

Abstract
The anticoagulant activated protein C (APC) protects neurons and vascular cells from injury through its direct cytoprotective effects that are independent of its anticoagulant action. Wild-type recombinant murine APC (wt-APC) exerts significant neuroprotection in mice if administered early after traumatic brain injury (TBI). Here, we compared efficacy and safety of a late therapy for TBI with wt-APC and 3K3A-APC, an APC analog with ~80% reduced anticoagulant activity but normal cytoprotective activity, using a controlled cortical impact model of TBI. Mice received 0.8 mg/kg intraperitoneally of recombinant murine 3K3A-APC, wt-APC or saline at 6, 12, 24 and 48 h after injury. 3K3A-APC (n=15) relative to wt-APC (n=15) improved motor and sensorimotor recovery within the first three days post-trauma as demonstrated by rotarod (p<0.05) and beam balance test (p<0.05), respectively. Both, wt-APC and 3K3A-APC reduced the lesion volume seven days after injury by 36% (n=8; p<0.01) and 56% (n=8; p<0.01), respectively, compared to saline (n=8). Three days post-TBI, the hemoglobin levels in the injured brain were increased by ~3-fold after wt-APC treatment compared to saline indicating an increased risk for intracerebral bleeding. In contrast, comparable levels of brain hemoglobin in 3K3A-APC-treated and saline-treated mice suggested that 3K3A-APC treatment did not increase risk for bleeding after TBI. Thus, compared to wt-APC, 3K3A-APC is more efficacious and safer therapy for TBI with no risk for intracerebral hemorrhage.
Keywords: Activated protein C analog, Traumatic brain injury, Neuroprotection, Hemorrhage
1. Introduction
Activated Protein C (APC) is an endogenous serine protease that participates in systemic anticoagulant activity (Griffin et al., 2002; Mosnier et al., 2007). This action is mediated by highly specific proteolytic degradation of factors Va and VIIIa, with contributions from various plasma cofactors (Mosnier et al., 2007). Independent of its anticoagulant pathway, APC exerts direct vasculoprotective and neuronal-protective activities that require proteolytic activation of protease-activated receptor-1 (PAR-1) and endothelial protein C receptor (EPCR) on endothelial cells (Cheng et al., 2003, 2006) and PAR-1 and PAR-3 on mouse cortical neurons (Guo et al., 2004, 2009a; Zhong et al., 2009) as well as PAR-1 and EPCR on rat hippocampal neurons (Gorbacheva et al., 2009, 2010). Cellular effects of APC include cytoprotective alterations in gene expression profiles, anti-apoptotic and anti-inflammatory activities and stabilization of endothelial barriers (Joyce et al., 2001; Riewald et al., 2002; Cheng et al., 2003; Domotor et al., 2003; Mosnier and Griffin, 2003; Feistritzer and Riewald, 2005; Finigan et al., 2005; Cheng et al., 2006). APC protects neurons and brain endothelial cells by inhibiting both the intrinsic and extrinsic apoptotic pathways (Cheng et al., 2003, 2006; Guo et al., 2004; Liu et al., 2004; Gorbacheva et al., 2010).
APC therapy has been shown to be neuroprotective in rodent models of transient brain ischemia (Shibata et al., 2001; Cheng et al., 2003, 2006) and embolic stroke (Zlokovic et al., 2005), neonatal hypoxic/ischemic brain injury (Yesisilirmak et al., 2008), compression-induced spinal cord injury (Taoka et al., 1998), ischemic spinal cord injury (Hirose et al., 2000; Yamauchi et al., 2006), multiple sclerosis (Han et al., 2008) and amyothrophic lateral sclerosis (Zhong et al., 2009). Following cerebral ischemia, APC can also promote angiogenesis and neurogenesis independently of its neuroprotective effects (Thiyagarajan et al., 2008). By using a controlled cortical impact (CCI) model of traumatic brain injury (TBI) in mice, we have recently shown that wild-type recombinant APC (wt-APC) given immediately after trauma improved functional outcome and reduced the lesion volume (Petraglia et al., 2010).
Although our recent study demonstrated potential of APC as a therapy for TBI (Petraglia et al., 2010), the anticoagulant properties of wt-APC may carry a significant risk for intracerebral bleeding, as shown in patients with severe sepsis (Bernard et al., 2001) and in rodent models of stroke when APC was administered at later time points after ischemia (Wang et al., 2009; Guo et al., 2009a,b). In contrast to wt-APC, late post-ischemic therapy with 3K3A-APC, an APC analog with greatly reduced anticoagulant activity (Gale et al., 2002), had superior beneficial effects and no risk for intracerebral bleeding after stroke in rodents (Guo et al., 2009a; Wang et al., 2009). 3K3A-APC contains 3 alanine substitutions for 3 protease domain residues (Lysine 191–193) which reduce factor Va binding and inactivation (Gale et al., 2002), but do not affect APC exosites recognizing PAR-1 and EPCR, resulting in normal cytoprotection (Mosnier et al., 2004). In the present study, we compared efficacy and safety of a late therapy for TBI with wt-APC and 3K3A-APC.
