

Abstract
Zymomonas mobilis is a bacterium that can produce ethanol by fermentation. Due to its unique metabolism and efficient ethanol production, Z. mobilis has attracted special interest for biofuel energy applications; an important area of study is the regulation of those specific metabolic pathways. Small RNAs (sRNAs) have been studied as molecules that function as transcriptional regulators in response to cellular stresses. While sRNAs have been discovered in various organisms by computational prediction and experimental approaches, their discovery in Z. mobilis has not yet been reported. In this study, we have applied transcriptome analysis and computational predictions to facilitate identification and validation of 15 novel sRNAs in Z. mobilis. We furthermore characterized their expression in the context of high and low levels of intracellular ethanol. Here, we report that 3 of the sRNAs (Zms2, Zms4, and Zms6) are differentially expressed under aerobic and anaerobic conditions, when low and high ethanol productions are observed, respectively. Importantly, when we tested the effect of ethanol stress on the expression of sRNAs in Z. mobilis, Zms2, Zms6, and Zms18 showed differential expression under 5% ethanol stress conditions. These data suggest that in this organism regulatory RNAs can be associated with metabolic functions involved in ethanol stress responses.
INTRODUCTION
High tolerance to ethanol is a desirable feature for ethanologenic strains used in industry. Given that ethanol is toxic to cells by inhibiting cell growth and metabolism, production of ethanol itself represents a bottleneck for the industrial use of biological systems (1, 2). Zymomonas mobilis is a Gram-negative bacterium that can efficiently produce ethanol from several carbon sources, including glucose, fructose, and sucrose, via the Entner-Doudoroff pathway (3). In addition, Z. mobilis maintains cell viability anaerobically when yielding high levels of ethanol (4). In fact, several reports have indicated that the presence of oxygen during fermentation affects ethanol production due to increased number of inhibitors (e.g., acetaldehyde and acetate) under aerobic conditions (5, 6). On the other hand, anaerobic growth of Z. mobilis can facilitate rapid glucose consumption with an increase in ethanol production relative to aerobic fermentation (5, 7).
Z. mobilis has a number of desirable characteristics that make it attractive as a biofuel organism (8). For instance, Z. mobilis can efficiently produce ethanol up to 12% (wt/vol) from carbohydrates at a higher rate and a 3- to 5-fold-higher yield than yeast (9). In addition to its high ethanol-producing capability with relatively low biomass, its rates of sugar uptake and processing are also high. Other advantages include the following: (i) Z. mobilis can tolerate up to 16% (wt/vol) ethanol, (ii) Z. mobilis is easy to handle for genetic manipulation and therefore amenable for developing recombinant strains with enhanced ethanol productivity, and (iii) the complete genome sequence of Z. mobilis is available for metabolic engineering (3, 10–12).
An intriguing aspect of Z. mobilis is the potential shifts in metabolism that likely occur as the organism transitions from high to low oxygen, where it is the most efficient at accumulating ethanol. In this work, we wanted to examine the potential role of regulatory small RNAs (sRNAs) in this process. These regulators are relatively short in prokaryotes (∼50 to 300 nucleotides) and are not translated (13, 14), although a possibility is that they produce small (functional or nonfunctional) peptides. Therefore, sRNAs represent a subset of noncoding RNAs that can be both activators and repressors for regulating proteins and mRNAs via a variety of mechanisms. For instance, (i) antisense sRNAs affect translation and mRNA stability of the complementary target gene, and (ii) trans-acting sRNAs regulate mRNAs by imperfect base-pairing with distal mRNA targets (15–17). sRNAs have been known to regulate various metabolic pathways under cellular stress conditions such as oxidative stress, ethanol, and temperature or pH change (18–20). When cells encounter environmental changes, regulatory sRNAs help to modulate gene expression by optimizing cellular metabolism for survival. Our motivation in this work was rooted by the ubiquitous discovery and validation of these regulatory elements in bacteria using many computational and experimental strategies (14, 21–23). Interestingly, recent data have shown higher expression of Hfq under anaerobic conditions in Z. mobilis, with higher ethanol production than under aerobic conditions (4). Hfq is a conserved bacterial Sm-like family of RNA-binding proteins, particularly in Gram-negative bacteria, that can bind sRNAs and their target mRNAs to direct functionality (24, 25). In addition, Hfq has been shown to play an important role in tolerance to multiple biomass pretreatment inhibitors such as acetate, vanillin, and furfural (26) in Z. mobilis. Collectively, these findings supported our initial hypothesis regarding the possibility that sRNAs play important mechanistic roles under differential oxygen (and thereby ethanol) conditions in this bacterium.
The study of potential sRNA regulation in the context of bacterial strains that are capable of producing and tolerating high levels of biofuels (and precursors) dates back to previous studies. For instance, small RNAs have been confirmed in Clostridium acetobutylicum, another important strain in the production of acetone and biobutanol from carbohydrates (27–29). In the case of Z. mobilis, although its genome has been completely sequenced (30), most research has focused on describing membrane composition, understanding patterns of gene expression, and characterizing lipid composition. In this study, we focused on investigating the potential presence of regulatory sRNAs in Z. mobilis. We furthermore characterized the expression of these newly uncovered RNA elements under anaerobic and aerobic conditions (known to result in differential levels of ethanol accumulation).
MATERIALS AND METHODS
Strains and culture condition.
Zymomonas mobilis ZM4 (ATCC 31821) was cultured in RM medium (glucose, 20.0 g/liter; yeast extract, 10.0 g/liter; KH2PO4, 2.0 g/liter [pH 6.0]) (55) at 30°C (pH 6.0). A single colony was inoculated into 5 ml of RM medium and cultured aerobically or anaerobically at 30°C overnight. A 1/100 of initial culture was added to 1 liter of prewarmed RM broth and then cultured overnight at 30°C with shaking at 150 rpm. The inoculum was added to each culture so that the initial optical density at 600 nm (OD600) was around 0.17. Each culture was grown aerobically or anaerobically and then collected at 13 h (late exponential/early stationary phase) or 26 h (late stationary phase) postinoculation as pH was adjusted every 4 h. The experiments were done in triplicate. For anaerobic culture, medium was nitrogen purged and tightly capped in a completely sealed flask. Cell density was measured at 600 nm using a spectrophotometer (SmartSpec Plus; Bio-Rad).
