry in comparison to 16S rRNA gene sequencing for species identification of nonfermenting bacteria. J. Clin. Microbiol. 46:1946–1954. 10.1128/JCM.00157-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wensing A, Zimmermann S, Geider K. 2010. Identification of the corn pathogen Pantoea stewartii by mass spectrometry of whole-cell extracts and its detection with novel PCR primers. Appl. Environ. Microbiol. 76:6248–6256. 10.1128/AEM.01032-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghyselinck J, Van Hoorde K, Hoste B, Heylen K, De Vos P. 2011. Evaluation of MALDI-TOF MS as a tool for high-throughput dereplication. J. Microbiol. Methods 86:327–336. 10.1016/j.mimet.2011.06.004 [DOI] [PubMed] [Google Scholar]
- 24.Pavlovic M, Huber I, Konrad R, Busch U. 2013. Application of MALDI-TOF MS for the identification of food borne bacteria. Open Microbiol. J. 7:135–141. 10.2174/1874285801307010135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reich M, Bosshard PP, Stark M, Beyser K, Borgmann S. 2013. Species identification of bacteria and fungi from solid and liquid culture media by MALDI-TOF mass spectrometry. J. Bacteriol. Parasitol. 10.4172/2155-9597.S5-002 [DOI] [Google Scholar]
- 26.Sparbier K, Weller U, Boogen C, Kostrzewa M. 2012. Rapid detection of Salmonella sp. by means of a combination of selective enrichment broth and MALDI-TOF MS. Eur. J. Clin. Microbiol. Infect. Dis. 31:767–773. 10.1007/s10096-011-1373-0 [DOI] [PubMed] [Google Scholar]
- 27.Brankatschk K, Blom J, Goesmann A, Smits THM, Duffy B. 2011. Genome of a European fresh-vegetable food safety outbreak strain of Salmonella enterica subsp. enterica serovar Weltevreden. J. Bacteriol. 193:2066. 10.1128/JB.00123-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Madonna AJ, Basile F, Ferrer I, Meetani MA, Rees JC, Voorhees KJ. 2000. On-probe sample pretreatment for the detection of proteins above 15 KDa from whole cell bacteria by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 14:2220–2229. [DOI] [PubMed] [Google Scholar]
- 29.Dieckmann R, Helmuth R, Erhard M, Malorny B. 2008. Rapid classification and identification of salmonellae at the species and subspecies levels by whole-cell matrix-assisted laser desorption ionization-time of flight mass spectrometry. Appl. Environ. Microbiol. 74:7767–7778. 10.1128/AEM.01402-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dieckmann R, Malorny B. 2011. Rapid screening of epidemiologically important Salmonella enterica subsp. enterica serovars by whole-cell matrix-assisted laser desorption ionization-time of flight mass spectrometry. Appl. Environ. Microbiol. 77:4136–4146. 10.1128/AEM.02418-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fagerquist CK, Miller WG, Harden LA, Bates AH, Vensel WH, Wang G, Mandrell RE. 2005. Genomic and proteomic identification of a DNA-binding protein used in the “fingerprinting” of Campylobacter species and strains by MALDI-TOF-MS protein biomarker analysis. Anal. Chem. 77:4897–4907. 10.1021/ac040193z [DOI] [PubMed] [Google Scholar]
- 32.Fagerquist CK, Bates AH, Heath S, King BC, Garbus BR, Harden LA, Miller WG. 2006. Sub-speciating Campylobacter jejuni by proteomic analysis of its protein biomarkers and their post-translational modifications. J. Proteome Res. 5:2527–2538. 10.1021/pr050485w [DOI] [PubMed] [Google Scholar]
- 33.Fagerquist CK, Garbus BR, Miller WG, Williams KE, Yee E, Bates AH, Boyle S, Harden LA, Cooley MB, Mandrell RE. 2010. Rapid identification of protein biomarkers of Escherichia coli O157:H7 by matrix-assisted laser desorption ionization-time-of-flight-time-of-flight mass spectrometry and top-down proteomics. Anal. Chem. 82:2717–2725. 10.1021/ac902455d [DOI] [PubMed] [Google Scholar]
- 34.Syrmis MW, Moser RJ, Whiley DM, Vaska V, Coombs GW, Nissen MD, Sloots TP, Nimmo GR. 2011. Comparison of a multiplexed MassARRAY system with real-time allele-specific PCR technology for genotyping of methicillin-resistant Staphylococcus aureus. Clin. Microbiol. Infect. 17:1804–1810. 10.1111/j.1469-0691.2011.03521.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.