2. Results
2.1. Motor and sensorimotor recovery following CCI
A series of tests were performed on consecutive days following CCI to evaluate functional motor recovery in all groups. In the rotarod assessment of motor activity (Hamm et al., 1994), both 3K3A-APC-treated (n=15 mice) and wt-APC-treated (n=15 mice) groups demonstrated significant improvements relative to vehicle-treated mice (n=10 mice) until post-injury day 7 when performances leveled off (p<0.05, ANOVA; Fig. 1A). This finding was consistent with a previous report using an early post-TBI treatment with wt-APC (Petraglia et al., 2010). On day 1 post-TBI, mice treated with 3K3A-APC had an average pre-injury baseline latency of 69% compared to 52% in mice treated with wt-APC, and 27% in controls, suggesting that 3K3A-APC relative to wt-APC enhances motor performance (p<0.05, Tukey post hoc test). On days 2 and 3 post-TBI, similar superior activity of 3K3A-APC was demonstrated compared to wt-APC (p<0.05, Tukey post hoc test). After day 4, 3K3A-APC normalized rotarod performance to the original baseline values, while it took until 6 and 7 days after injury for wt-APC and saline to achieve the same level of protection, respectively.
Fig. 1.
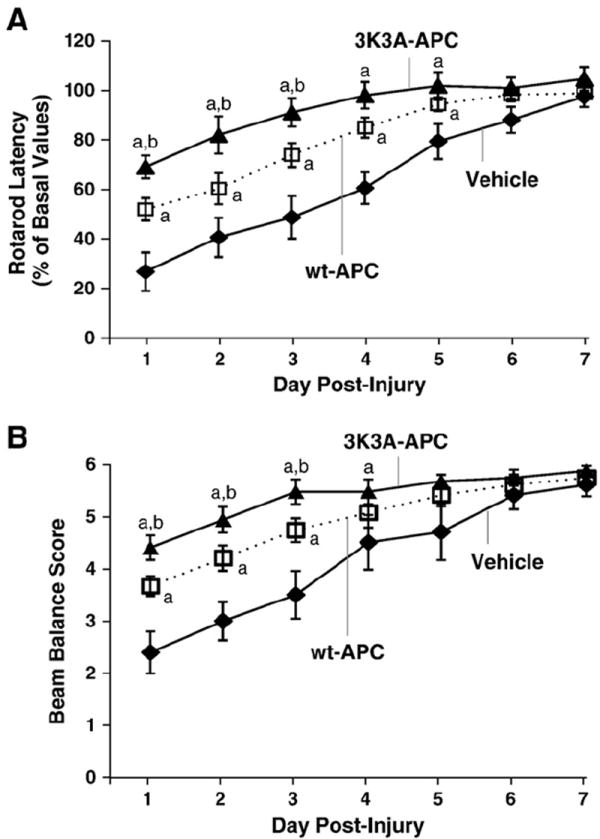
Effects of 3K3A-APC and wt-APC therapies on motor neurological and complex sensorimotor outcomes determined by rotarod neuromotor (A) and beam balance (B) tests, respectively. 3K3A-APC or wt-APC were administered at 0.8 mg/kg I.P. 6 h after injury and subsequently at 12, 24 and 48 h after injury. Control mice were treated with saline (vehicle). Data are means±S.E.M., n=15 for APC-treated mice/groups, and n=10 for saline-treated mice; ap<0.05 for 3K3A-APC or wt-APC compared to saline; bp<0.05 for 3K3A-APC vs. wt-APC, Tukey post hoc test.
Similar results were obtained on the beam balance task, when again both APC-treated groups of mice outperformed control mice throughout first three days after injury (p<0.001, ANOVA; Fig. 1B). Subgroup analysis revealed that 3K3A-APC-treated animals (n=15 mice) performed significantly better than wt-APC-treated mice (n=15 mice) over the first three days (p<0.05, Tukey post hoc test, respectively). This data suggests that 3K3A-APC provides significantly better sensorimotor recovery in the early phase after injury (Dixon et al., 1987) than wt-APC.
2.2. 3K3A-APC exerts greater neuroprotection compared to wt-APC
All mice used for behavioral testing were sacrificed on day 7. In each group, eight mice were randomly selected for the lesion volume analysis. Cresyl violet staining was performed on brain tissue sections to determine the lesion volume After 7 days, both APC-treated groups had greatly reduced lesion volume relative to controls (p<0.001, ANOVA; Fig. 2A). Specifically, 3K3A-APC lessened the lesion volume by 56% (p<0.01, Tukey post hoc test) and wt-APC by 36% (p<0.01, Tukey post hoc test). Moreover, 3K3A-APC relative to wt-APC diminished the lesion volume by 31% (p<0.05, Tukey post hoc test; Fig. 2B), thereby demonstrating a superior neuroprotection.
Fig. 2.