Measurement of glucose and ethanol concentrations.
Glucose concentrations were measured using the YSI 7100 multiparameter bioanalytical system (YSI Life Sciences, Yellow Springs, OH). Ethanol concentrations were measured using a UV-based ethanol assay kit (R-Biopharm, Darmstadt, Germany) according to the manufacturer's protocol.
Total RNA preparation.
Total RNA was prepared according to a protocol previously published (31) for all the growth conditions tested. Briefly, cells were grown aerobically or anaerobically and collected at 16 h after inoculation for deep sequencing. All centrifugation was performed at 4°C. Cells were pelleted and resuspended in 1 ml of TRIzol reagent (Invitrogen). Following pelleting, cells were transferred to screw-cap tubes containing glass beads (Sigma) and incubated at 25°C for 5 min. Cells were lysed using a mini-beadbeater (Biospec), with 100-s pulses three times. Cells were kept on ice for 10 min between each 100-s treatment. The beads and cellular debris were centrifuged at 4°C for 2 min. The supernatant was transferred to a clean siliconized 2-ml tube. After addition of 300 μl of chloroform-isoamyl alcohol mixture (24:1, vol/vol), the samples were inverted for 15 s and then incubated at 25°C for 3 min. Then, tubes were centrifuged at 13,000 rpm for 10 min, and the aqueous top phase was transferred to a clean siliconized 1.5-ml tube. Following this step, 270 μl of isopropanol and 270 μl of a mixture of 0.8 M sodium citrate and 1.2 M sodium chloride were added. The samples were mixed well and then incubated on ice for 10 min. The RNA was pelleted by centrifugation at 13,000 rpm for 15 min. The pellet was washed with 1 ml of 95% cold ethanol and centrifuged for 5 min. The pelleted RNA was allowed to air dry for ∼5 min and was resuspended in 30 μl of RNase-free water (Ambion). The RNA concentration was measured with a NanoDrop ND-1000 spectrophotometer (Thermo). Samples were stored at −20°C. Total RNAs were validated on 10% urea gels to verify the quality of the RNAs and make sure that the RNAs did not undergo any degradation.
Deep sequencing for RNA and data processing.
Prepared RNA was quantified and qualified using a bioanalyzer before sequencing. The NEBNext Small RNA Library Prep Set for Illumina (New England BioLabs Inc.) was used for generating small RNA libraries. Sequencing was performed using an Illumina HiSeq 2000 with paired-end 100-base reads (Genomic Sequencing and Analysis Facility at the University of Texas at Austin). Prior to analyzing sequencing results, the adapter sequences were trimmed to remove low-quality bases at the ends of the reads. Data were processed using BWA (Burrows Wheeler Aligner) (32) and mapped onto the Z. mobilis ZM4 complete genome (GenBank accession number NC_006526). The mapped sequencing reads were visualized in Integrated Genome Viewer (IGV) (33).
Computational analysis of predicted sRNA by Blast.
Sequence conservation analysis of intergenic regions was implemented using WU-BLAST (blastn 2.0MP-WashU [4 May 2006]; W. Gish, personal communication). WU BLAST output was filtered with a PERL script to a stringent threshold of at least 50% query sequence coverage with 50% identity in the conserved region. These parameters were selected according to search criteria that have been developed to analyze the conservation levels of protein-encoding sequences, where the expected level of conservation is much higher. We categorized within a genus and outside a genus for the data analysis.
Northern blotting.
Small RNA Northern blotting was performed as described in reference 31. Briefly, Northern blotting analysis was performed to verify expression of potential sRNAs candidates that resulted from computational predictions and transcriptomic analysis. DNA oligonucleotide probes specific for each candidate sRNA (see Table S1 in the supplemental material) were labeled using 20 pmol of oligonucleotide in a 20-μl kinase reaction mixture containing 25 μM [γ-32P]ATP and 20 U of T4 polynucleotide kinase (NEB) at 37°C for 1 h. A ladder (ΦX174 DNA/HinfI [Promega]) was labeled in the same manner. Total RNA (50 μg to ∼100 μg) was separated on a 10% denaturing polyacrylamide gel that was then transferred to a positively charged membrane (Hybond N+; GE Life Sciences) for blotting. Hybridization was performed using Amersham Rapid-hyb buffer (GE Healthcare) by following their recommended protocol for oligonucleotide probes, with a 3-h incubation or overnight incubation at 42°C. After three washing steps with washing buffer (5× SSC–0.1% SDS [1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate] for the 1st washing and 1× SSC–0.1% SDS for the 2nd and 3rd washing steps), membranes were exposed to a phosphor screen overnight and visualized with a phosphorimager (Typhoon imager; Amersham Biosciences).
Deep 5′ and 3′ RACE.
Deep rapid amplification of cDNA ends (RACE) was performed using total RNA samples from both aerobic and anaerobic cultures. 5′ deep RACE was performed using the Ion Torrent 316 chip (Wadsworth Center Applied Genomic Technologies Core Facility) as previously described (23, 34). Briefly, the FirstChoice RLM-RACE kit (Ambion) was used with minor modifications to the protocol. A total of 8 μg of RNA was treated with tobacco acid pyrophosphatase (TAP) at 37°C for 1 h, followed by ligation of the 5′ RACE kit adapter at 37°C for 1 h. The resulting RNA was then reverse transcribed according to the manufacturer's protocol. PCR was performed on the resulting cDNA. All primer sequences used for deep RACE are listed in Table S2 in the supplemental material. To increase the yield of some sRNAs, PCRs were reamplified using the product from the original reaction as a template and the same primers. Resulting PCR products were purified using the QIAquick PCR purification kit (Qiagen) and RNase-free water (Ambion) for final elution. All products were pooled.
For 3′ RACE, a published protocol (34) was followed, using a miScript reverse transcription kit (Qiagen) to perform reverse transcription. PCR was performed on the resulting cDNA. Resulting PCR products were purified using a QIAquick PCR purification kit (Qiagen) and eluted in RNase-free water (Ambion). Sequences of all primers used were listed (see Table S2 in the supplemental material). All products were pooled and sequenced using an Ion Torrent 316 chip at the Wadsworth Center Applied Genomic Technologies Core Facility. Data analysis was done using public resources in the Galaxy website (http://usegalaxy.org/). To analyze the sequencing results for the 5′ and 3′ RACE, adapter sequences were first removed for each sample and then sequences lacking 5′ or 3′ adapter sequences were removed. After analysis of the sequencing results, data were mapped onto the Z. mobilis ZM4 complete genome sequence (NC_006526) using Bowtie2 (35) with default parameters and visualized with IGV (33).
Nucleotide sequence accession number.
Newly determined sequencing data reported in this manuscript are available from Gene Expression Omnibus (GEO) under accession number GSE57773.
RESULTS
Transcriptome analysis of Zymomonas mobilis for identifying putative small RNAs.
A combination of computational and experimental methods was used in this work to identify novel sRNAs in Z. mobilis. First, we isolated total RNA from cells cultured under anaerobic conditions (as higher growth rates are observed under these conditions for Z. mobilis) and conducted a high-throughput transcriptome sequencing analysis using Illumina Hiseq. Prior to sequencing, RNA quantification and quality assessment were performed with a bioanalyzer. Following mapping of sequencing results to the Z. mobilis complete genome (NCBI reference sequence NC_006526.2), all hits were visualized using Integrative Genomics Viewer (IGV; http://www.broadinstitute.org/igv/). The experimental search scheme is outlined in Fig. 1A. Importantly, we identified a total of 95 candidates that mostly represented highly expressed transcripts (having over 100 mapped sequence reads; at least 10% of the average number of reads observed in tRNAs). Although we expected that slightly expressed sRNA candidates could also have an important role in regulation (36), our initial focus on highly expressed candidates stemmed from our interest in further confirming expression of these sRNAs via Northern blotting and in fully mapping the transcript ends. These sRNA characterization techniques are known to be more robust with larger sRNA quantities (37). It is worthwhile to note that in this study, we narrowed our search to intergenic sRNA candidates. Our rationale for excluding sequences that even partially overlapped known open reading frames is that intergenic candidates have a lower possibility of representing fragmented mRNA transcripts or other degradation products.
FIG 1.

Experimental scheme for sRNA candidate selection. (A) This schematic shows the strategy for selecting sRNA candidates from deep-sequencing methods. (B) Each number of candidate sRNAs from experimental and computational approaches is shown in the Venn diagram.
Computationally predicted sRNAs in Zymomonas mobilis.
As a complementary technique to sRNA identification in Z. mobilis, we used a combination of computational approaches that have proven successful in our previous work (23). Our interest in complementing our experimental search with such approaches stemmed from the fact that even though RNA sequencing is a powerful transcriptome analysis technique, it can only capture transcripts expressed during the particular experimental condition under which cells are collected for RNA preparation. It is therefore not surprising that computational predictions have also become widely used for the discovery of small regulatory RNAs in bacteria (14, 21). We performed two specific computational prediction approaches to identify novel sRNA candidates in Z. mobilis: (i) SIPHT (sRNA identification protocol using high-throughput technologies) (38) and (ii) a bioinformatics analysis recently developed in our laboratory (unpublished data) based on the search of long and conserved intergenic regions. Using SIPHT, we identified 4 novel sRNA candidates. As a side note, SIPHT predicts intergenic loci in any of the over 1,500 bacterial replicons in the NCBI database guided by sequence conservation upstream of putative Rho-independent terminators (38).
In addition to using SIPHT to identify potential sRNA targets, we performed a genome-wide BLAST conservation and size analysis of all 1,011 intergenic regions that have not been annotated to be gene encoding in Z. mobilis and predicted 20 additional candidates (see Materials and Methods for a detailed description). These predictions take advantage of sRNA enrichment trends that we have previously established in long and highly conserved regions of multiple bacterial genomes (we plan to publish the results in the future). Results from all bioinformatics studies are listed in Table S3 in the supplemental material. Collectively, 106 sRNA candidates were identified from computational analysis and experimental approaches (see Table S4 in the supplemental material).
When comparing results from our computational analysis, we found that 3 out of 4 candidates predicted by SIPHT were also identified in our bioinformatics analysis. However, only 10 of the 85 candidates that were selected from the analysis of deep sequencing data were also predicted computationally. The combined experimental and computational scheme for selecting sRNA candidates is summarized in Fig. 1A. Figure 1B shows the overlap in sRNA predictions from all methods used in this work. Strikingly, one only candidate was identified by all prediction methods; this further highlights the different pools of potential sRNA candidates that were tapped into by these methods.
High-throughput validation of sRNAs using Northern blotting.
To validate sRNA expression from the pool of all candidates, we performed large-scale Northern blotting. Cells were grown anaerobically and collected for RNA extraction in stationary phase given that Z. mobilis Hfq has been shown to be more abundantly expressed in anaerobic, stationary phase than in aerobic, stationary phase (26). As Hfq is known as a global sRNA regulator (39), we reasoned that there was a higher chance to identify (and experimentally validate) sRNAs under this condition. A list of all the probes used for Northern blotting is included in Table S1 in the supplemental material. Given that deep-sequencing data did not provide strand information, sRNAs were probed on both the plus strand and the minus strand. In addition, each candidate was probed with at least two different probes. Importantly, expression of a total of 15 candidates was confirmed with multiple probes, designed to bind different regions of the putative sRNA transcript.
Figures 2 and 3 show a summary of all the confirmed sRNAs as well as an image of the positive signal obtained by Northern blotting using their corresponding probes. Confirmed sRNAs were originally enumerated with a designated Zms (Zymomonas mobilis sRNA) nomenclature, but they were then annotated according to a published system for bacterial sRNAs (40). As indicated in Fig. 2, 12 of the confirmed sRNAs were identified from the high-throughput sequencing analysis and 3 were identified computationally; 3 sRNAs were found from both prediction methods. It is worthwhile pointing out the presence of multiple bands in some of our samples; these could represent degradation products or several transcription products from the same region. Importantly, the same patterns were observed despite the use of different probes for each region.