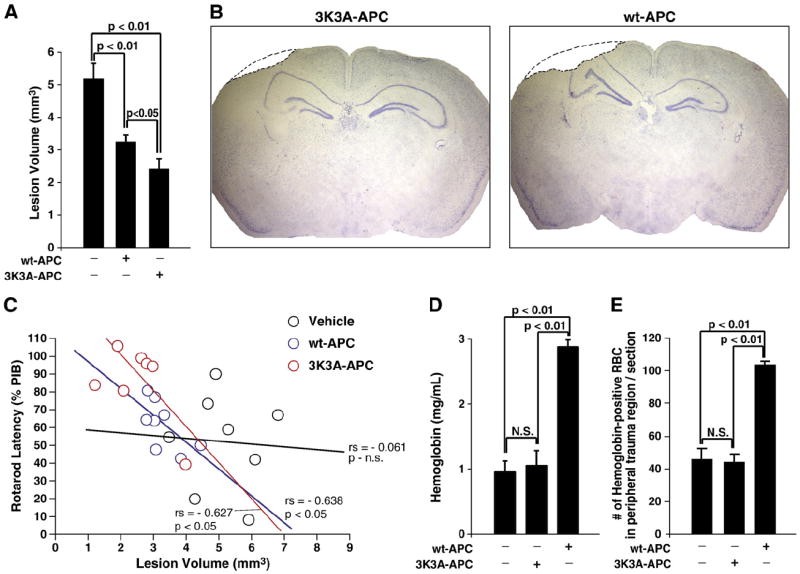
Effects of 3K3A-APC and wt-APC therapies on lesion volume at 7 days (A) and representative images of the lesions seven days after TBI in mice treated with 3K3A-APC or wt-APC (B). Regression analysis of functional outcome determined by rotarod performance at day 3 versus lesion volume at day 7. (C). A hemoglobin assay performed at 72 h post-injury demonstrates ~3-fold increase of intracerebral bleeding in wt-APC-treated mice compared to controls, whereas 3K3A-APC does not show a significant difference from the vehicle-treated controls (D). Hemoglobin immunostaining corroborates findings of the hemoglobin assay in C. Hemoglobin-positive extravasated red blood cells (RBC) were found within the peripheral trauma area only, and not in other brain regions or the contralateral hemisphere. Data are means±S.E.M., n=8 mice per group for all groups in lesion volume data; n=4 mice per group in all groups for hemoglobin assay and immunostaining. Mice were administered with 0.8 mg/kg I.P. 3K3A-APC or wt-APC or saline (vehicle) at 6, 12, 24 and 48 h after injury.
Subsequent statistical analysis was performed to compare how the lesion volumes at day 7 relate to functional recovery on day 3. Regression analysis using functional outcome on rotarod performance against lesion volumes showed no correlation between the two variables in control mice. However, there was a significant inverse relationship in wt-APC-treated and 3K3A-APC-treated group (p<0.05, Pearson’s Correlation; Fig. 2C). Moreover, the slope for 3K3A-APC treatment was slightly more negative (steeper) than that of wt-APC.
2.3. 3K3A-APC reduces bleeding risk compared to wt-APC
In a separate set of experiments (n=4 mice per group), hemoglobin levels were determined in the injured brains 72 h after injury, as described (Choudhri et al., 1997). Mice received treatment with vehicle, wt-APC or 3K3A-APC as above. In these experiments we initially selected 72 h after CCI because this was the last point after injury when a significant difference was seen between the 3K3A-APC and wt-APC on rotarod and beam balance tests. Hemoglobin levels were not significantly altered by 3K3A-APC compared to saline (Fig. 2D). However, hemoglobin levels were nearly 3 times greater after wt-APC treatment compared to 3K3A-APC treatment (p<0.01, Tukey post hoc test). Therefore, the risk for intracerebral bleeding was significantly higher in wt-APC-treated mice than in 3K3A-APC-treated mice.
Following these results, a double immunostaining for hemoglobin-positive red blood cells (RBC) and lectin-positive vascular profiles was additionally performed on tissue sections within the lesion area (n=4 mice per group) at day 7 after injury to localize where the bleeding occurred. It was found in all groups that the extravascular RBC were confined only to the peripheral trauma area surrounding immediately the cortical lesion cavity. There was no RBC extravasation in the contralateral hemisphere nor in the deeper brain regions of the ipsilateral lesioned hemisphere. This additional analysis has confirmed independently that 3K3A-APC does not increase the risk for bleeding compared to saline (Fig. 2E). However, as shown by day 3 (Fig. 2D), wt-APC-treated mice had ~2.5-fold increase in the number of exravasated RBCs in the peripheral trauma area compared to either 3K3A-APC or vehicle-treated animals (p<0.01, Tukey post hoc test; Fig. 2E).