FIG 2.

Summary of validated sRNA candidates. Properties of Zms1 to Zms24 are shown. The approximate sRNA size observed by Northern blotting corresponds with 5′ and 3′ deep RACE results. Coordinates in bold were verified with 5′ and 3′ deep RACE. Other coordinates are from predicted coordinates from a computational search or calculated from Northern blotting. Arrows between coding genes are represented sRNAs, and the direction of arrows shows the orientation of each sRNA. All prediction methods are shown. Identified sRNAs are classified into two categories: entirely intergenic or overlapping adjacent genes.
FIG 3.

Representative Northern blots for confirmed sRNA in Z. mobilis. Northern blotting was performed to examine the expression of candidate sRNAs. Representative blots were confirmed with at least two different probes (see Table S1 in the supplemental material). A black arrowhead indicates the sRNA band for each candidate. Lane 1, ΦX174 DNA-HindIII-digested ladder; lane 2, sRNA.
Mapping of transcription start and end site by 5′ and 3′ deep RACE.
Given that high-throughput transcriptome analysis does not provide precise information of transcriptional start and end sites, we adapted deep 5′ and 3′ RACE analysis for precise mapping of transcript ends and for further confirmation of sequencing results. This method combines conventional RACE technique with deep-sequencing technology for the efficient verification of transcription start and end site in sRNA candidates (34, 41). Coordinates for the 5′ and 3′ ends of each sRNA from deep RACE analysis are shown in Fig. 2. Figure 4 shows representative data for mapping transcription start and end sites from 5′ and 3′ deep RACE for 2 selected sRNA candidates. As determined by comparing the lengths of confirmed sRNAs, the results are in agreement with previous results confirmed by Northern blotting. Mapping results of all sRNAs by deep RACE with adjacent genes are shown in Fig. S1 in the supplemental material.
FIG 4.

Representative 5′ and 3′ deep RACE data. Two representative (Zms2 and Zms6) deep RACE graphs are shown. Blue lines show the number of 5′ RACE reads mapped to respective genome, while red lines show the number of 3′ RACE reads. The black arrow under the chart shows where the sRNA is located, and the gray arrows represented the adjacent annotated coding regions.
Differential expression of sRNA candidates under different conditions of ethanol production.
To understand how expression of the confirmed sRNAs could change under different conditions of ethanol accumulation, we cultured cells under anaerobic and aerobic conditions. We pursued the analysis of all confirmed sRNAs under both aerobic and anaerobic conditions, as these could be important to basic cellular functions and to the regulation of ethanol production and/or tolerance, respectively. It has been known that the lack of oxygen positively affects glucose consumption, ethanol accumulation, and growth in Z. mobilis (4). To achieve conditions that show differential production of ethanol, Z. mobilis was grown aerobically and anaerobically. As shown in Fig. 5A, the maximal growth rates of Z. mobilis under aerobic and anaerobic conditions (estimated as 0.26 h−1 and 0.28 h−1, respectively) did not show a significant difference. In addition, we verified established trends in glucose consumption and ethanol production under these conditions. After 26 h of culture, 84.23 mM and 169.74 mM ethanol were measured under aerobic and anaerobic conditions, respectively. As shown in Fig. 5B, glucose is consumed faster under the anaerobic conditions and the corresponding production of ethanol is more rapid under anaerobic conditions. These trends were also consistent with previously published reports (4) and confirmed that the desired culturing conditions were achieved. After screening all confirmed sRNAs, one of the most interesting aspects of this work was the finding that 3 sRNAs (Zms2, Zms4, and Zms6) showed differential expression under aerobic and anaerobic culture conditions (Fig. 5C). Zms2 and Zms6 showed 0.8-fold and 0.64-fold decreases, respectively, in expression level under anaerobic culture condition. Inversely, Zms4 showed 1.5-fold-increased expression under anaerobic culture conditions. These results suggested the possibility that these sRNAs could be functionally associated with the metabolic regulation of ethanol production and/or ethanol tolerance.
FIG 5.

Growth of Z. mobilis and differential expression of sRNAs under anaerobic and aerobic conditions. (A) Growth curves under each condition are shown. Mean values ± standard deviations (bars) for triplicates are shown for each condition. The OD600 of cells was measured every 2 h. (B) Ethanol production and glucose consumption are shown. Mean values ± standard deviations (bars) for triplicates are shown for each condition. (C) The expression of sRNA for each candidate is shown with the corresponding intensity of each band detected by Northern blotting. Zms2 (∼90 bp), Zms4 (∼271 bp), and Zms6 (∼290 bp) showed differential expression between aerobic and anaerobic conditions. Band intensities were normalized based on those of tRNA.
Differential expression of sRNA candidates is responsive to environmental growth conditions.
After confirming differentially expressed sRNAs under different levels of ethanol, we next tested directly the effect of ethanol stress on the expression of all identified sRNAs. Previous work had shown that coordinated changes in expression of specific heat shock proteins and metabolic enzymes (e.g., alcohol dehydrogenase) are important under high ethanol stress (42, 43). This supported the possibility that sRNAs could also be differentially expressed as potential posttranscriptional regulators under high ethanol stress conditions. Therefore, we systematically tested expression levels of all identified sRNA candidates under the ethanol stress conditions. We chose a 5% (vol/vol) ethanol supplement to the medium as the ethanol stress conditions given that 6% (vol/vol) ethanol was previously shown to affect cell viability dramatically (44). We confirmed that Zms2, Zms6, and Zms18 showed differential expression under ethanol stress conditions (Fig. 6A). Interestingly, Zms2 and Zms6 also exhibited differential expression between aerobic and anaerobic conditions. In contrast, Zms18 only showed differential expression between 0% ethanol-supplemented growth conditions and 5% ethanol-supplemented growth conditions, indicating its potential involvement in the regulation of the ethanol tolerance in Z. mobilis. In the case of Zms4, even though it was observed to be expressed at higher levels under anaerobic conditions (relative to aerobic conditions), it was not observed to be differentially expressed between 0% and 5% ethanol stress conditions (data not shown). A plausible possibility is that Zms4 is more involved in managing oxygen stress.
FIG 6.
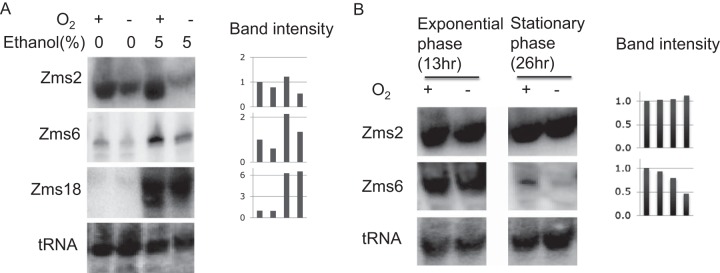
Expression patterns of sRNAs under ethanol stress and various growth conditions. (A) Addition of ethanol affects the expression levels of sRNAs. The first two lanes show results from medium with no ethanol supplementation, and last two lanes show sRNA expression under 5% ethanol supplementation. All samples were collected under 13 h (late exponential phase) after inoculation. Aerobic and anaerobic conditions are shown as O2 + and −, respectively. Band intensities were normalized based on tRNA expression levels. (B) Different growth phase samples were collected 13 h (late exponential phase) or 26 h (late stationary phase) after inoculation for Northern blotting. Band intensities were normalized based on tRNA expression levels.
Lastly, all experiments described above were done under conditions of late exponential phase. Given variations in gene expression levels that have been confirmed under different growth phases in Yersinia and Mycobacterium (45, 46), we reasoned that functional sRNAs could also be differentially expressed under different growth phases in Zymomonas. Interestingly, Hfq, which is known as an RNA chaperone in bacteria (25, 47), has been identified in Z. mobilis and showed greater expression in anaerobic stationary phase (4). To test for differential sRNA expression as a result of different growth phases, we harvested total RNA samples from cells collected at 13 h postinoculation (late exponential phase) and 26 h postinoculation (late stationary phase). Importantly, Zms2 and Zms6 also showed differential expression between early and late stationary phase (Fig. 6B). Both sRNAs accumulate until late exponential phase and then decrease in late stationary phase.
Target prediction of verified sRNAs.
As the underlying mechanism of sRNAs depends on base-pairing, many computational resources for sRNA target prediction are available. Given the importance of identifying sRNA targets for unraveling their underlying mechanistic roles, we performed an initial target prediction analysis using TargetRNA2 (http://cs.wellesley.edu/∼btjaden/TargetRNA2/) as a preliminary study. We focused on Zms2, Zms4, Zms6, and Zms18, which were differentially expressed under different stress conditions. Table 1 shows the list of target genes obtained using TargetRNA2. All listed targets have a P value of <0.001. In case of Zms2, there is an ABC (ATP-binding cassette) transporter gene in the list. Recently, an ABC transporter gene has been found as a target gene for sRNAs in Clostridium, and it could be related to antibiotic resistance (27); however, we blasted the sequence of this sRNA and that of Zms2, and there is no sequence homology (data not shown). Further analysis will focus on validating these predictions.
TABLE 1.
Genes for sRNAs from target analysis
| sRNA | Target prediction result (P < 0.001) |
|---|---|
| Zms2 | ABC transporter (ZMO0175) |
| Outer membrane autotransporter barrel domain-containing protein (ZMO0213) | |
| Hydroxylamine reductase (ZMO0117) | |
| Citrate synthase (ZMO1963) | |
| TonB-dependent receptor (ZMO0031) | |
| Zms4 | Glutaredoxin-like protein (ZMO1873) |
| CheX protein (ZMO0084) | |
| Zms6 | Chemotaxis protein CheD (ZMO0080) |
| Nitrogen fixation protein (ZMO1829) | |
| 7-Cyano-7-deazaguanine reductase (ZMO0326) | |
| Ppx/GppA phosphatase (ZMO0403) | |
| Zms18 | Cation efflux protein (ZMO0204) |
DISCUSSION
Recent research on Z. mobilis has unraveled changes in its transcriptomic and metabolic pathways associated with ethanol metabolism. In this study, we successfully discovered 15 novel sRNAs in Z. mobilis utilizing experimental and computational approaches. Although 106 candidates selected from our combinatorial methods were tested by Northern blotting, expression was confirmed for only 15 sRNAs. It is worth noting the possibility that many of the candidates, identified by transcriptomic or computational analysis, were below the detectable threshold by Northern blotting under the experimental conditions used in this study. Compared to mapped reads of tRNAs in deep sequencing, which were mapped with an average of 1,500 reads, identified sRNA candidates showed very low read numbers. This could partially explain why we detected only 14% of sRNAs by Northern blotting even though we used an excessive amount of total RNA (up to 100 μg) for detection. Despite high levels of total RNA used for testing, the intensity of some detected sRNAs is very low; this attests to the limitation of Northern blotting as an experimental tool for sRNA validation. Furthermore, total RNA, used for deep sequencing and Northern blotting, was extracted from cells only under limited physiological conditions. This limited number of growth conditions can also explain why some predicted sRNAs were not detected experimentally, since they could still be transcribed under different conditions that we have not tested.
Besides the analysis of sRNA candidates from deep-sequencing data, we also analyzed expression of mRNAs under aerobic and anaerobic conditions. When we compared our results with published transcriptomic analysis data (4), we found that most upregulated and downregulated genes under aerobic conditions were also identified in our study (see Table S5 in the supplemental material). We confirmed that Entner-Doudoroff pathway genes were more abundant under anaerobic conditions. We also observed that several transcription and response regulators are upregulated in aerobic conditions. Additionally, we detected about 200 upregulated genes and 62 downregulated genes under aerobic conditions. Most of the newly found genes in our study are related to metabolism and cellular processes. Our analysis showed that alcohol dehydrogenase, which was downregulate under ethanol treatment conditions (44), was less abundant in late exponential phase under aerobic conditions (see Table S5). We also confirmed that the Hfq gene (ZMO0347) was more abundant under anaerobic conditions. Differences in our data relative to previously published microarray data (4) (particularly in the fold changes detected) could be explained by the increase sensitivity of deep-sequencing methods and by the collection of samples under different growth phases.