3. Discussion
This study shows that late administration after TBI of both wt-APC and 3K3A-APC exerts beneficial effects. However, 3K3A-APC, an APC mutant with greatly reduced anticoagulant activity (Gale et al., 2002) but preserved cytoprotective activity (Mosnier et al., 2004), protected brain after TBI with superior beneficial effects compared to wt-APC. This has been demonstrated by both enhanced motor and sensorimotor performance within the first three days of injury. It should be noted that, with the current CCI model, by day 7, even control mice have returned to their basal levels of functional performance. Though significant improvement can be witnessed by treatment groups in the first 3 days with this model with 3K3A-APC, the future studies should test whether the beneficial effects of the 3K3A-APC therapy remain also in a more severe injury model which precludes a spontaneous functional recovery by day 7.
Improved neuroprotection of 3K3A-APC over wt-APC is also demonstrated by reduced lesion volume seven days after injury in a mouse model of TBI. It is of note that non-treated animals did not show any correlation between the functional outcome and the lesion volume consistent with earlier findings in this brain injury model (Petraglia et al., 2010). In contrast, wt-APC-treated or 3K3A-APC-treated mice showed a strong negative correlation indicating that better functional outcome on day 3 was predictive of the lesser volume of injury at day 7. In addition, comparable post-TBI hemoglobin levels in brains of saline-treated and 3K3A-APC-treated mice and comparable extravasations of red blood cells in the peripheral trauma area immediately surrounding the lesion cavity show that 3K3A-APC does not increase risk for intracerebral bleeding. On the other hand, late application of wt-APC increased by about 3-fold the risk for brain hemorrhage as determined 3 days post-injury and by ~2.5-fold as determined by red blood cells extravasation 7 days after injury. Moreover, the remaining bleeding noticeable 7 days after the injury was confined to the peripheral trauma region, and was, as expected, most pronounced in wt-APC group.
It has been shown that the mutations affecting the Va binding site on the protease domain of 3K3A-APC do not affect the N-terminal Gla domain of APC, which binds to EPCR on the surface of endothelial cells, and do not alter the proteolytic active site mediating the activation of PAR-1 (Mosnier et al., 2007). Therefore, both neuronal-protective and vascular-protective effects are fully preserved in 3K3A-APC mutant. In fact, recent studies using a permanent distal middle cerebral occlusion model of stroke in mice have demonstrated that 3K3A-APC has superior neuroprotection and reduced risk for bleeding (Guo et al., 2009a; Wang et al., 2009). Although, it is difficult to determine the exact contributions of direct neuroprotection and vasculoprotection versus reduced risk for bleeding in the overall beneficial effects of 3K3A-APC therapy compared to wt-APC in stroke and TBI models in vivo, it has been reported that 3K3A-APC has superior cytoprotective effects compared to wt-APC in cultured neurons challenged by N-methy-d-aspratate and brain endothelial cells subjected to oxygen/glucose deprivation (Guo et al., 2009a). The mechanism(s) for the augmented cytoprotection of 3K3A-APC versus wt-APC remains to be elucidated. In addition to the altered amino acid composition in the loop 37, different post-translational modifications of the mutant protein can occur that could change contents of negatively charged sialic acid and/or the distribution and branching of the N-linked glycan species causing an increased ability of mutant 3K3A-APC to interact with PAR-1 and/or other putative APC receptors (Wang et al., 2009; Guo et al., 2009a). Thus, enhanced neuroprotective and/or vasculoprotective direct activities may contribute to the observed protection in vivo after TBI in addition to elimination of the bleeding risk.
In summary, our findings suggest that i) late APC therapy for TBI has beneficial effects and ii) 3K3A-APC compared to wt-APC has superior neuroprotective activity resulting in significantly improved functional and neuropathological outcomes compared to the benefits established by wt-APC and no risk for increased intracerebral bleeding. The follow-up study will focus on the mechanism of action in the enhancement of cytoprotection of APC therapy after TBI.
4. Experimental procedures
4.1. Murine APC preparations
Murine protein C (PC), wild-type (wt) and 3K3A (KKK191-193AAA), stable cell lines were generated in Chinese hamster ovary (CHO) cells. The recombinant activated PCs were prepared as described (Gale et al., 2002; Mosnier et al., 2004) with modifications (Guo et al., 2009a; Wang et al., 2009). The cells were maintained in suspension in CD OptiCHO medium (Invitrogen, Carlsbad, CA) containing 2 mM CaCl2, 10 μg/ml vitamin K and 2 mM GlutaMAX (Invitrogen). For production, the cells were grown in the same medium in a 10 L Biowave bioreactor (5 L working volume) and fed with CD CHO Efficient Feed™ A (Invitrogen, Carlsbad, CA). A four-step purification procedure was used: capturing PC using FFQ resin (GE Healthcare, Piscataway, NJ), further purification of PC using Uno Q column (BioRad, Richmond, CA), activation with recombinant human thrombin (ZymoGenetics, Seattle, WA), and removal of thrombin using Uno Q column. The purity of 3K3A-APC and wt-APC were determined by reduced SDS-PAGE/silver staining. The residual level of thrombin in the purified APC preparations was ~0.018% based on thrombin time clotting assays using purified fibrinogen.
4.2. Controlled cortical impact model
The Animal Care Committee at the University of Rochester using National Institute of Health guidelines approved all procedures. As previously described (Whalen et al., 1999), CCI was performed on twelve-week-old C57Bl6 mice. Mice were anesthetized using 2% isoflurane and a 2:1 mixture of nitrous oxide and oxygen, respectively, by means of a nose cone. Under these conditions, breathing was monitored and mice were placed in the stereotaxic frame. A craniotomy was made using a portable drill and a 5-mm trephine over the left parietotemporal cortex. The resulting bone flap was removed. CCI was implemented using a pneumatic cylinder with a 3-mm flat-tip impounder, at a velocity of 6 m/s, depth of 1 mm and duration of 100 ms. All mice received multiple-dose injections at 6, 12, 24 and 48 h after injury. Control mice (n=10) received intraperitoneal (IP) saline injections at these time points, and the remaining mice were dosed (IP) with recombinant murine wt-APC (0.8 mg/kg, n=15) or murine 3K3A-APC (KKK191-193AAA) (0.8 mg/kg, n=15). It has been shown that IP administration of 0.8 mg/kg wt-APC produces a transient rise in APC circulating levels with a plateau of ~6 nM over 30 min, which was about two orders of magnitude greater than the basal levels of mouse endogenous plasma APC, and similar to APC plasma profile seen after intravenous administration of 0.2 mg/kg wt-APC via the femoral vein (Thiyagarajan et al., 2008). It has been also shown that wt-APC and its analogs with reduced anticoagulant activity have comparable arterial plasma profiles after systemic administration (Zhong et al., 2009).
4.3. Behavioral motor function evaluation
4.3.1. Rotarod
A standardized rotarod test was employed each consecutive day to assess behavioral function following CCI (Zlokovic et al., 2005; Hamm et al., 1994). All mice were trained for 5 days prior to injury, and the performance of each mouse the day before injury was recorded in order to establish a pre-injury baseline. An initial velocity of 5 rpm was used for the first 60 s, a linear increase from 5 to 10 rpm for the next 100 s, and a linear increase from 10 to 25 rpm over the time frame 160 to 240 s. Four trials were measured with each mouse and the best score was taken. Latencies for each group of mice were averaged and expressed as a percentage of their respective basal values for days 1 to 7 after injury.
4.3.2. Beam balance
The beam balance test was used to measure the more complex components of sensorimotor function and coordination for mice (Zlokovic et al., 2005). Mice were placed on a 1.0-cm wooden beam, and the ability to stabilize was rated on a scale from 0 to 6 using the scoring scheme: 0, hung on less than 20 s; 1, hung on for greater than 20 s, but less than 40 s; 2, hung on for greater than 40 s, but less than 60 s; 3, hung for at least 60 s, but hugs beam with both hind paws hanging; 4, hung for at least 60 s, but hugs beam and one hind paw hangs; 5, moved along beam with unsteady movements or slipping; 6, moved along beam with no demonstrated unsteadiness. Mice were pretrained 24 h before injury to ensure that they could balance on the beam with a steady posture for 1 min. A composite group score was calculated as the mean of these daily scores and used for analysis.
4.4. Lesion volume analysis
In each group of mice used for behavioral tests, eight mice were randomly selected 7 days after TBI for the lesion volume analysis. These mice were anesthetized with 100 mg/kg ketamine and 10 mg/kg xylazine, and subsequently transcardially perfusedwith heparin in saline. Brains were then removed and frozen in Optimal Cutting Temperature (O.C.T.) embedding media (Fisher Scientific, Houston, TX) with powdered dry ice for cryostat sectioning. Morphometric image analysis (Image-Pro Plus 4.5.1; Media Cybernetics Inc, Bethesda, MD) was used to determine the lesion volume, as reported (Wang et al., 1997). At each 0.5-mm interval, 25-μm sections were mounted on slides and stained with cresyl violet solution (FD NeuroTechnologies Inc, Baltimore, MD). The areas of the lesions were measured using image analysis (Image-Pro Plus 4.5.1) and then integrated to calculate the corresponding lesion volumes.
4.5. Hemoglobin assay
In a separate set of experiments (n=4 mice per group), hemoglobin levels were determined in the injured brain hemisphere by using a spectrophotometric assay, as previously described (Choudhri et al., 1997; Shibata et al., 2001; Cheng et al., 2006). Treatment with 3K3A-APC, wt-APC or saline was as above. Mice were sacrificed under anesthesia and transcardially perfused at 72 h. Hemisections of the ipsilateral hemisphere were homogenized and treated with Drabkin’s reagent (Sigma) to measure hemoglobin content. Purified hemoglobin (Sigma) diluted in Drabkin’s reagent was used for creating a standard curve at a wavelength of 540 nm.