It is also worthwhile to point out that although we initially selected sRNA candidates from the intergenic regions, upon confirmation by Northern blotting, some sRNAs were detected to be longer than predicted. Our 5′ and 3′ deep RACE results further confirmed that some sRNAs overlapped with 5′ or 3′ ends of adjacent genes. Thus, we categorized our identified sRNAs into two groups based on their location: intergenic sRNAs and overlapping sRNAs. Intergenic sRNAs are transcribed from intergenic regions between adjacent genes. On the other hand, overlapping sRNAs can be located at the 5′ untranslated regions (UTRs) of adjacent genes to function as riboswitches (48) or can also be generated from mRNA posttranslational processing if encoded from the 3′ end of the adjacent gene. It has been known that sRNAs can be transcribed from independent promoters or derived from processing of mRNA UTRs (49, 50). There are several pieces of evidence that the sRNAs identified in this study are not fragments of mRNAs: (i) many sRNAs are transcribed in different orientations from adjacent genes, and (ii) our several Northern blots showed no larger bands that could correspond to preprocessed mRNAs. Even though it is unlikely that any of the sRNAs we identified in this study were generated by mRNA processing, it is well established that regulatory sRNAs can be derived from processing of mRNA UTRs (51, 52).
To further characterize the uncovered sRNAs, we confirmed their expression levels under ethanol levels (5%) that have been reported to stress cell growth and decrease ethanol productivity (53). Three sRNAs (Zms2, Zms6, and Zms18) were expressed differentially under ethanol stress, suggesting that they could be related to regulatory mechanisms of ethanol production or tolerance in Z. mobilis. Analysis of comprehensive comparison with transcriptomic and proteomic data under this condition might be the next step for defining targets of sRNAs to understand regulatory mechanisms. Likewise, we uncovered in our studies two sRNAs (Zms2 and Zms6) that accumulate until late exponential phase and then decrease in late stationary phase. In Yersinia, some sRNAs showed the same pattern of expression under late exponential and stationary phase, and these differential levels of sRNA expression correlated with Hfq expression (45). Therefore, we speculate that function of these sRNAs might be Hfq dependent. Further analysis should be performed to understand the role of Hfq in Zymomonas mobilis. Lastly, it is noteworthy that preliminary target prediction analysis shows ABC transporter genes as a putative target of Zms2. However, there is no conservation in sequence of Zms2 in other organisms. Ongoing studies are focusing on elucidating the metabolic roles of Zms2 and other differentially expressed sRNAs under ethanol. As part of these future efforts, we are focusing on experimental validation of the targets identified by computational prediction methods.
This study reinforces the importance of sRNA-associated mechanisms for engineering of microbes that are relevant to the production of biofuels. Interestingly, sRNA regulation could also be exploited in the metabolic synchronization of ethanologenic organisms within consortia. This strategy is already being explored to increase levels of ethanol production involving cocultures of bacteria and yeast (54).
Supplementary Material
ACKNOWLEDGMENTS
We thank Ricardo De La Pena for his assistance with RACE sample preparation and Sang Hyun Ju for his assistance with data confirmation. We thank the Genomic Sequencing and Analysis Facility at the University of Texas at Austin for Illumina sequencing and the Wadsworth Center Applied Genomic Technologies Core Facility for Ion Torrent sequencing.
Our research was supported by funds from the NSF Career Award program (grant CBET-1254754) and Welch Foundation (grant F-1756) to L.M.C.
Footnotes
Published ahead of print 2 May 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00429-14.
REFERENCES
- 1.Osman YA, Ingram LO. 1985. Mechanism of ethanol inhibition of fermentation in Zymomonas mobilis CP4. J. Bacteriol. 164:173–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stanley D, Bandara A, Fraser S, Chambers PJ, Stanley GA. 2010. The ethanol stress response and ethanol tolerance of Saccharomyces cerevisiae. J. Appl. Microbiol. 109:13–24. 10.1111/j.1365-2672.2009.04657.x [DOI] [PubMed] [Google Scholar]
- 3.Rogers PL, Jeon YJ, Lee KJ, Lawford HG. 2007. Zymomonas mobilis for fuel ethanol and higher value products. Adv. Biochem. Eng. Biotechnol. 108:263–288. 10.1007/10_2007_060 [DOI] [PubMed] [Google Scholar]
- 4.Yang S, Tschaplinski TJ, Engle NL, Carroll SL, Martin SL, Davison BH, Palumbo AV, Rodriguez M, Jr, Brown SD. 2009. Transcriptomic and metabolomic profiling of Zymomonas mobilis during aerobic and anaerobic fermentations. BMC Genomics 10:34. 10.1186/1471-2164-10-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moreau RA, Powell MJ, Fett WF, Whitaker BD. 1997. The effect of ethanol and oxygen on the growth of Zymomonas mobilis and the levels of hopanoids and other membrane lipids. Curr. Microbiol. 35:124–128. 10.1007/s002849900224 [DOI] [PubMed] [Google Scholar]
- 6.Swings J, De Ley J. 1977. The biology of Zymomonas. Bacteriol. Rev. 41:1–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bringer S, Finn R, Sahm H. 1984. Effect of oxygen on the metabolism of Zymomonas mobilis. Arch. Microbiol. 139:376–381. 10.1007/BF00408383 [DOI] [Google Scholar]