4.6. Immunohistochemical staining
To localize the excess of red blood cells (RBC) extravasated from blood vessels in untreated and the two APC-treated groups, a double immunostaining for hemoglobin-positive RBC and lectin-positive brain microvessels was additionally performed at day 7 after CCI. Coronal cryostat sections (14 μm) were mounted on slides and post-fixed with 4% paraformaldehyde in phosphate-buffered saline for 10 min and then stored at −20 °C until immunostaining. Initially, sections were hydrated 3 times with Tris-buffered saline (TBS) for 5 min each rinse. Then, sections were washed with TBS containing 0.1% Triton X-100 (TBS-T). Brain sections were incubated with an antibody to hemoglobin-α (F-14) (Santa Cruz Biotechnology) overnight at 4 °C followed by incubation with the secondary anti-goat immunoglobulin G-Cy3 antibody and fluorescein-conjugated Lycopersicon esculentum (tomato) lectin (Vector Laboratories). Secondary antibodies were incubated for 75 min at 37 °C. Slides were imaged using an Olympus AX70 epifluorescent microscope coupled to a Mercury Short ARC HBO light source passed through green fluorescent protein and rhodamine excitation fluorescent cubes for lectin and hemoglobin immunodetection, respectively. All images were acquired using an Olypmus UPlan Apo 40× objective.
At day 7 most of the lesion was resolved forming a cavity and all extravascular RBC were localized to the peripheral trauma area. The numbers or extravasated RBC per section were determined typically from 3 to 4 tissue sections within the lesion area separated by 500 μm from each other and the numbers per animal per section averaged. It is of note, the extravascular RBC were not found on brain sections on the contralateral hemisphere nor in the deeper brain regions of the ipsilateral lesioned hemisphere, and were confined exclusively to the immediate area surrounding the cortical lesion void.
4.7. Statistical analysis
All data are presented as the mean and standard error of the mean. Analysis of variance (ANOVA) for repeated measures was used to determine statistically significant differences for all behavioral tests. ANOVA followed by the Tukey post hoc test was used for all subgroup comparisons in Microsoft Excel. Differences were considered statistically significant if p <0.05. Pearson’s correlation was used for regressions analysis of functional performance versus lesion volume using statistical software (GraphPad Prism 3.02).
Acknowledgments
We are thankful to Dr. M. Thiyagarajan for assistance in the hemoglobin level studies. We thank Dayle Schweibert for technical assistance with the WAVE bioreactor.
This work is supported by the National Institute of Health grants HL63290 and HL81528 and ZZ Biotech.
Abbreviations
- TBI
traumatic brain injury
- APC
activated protein C
- PC
protein C
- PAR-1
protease-activated receptor-1
- EPCR
endothelial protein C receptor
- BBB
blood brain barrier
- CCI
controlled cortical impact
- wt
wild-type
- IP
intraperitoneal
- PIB
pre-injury baseline
- ANOVA
analysis of variance
References
- Bernard GR, Ely EW, Wright TJ, Fraiz J, Stasek JE, Jr, Russell JA, Mayers I, Rosenfeld BA, Morris PE, Yan SB, Helterbrand JD. Safety and dose relationship of recombinant human activated protein C for coagulopathy in severe sepsis. Crit Care Med. 2001;29(11):2051–2059. doi: 10.1097/00003246-200111000-00003. [DOI] [PubMed] [Google Scholar]
- Cheng T, Liu D, Griffin JH, Fernandez JA, Castellino F, Rosen ED, Fukudome K, Zlokovic BV. Activated protein C blocks p53-mediated apoptosis in ischemic human brain endothelium and is neuroprotective. Nat Med. 2003;9:338–342. doi: 10.1038/nm826. [DOI] [PubMed] [Google Scholar]
- Cheng T, Petraglia AL, Li Z, Thiyagarajan M, Zhong Z, Wu Z, Liu D, Maggirwar SB, Deane R, Fernandez JA, LaRue B, Griffin JH, Chopp M, Zlokovic BV. Activated protein C inhibits tissue plasminogen activator-induced brain hemorrhage. Nat Med. 2006;12:1278–1285. doi: 10.1038/nm1498. [DOI] [PubMed] [Google Scholar]
- Choudhri TF, Hoh BL, Solomon RA, Connolly ES, Jr, Pinsky DJ. Use of a spectrophotometric hemoglobin assay to objectively quantify intracerebral hemorrhage in mice. Stroke. 1997;28(11):2296–2302. doi: 10.1161/01.str.28.11.2296. [DOI] [PubMed] [Google Scholar]
- Dixon CE, Lyeth BG, Povlishock JT. A fluid percussion model of experimental brain injury in the rat. J Neurosurg. 1987;67:110–119. doi: 10.3171/jns.1987.67.1.0110. [DOI] [PubMed] [Google Scholar]