- 8.Widiastuti H, Kim JY, Selvarasu S, Karimi IA, Kim H, Seo JS, Lee DY. 2011. Genome-scale modeling and in silico analysis of ethanologenic bacteria Zymomonas mobilis. Biotechnol. Bioeng. 108:655–665. 10.1002/bit.22965 [DOI] [PubMed] [Google Scholar]
- 9.Jeffries TW. 2005. Ethanol fermentation on the move. Nat. Biotechnol. 23:40–41. 10.1038/nbt0105-40 [DOI] [PubMed] [Google Scholar]
- 10.Carey VC, Ingram LO. 1983. Lipid composition of Zymomonas mobilis: effects of ethanol and glucose. J. Bacteriol. 154:1291–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang S, Pappas KM, Hauser LJ, Land ML, Chen GL, Hurst GB, Pan C, Kouvelis VN, Typas MA, Pelletier DA, Klingeman DM, Chang YJ, Samatova NF, Brown SD. 2009. Improved genome annotation for Zymomonas mobilis. Nat. Biotechnol. 27:893–894. 10.1038/nbt1009-893 [DOI] [PubMed] [Google Scholar]
- 12.Zhang M, Eddy C, Deanda K, Finkelstein M, Picataggio S. 1995. Metabolic engineering of a pentose metabolism pathway in ethanologenic Zymomonas mobilis. Science 267:240–243. 10.1126/science.267.5195.240 [DOI] [PubMed] [Google Scholar]
- 13.Wassarman KM. 2002. Small RNAs in bacteria: diverse regulators of gene expression in response to environmental changes. Cell 109:141–144. 10.1016/S0092-8674(02)00717-1 [DOI] [PubMed] [Google Scholar]
- 14.Livny J, Waldor MK. 2007. Identification of small RNAs in diverse bacterial species. Curr. Opin. Microbiol. 10:96–101. 10.1016/j.mib.2007.03.005 [DOI] [PubMed] [Google Scholar]
- 15.Aiba H. 2007. Mechanism of RNA silencing by Hfq-binding small RNAs. Curr. Opin. Microbiol. 10:134–139. 10.1016/j.mib.2007.03.010 [DOI] [PubMed] [Google Scholar]
- 16.Gudapaty S, Suzuki K, Wang X, Babitzke P, Romeo T. 2001. Regulatory interactions of Csr components: the RNA binding protein CsrA activates csrB transcription in Escherichia coli. J. Bacteriol. 183:6017–6027. 10.1128/JB.183.20.6017-6027.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Storz G, Vogel J, Wassarman KM. 2011. Regulation by small RNAs in bacteria: expanding frontiers. Mol. Cell 43:880–891. 10.1016/j.molcel.2011.08.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Altuvia S, Weinstein-Fischer D, Zhang A, Postow L, Storz G. 1997. A small, stable RNA induced by oxidative stress: role as a pleiotropic regulator and antimutator. Cell 90:43–53. 10.1016/S0092-8674(00)80312-8 [DOI] [PubMed] [Google Scholar]
- 19.Gottesman S, McCullen CA, Guillier M, Vanderpool CK, Majdalani N, Benhammou J, Thompson KM, FitzGerald PC, Sowa NA, FitzGerald DJ. 2006. Small RNA regulators and the bacterial response to stress. Cold Spring Harbor Symp. Quant. Biol. 71:1–11. 10.1101/sqb.2006.71.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Georg J, Hess WR. 2011. Regulatory RNAs in cyanobacteria: developmental decisions, stress responses and a plethora of chromosomally encoded cis-antisense RNAs. Biol. Chem. 392:291–297. 10.1515/BC.2011.046 [DOI] [PubMed] [Google Scholar]
- 21.Sridhar J, Gunasekaran P. 2013. Computational small RNA prediction in bacteria. Bioinform. Biol. Insights 7:83–95. 10.4137/BBI.S11213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Altuvia S. 2007. Identification of bacterial small non-coding RNAs: experimental approaches. Curr. Opin. Microbiol. 10:257–261. 10.1016/j.mib.2007.05.003 [DOI] [PubMed] [Google Scholar]
- 23.Tsai CH, Baranowski C, Livny J, McDonough KA, Wade JT, Contreras LM. 2013. Identification of novel sRNAs in mycobacterial species. PLoS One 8:e79411. 10.1371/journal.pone.0079411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vogel J, Luisi BF. 2011. Hfq and its constellation of RNA. Nat. Rev. Microbiol. 9:578–589. 10.1038/nrmicro2615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sittka A, Lucchini S, Papenfort K, Sharma CM, Rolle K, Binnewies TT, Hinton JC, Vogel J. 2008. Deep sequencing analysis of small noncoding RNA and mRNA targets of the global post-transcriptional regulator, Hfq. PLoS Genet. 4:e1000163. 10.1371/journal.pgen.1000163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang S, Pelletier DA, Lu TY, Brown SD. 2010. The Zymomonas mobilis regulator hfq contributes to tolerance against multiple lignocellulosic pretreatment inhibitors. BMC Microbiol. 10:135. 10.1186/1471-2180-10-135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen Y, Indurthi DC, Jones SW, Papoutsakis ET. 2011. Small RNAs in the genus Clostridium. mBio 2:e00340-10. 10.1128/mBio.00340-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Borden JR, Jones SW, Indurthi D, Chen Y, Terry Papoutsakis E. 2010. A genomic-library based discovery of a novel, possibly synthetic, acid-tolerance mechanism in Clostridium acetobutylicum involving non-coding RNAs and ribosomal RNA processing. Metab. Eng. 12:268–281. 10.1016/j.ymben.2009.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Venkataramanan KP, Jones SW, McCormick KP, Kunjeti SG, Ralston MT, Meyers BC, Papoutsakis ET. 2013. The Clostridium small RNome that responds to stress: the paradigm and importance of toxic metabolite stress in C. acetobutylicum. BMC Genomics 14:849. 10.1186/1471-2164-14-849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seo JS, Chong H, Park HS, Yoon KO, Jung C, Kim JJ, Hong JH, Kim H, Kim JH, Kil JI, Park CJ, Oh HM, Lee JS, Jin SJ, Um HW, Lee HJ, Oh SJ, Kim JY, Kang HL, Lee SY, Lee KJ, Kang HS. 2005. The genome sequence of the ethanologenic bacterium Zymomonas mobilis ZM4. Nat. Biotechnol. 23:63–68. 10.1038/nbt1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DiChiara JM, Contreras-Martinez LM, Livny J, Smith D, McDonough KA, Belfort M. 2010. Multiple small RNAs identified in Mycobacterium bovis BCG are also expressed in Mycobacterium tuberculosis and Mycobacterium smegmatis. Nucleic Acids Res. 38:4067–4078. 10.1093/nar/gkq101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li H, Durbin R. 2010. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26:589–595. 10.1093/bioinformatics/btp698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat. Biotechnol. 29:24–26. 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beauregard A, Smith EA, Petrone BL, Singh N, Karch C, McDonough KA, Wade JT. 2013. Identification and characterization of small RNAs in Yersinia pestis. RNA Biol. 10:397–405. 10.4161/rna.23590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9:357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gottesman S. 2005. Micros for microbes: non-coding regulatory RNAs in bacteria. Trends Genet. 21:399–404. 10.1016/j.tig.2005.05.008 [DOI] [PubMed] [Google Scholar]
- 37.Várallyay E, Burgyan J, Havelda Z. 2008. MicroRNA detection by northern blotting using locked nucleic acid probes. Nat. Protoc. 3:190–196. 10.1038/nprot.2007.528 [DOI] [PubMed] [Google Scholar]
- 38.Livny J. 2012. Bioinformatic discovery of bacterial regulatory RNAs using SIPHT. Methods Mol. Biol. 905:3–14. 10.1007/978-1-61779-949-5_1 [DOI] [PubMed] [Google Scholar]
- 39.Vogel J, Sharma CM. 2005. How to find small non-coding RNAs in bacteria. Biol. Chem. 386:1219–1238. 10.1515/BC.2005.140 [DOI] [PubMed] [Google Scholar]
- 40.Lamichhane G, Arnvig KB, McDonough KA. 2013. Definition and annotation of (myco)bacterial non-coding RNA. Tuberculosis (Edinb.) 93:26–29. 10.1016/j.tube.2012.11.010 [DOI] [PubMed] [Google Scholar]
- 41.Olivarius S, Plessy C, Carninci P. 2009. High-throughput verification of transcriptional starting sites by Deep-RACE. Biotechniques 46:130–132. 10.2144/000113066 [DOI] [PubMed] [Google Scholar]
- 42.Thanonkeo P, Sootsuwan K, Leelavacharamas V, Yamada M. 2007. Cloning and transcriptional analysis of groES and groEL in ethanol-producing bacterium Zymomonas mobilis TISTR 548. Pak. J. Biol. Sci. 10:13–22. 10.3923/pjbs.2007.13.22 [DOI] [PubMed] [Google Scholar]
- 43.An H, Scopes RK, Rodriguez M, Keshav KF, Ingram LO. 1991. Gel electrophoretic analysis of Zymomonas mobilis glycolytic and fermentative enzymes: identification of alcohol dehydrogenase II as a stress protein. J. Bacteriol. 173:5975–5982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang S, Pan C, Tschaplinski TJ, Hurst GB, Engle NL, Zhou W, Dam P, Xu Y, Rodriguez M, Jr, Dice L, Johnson CM, Davison BH, Brown SD. 2013. Systems biology analysis of Zymomonas mobilis ZM4 ethanol stress responses. PLoS One 8:e68886. 10.1371/journal.pone.0068886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koo JT, Alleyne TM, Schiano CA, Jafari N, Lathem WW. 2011. Global discovery of small RNAs in Yersinia pseudotuberculosis identifies Yersinia-specific small, noncoding RNAs required for virulence. Proc. Natl. Acad. Sci. U. S. A. 108:E709–E717. 10.1073/pnas.1101655108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.DiChiara JM, Contreras-Martinez LM, Livny J, Smith D, McDonough KA, Belfort M. 2010. Multiple small RNAs identified in Mycobacterium bovis BCG are also expressed in Mycobacterium tuberculosis and Mycobacterium smegmatis. Nucleic Acids Res. 38:4067–4078. 10.1093/nar/gkq101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang A, Wassarman KM, Rosenow C, Tjaden BC, Storz G, Gottesman S. 2003. Global analysis of small RNA and mRNA targets of Hfq. Mol. Microbiol. 50:1111–1124. 10.1046/j.1365-2958.2003.03734.x [DOI] [PubMed] [Google Scholar]
- 48.Loh E, Dussurget O, Gripenland J, Vaitkevicius K, Tiensuu T, Mandin P, Repoila F, Buchrieser C, Cossart P, Johansson J. 2009. A trans-acting riboswitch controls expression of the virulence regulator PrfA in Listeria monocytogenes. Cell 139:770–779. 10.1016/j.cell.2009.08.046 [DOI] [PubMed] [Google Scholar]
- 49.Vogel J, Bartels V, Tang TH, Churakov G, Slagter-Jager JG, Huttenhofer A, Wagner EG. 2003. RNomics in Escherichia coli detects new sRNA species and indicates parallel transcriptional output in bacteria. Nucleic Acids Res. 31:6435–6443. 10.1093/nar/gkg867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kawano M, Reynolds AA, Miranda-Rios J, Storz G. 2005. Detection of 5′- and 3′-UTR-derived small RNAs and cis-encoded antisense RNAs in Escherichia coli. Nucleic Acids Res. 33:1040–1050. 10.1093/nar/gki256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chao Y, Papenfort K, Reinhardt R, Sharma CM, Vogel J. 2012. An atlas of Hfq-bound transcripts reveals 3′ UTRs as a genomic reservoir of regulatory small RNAs. EMBO J. 31:4005–4019. 10.1038/emboj.2012.229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kawano M, Reynolds AA, Miranda-Rios J, Storz G. 2005. Detection of 5′- and 3′-UTR-derived small RNAs and cis-encoded antisense RNAs in Escherichia coli. Nucleic Acids Res. 33:1040–1050. 10.1093/nar/gki256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.He MX, Wu B, Shui ZX, Hu QC, Wang WG, Tan FR, Tang XY, Zhu QL, Pan K, Li Q, Su XH. 2012. Transcriptome profiling of Zymomonas mobilis under ethanol stress. Biotechnol. Biofuels 5:75. 10.1186/1754-6834-5-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zuroff TR, Xiques SB, Curtis WR. 2013. Consortia-mediated bioprocessing of cellulose to ethanol with a symbiotic Clostridium phytofermentans/yeast co-culture. Biotechnol. Biofuels 6:59. 10.1186/1754-6834-6-59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Goodman AE, Rogers PL, Skotnicki ML. 1982. Minimal medium for isolation of auxotrophic Zymomonas mutants. Appl. Environ. Microbiol. 44:496–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.