- Domotor E, Benzakour O, Griffin JH, Yule D, Fukudome K, Zlokovic BV. Activated protein C alters cytosolic calcium flux in human brain endothelium via binding to endothelial protein C receptor and activation of protease activated receptor-1. Blood. 2003;101:4797–4801. doi: 10.1182/blood-2002-12-3680. [DOI] [PubMed] [Google Scholar]
- Feistritzer C, Riewald M. Endothelial barrier protection by activated protein C through PAR1-dependent sphingosine 1-phosphate receptor-1 crossactivation. Blood. 2005;105:3178–3184. doi: 10.1182/blood-2004-10-3985. [DOI] [PubMed] [Google Scholar]
- Finigan JH, Dudek SM, Singleton PA, Chiang ET, Jacobson JR, Camp SM, Ye SQ, Garcia JG. Activated protein C mediates novel lung endothelial barrier enhancement: role of sphingosine 1-phosphate receptor transactivation. J Biol Chem. 2005;280:17286–17293. doi: 10.1074/jbc.M412427200. [DOI] [PubMed] [Google Scholar]
- Gale AJ, Tsavaler A, Griffin JH. Molecular characterization of an extended binding site for coagulation factor Va in the positive exosite of activated protein C. J Biol Chem. 2002;277(32):28836–28840. doi: 10.1074/jbc.M204363200. [DOI] [PubMed] [Google Scholar]
- Griffin JH, Zlokovic BV, Fernandez JA. Activated protein C: potential therapy for severe sepsis, thrombosis, and stroke. Semin Hematol. 2002;39(3):197–205. doi: 10.1053/shem.2002.34093. [DOI] [PubMed] [Google Scholar]
- Gorbacheva L, Davidova O, Sokolova E, Ishiwata S, Pinelis V, Strukova S, Reiser G. Endothelial protein C receptor is expressed in rat cortical and hippocampal neurons and is necessary for protective effect of activated protein C at glutamate excitotoxicity. J Neurochem. 2009;111(4):967–975. doi: 10.1111/j.1471-4159.2009.06380.x. [DOI] [PubMed] [Google Scholar]
- Gorbacheva L, Pinelis V, Ishiwata S, Strukova S, Reiser G. Activated protein C prevents glutamate- and thrombin-induced activation of nuclear factor-kappaB in cultured hippocampal neurons. Neuroscience. 2010;165(4):1138–1146. doi: 10.1016/j.neuroscience.2009.11.027. [DOI] [PubMed] [Google Scholar]
- Guo H, Liu D, Gelbard H, Cheng T, Insalaco R, Fernandez JA, Griffin JH, Zlokovic BV. Activated protein C prevents neuronal apoptosis via protease activated receptors 1 and 3. Neuron. 2004;41:563–572. doi: 10.1016/s0896-6273(04)00019-4. [DOI] [PubMed] [Google Scholar]
- Guo H, Singh I, Wang W, Deane R, Barrett T, Fernandez JA, Chow N, Griffin JH, Zlokovic BV. Neuroprotective activities of activated protein C mutant with reduced anticoagulant activity. Eur J Neurosci. 2009a;29(6):1119–1139. doi: 10.1111/j.1460-9568.2009.06664.x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Guo H, Wang Y, Singh I, Liu D, Fernandez JA, Griffin JH, Chow N, Zlokovic BV. Species-dependent neuroprotection by activated protein C mutants with reduced anticoagulant activity. J Neurochem. 2009b;109(1):116–124. doi: 10.1111/j.1471-4159.2009.05921.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamm RJ, Pike BR, O’Dell DM, Lyeth BG, Jenkins LW. The rotarod test: an evaluation of its effectiveness in assessing motor deficits following traumatic brain injury. J Neurotrauma. 1994;11(2):187–196. doi: 10.1089/neu.1994.11.187. [DOI] [PubMed] [Google Scholar]
- Han MH, Hwang SI, Roy DB, Lundgren DH, Price JV, Ousman SS, Fernald GH, Gerlitz B, Robinson WH, Baranzini SE, Grinnell BW, Raine CS, Sobel RA, Han DK, Steinman L. Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature. 2008;451(7182):1076–1081. doi: 10.1038/nature06559. [DOI] [PubMed] [Google Scholar]
- Hirose K, Okajima K, Taoka Y, Uchiba M, Tagami H, Nakano K, Utoh J, Okabe H, Kitamura N. Activated protein C reduces the ischemia/reperfusion-induced spinal cord injury in rats by inhibiting neutrophil activation. Ann Surg. 2000;232(2):272–280. doi: 10.1097/00000658-200008000-00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce DE, Gelbert L, Ciaccia A, DeHoff B, Grinnell BW. Gene expression profile of antithrombotic protein c defines new mechanisms modulating inflammation and apoptosis. J Biol Chem. 2001;276:11199–11203. doi: 10.1074/jbc.C100017200. [DOI] [PubMed] [Google Scholar]
- Liu D, Cheng T, Guo H, Fernandez JA, Griffin JH, Song X, Zlokovic BV. Tissue plasminogen activator neurovascular toxicity is controlled by activated protein C. Nat Med. 2004;10:1379–1383. doi: 10.1038/nm1122. [DOI] [PubMed] [Google Scholar]
- Mosnier LO, Griffin JH. Inhibition of staurosporine-induced apoptosis of endothelial cells by activated protein C requires protease-activated receptor-1 and endothelial cell protein C receptor. Biochem J. 2003;373:65–70. doi: 10.1042/BJ20030341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosnier LO, Gale AJ, Yegneswaran S, Griffin JH. Activated protein C variants with normal cytoprotective but reduced anticoagulant activity. Blood. 2004;104:1740–1744. doi: 10.1182/blood-2004-01-0110. [DOI] [PubMed] [Google Scholar]
- Mosnier LO, Zlokovic BV, Griffin JH. The cytoprotective protein C pathway. Blood. 2007;109(8):3161–3172. doi: 10.1182/blood-2006-09-003004. [DOI] [PubMed] [Google Scholar]
- Petraglia AL, Marky AH, Walker CT, Thiyagarajan M, Zlokovic BV. Activated protein C is neuroprotective and mediates new blood vessel formation and neurogenesis after controlled cortical impact. Neurosurgery. 2010;66(1):165–171. doi: 10.1227/01.NEU.0000363148.49779.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riewald M, Petrovan RJ, Donner A, Mueller BM, Ruf W. Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science. 2002;296(5574):1880–1882. doi: 10.1126/science.1071699. [DOI] [PubMed] [Google Scholar]
- Shibata M, Kumar SR, Amar A, Fernandez JA, Hofman F, Griffin JH, Zlokovic BV. Anti-inflammatory, antithrombotic, and neuroprotective effects of activated protein C in a murine model of focal ischemic stroke. Circulation. 2001;103:1799–1805. doi: 10.1161/01.cir.103.13.1799. [DOI] [PubMed] [Google Scholar]
- Taoka Y, Okajima K, Uchiba M, Murakami K, Harada N, Johno M, Naruo M. Activated protein C reduces the severity of compression-induced spinal cord injury in rats by inhibiting activation of leukocytes. J Neurosci. 1998;18(4):1393–1398. doi: 10.1523/JNEUROSCI.18-04-01393.1998. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Thiyagarajan M, Fernandez JA, Lane SM, Griffin JH, Zlokovic BV. Activated protein C promotes neovascularization and neurogenesis in postischemic brain via protease-activated receptor 1. J Neurosci. 2008;28:12788–12797. doi: 10.1523/JNEUROSCI.3485-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi T, Sakurai M, Abe K, Takano H, Sawa Y. Neuroprotective effects of activated protein C through induction of insulin-like growth factor-1 (IGF-1), IGF-1 receptor, and its downstream signal phosphorylated serine-threonine kinase after spinal cord ischemia in rabbits. Stroke. 2006;37(4):1081–1086. doi: 10.1161/01.STR.0000206280.30972.21. [DOI] [PubMed] [Google Scholar]
- Yesilirmak DC, Kumral A, Tugyan K, Cilaker S, Baskin H, Yilmaz O, Duman N, Ozkan H. Effects of activated protein C on neonatal hypoxic ischemic brain injury. Brain Res. 2008;1210:56–62. doi: 10.1016/j.brainres.2008.02.088. [DOI] [PubMed] [Google Scholar]
- Wang L, Kittaka M, Sun N, Schreiber SS, Zlokovic BV. Chronic nicotine treatment enhances focal ischemic brain injury and depletes free pool of brain microvascular tissue plasminogen activator in rats. J Cereb Blood Flow Metab. 1997;17(2):136–146. doi: 10.1097/00004647-199702000-00002. [DOI] [PubMed] [Google Scholar]
- Wang Y, Thiyagarajan M, Chow N, Singh I, Guo H, Davis TP, Zlokovic BV. Differential neuroprotection and risk for bleeding from activated protein C with varying degrees of anticoagulant activity. Stroke. 2009;40(5):1864–1869. doi: 10.1161/STROKEAHA.108.536680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalen MJ, Carlos TM, Dixon CE, Schiding JK, Clark RS, Baum E, Yan HQ, Marion DW, Kochanek PM. Effect of traumatic brain injury in mice deficient in intercellular adhesion molecule-1: assessment of histopathologic and functional outcome. J Neurotrauma. 1999;16(4):299–309. doi: 10.1089/neu.1999.16.299. [DOI] [PubMed] [Google Scholar]
- Zhong Z, Ilieva H, Hallagan L, Bell R, Singh I, Paquette N, Thiyagarajan M, Deane R, Fernandez JA, Lane S, Zlokovic AB, Liu T, Griffin JH, Chow N, Castellino FJ, Stojanovic K, Cleveland DW, Zlokovic BV. Activated protein C therapy slows ALS-like disease in mice by transcriptionally inhibiting SOD1 in motor neurons and microglia cells. J Clin Invest. 2009;119(11):3437–3449. doi: 10.1172/JCI38476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV, Zhang C, Liu D, Fernandez J, Griffin JH, Chopp M. Functional recovery after embolic stroke in rodents by activated protein C. Ann Neurol. 2005;58:474–477. doi: 10.1002/ana.20602. [DOI] [PubMed] [Google Scholar]