

 Anion-induced conformational switching provides insight into binding selectivity in a series of heteroaryl-urea receptors.
Anion-induced conformational switching provides insight into binding selectivity in a series of heteroaryl-urea receptors.
Abstract
Anion binding studies of 1,10-phenanthroline- and 2-pyridyl-substituted urea-based receptors reveal that guest-dependent conformations exist in structural variants related to a previously investigated bipyridyl-based receptor. Dynamic conformational switching persists in a monofunctional pyridyl-urea receptor, and the preorganization provided by a phenanthroline-based analogue promotes convergence of anion coordinating groups to a single guest. Despite this predisposition for anion coordination, the conformational flexibility of the bipyridyl-based receptor provides the most selective motif for H2PO4 – coordination. Furthermore, the two new phenanthrolyl- and pyridyl-receptors serve as models of the bipyridyl-based receptor, elucidating accurate stepwise association constants for 1 : 2 host/guest binding by this receptor, and suggest that oxoanions prefer the embrace of a “U” conformation in 1 : 1 complexes.
Introduction
Molecular switches and foldamers, particularly systems influenced by ionic stimuli, represent a growing area of supramolecular chemistry research.1 The non-covalent interactions between conformationally flexible sensing molecules and their ionic targets are often characterized by multiple competing equilibria and conformational states.2 A thorough understanding of these types of guest-induced molecular transformations is necessary for the improvement and practical implementation of molecular switches and stimuli-responsive hosts.2b
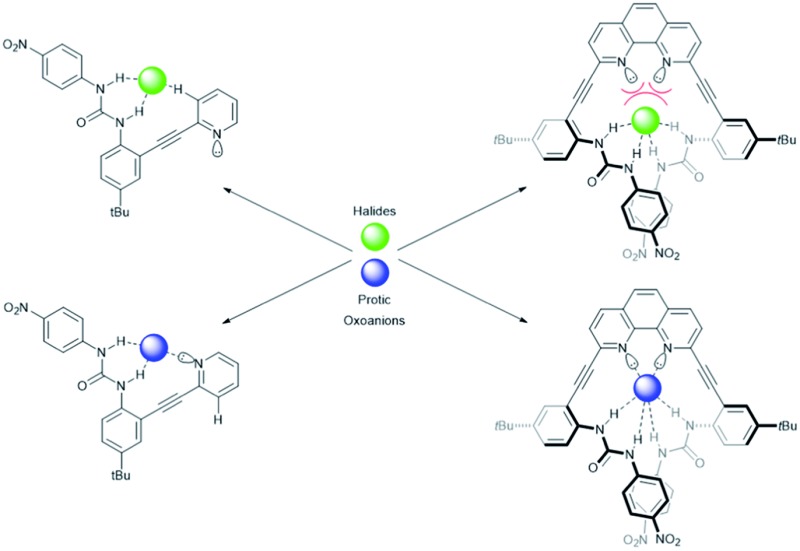
Recently, we have shown that a bipyridyl bisurea-based anion receptor (1, Fig. 1) is capable of adopting differing anion-dependent binding conformations due to limited rotation about the bipyridine bridging bond and unrestricted rotation of the arene–alkyne bonds.3 The conformational freedom in this system allows anionic guests the opportunity to experience a variety of different hydrogen bonding environments including those from bipyridyl nitrogens, ureas and even aryl C–Hs. Aryl C–H hydrogen bond donors have recently been shown to contribute significantly to anion binding in similar systems.4 As a result of the many available interactions with a potential guest, the bipyridine-based receptor demonstrated spectroscopically distinct conformational binding tendencies toward halides and oxoanions. The different anion-dependent, bound-states were shown to be reversible upon changing the anionic guest in excess from Cl– to HSO4 – and back.
Fig. 1. Receptor structures 1–3.

The conformational promiscuity and potential of receptor 1 to bind ditopically to two guests inspired further exploration into the nature of the conformational switching of this system. Herein we present the binding studies of structural analogues 2 and 3 (Fig. 1) and compare their effectiveness as anion receptors to 1. We hypothesized that the 1,10-phenanthroline receptor 2 (more conformationally restricted with the bipyridyl unit locked into planarity) and 2-pyridyl receptor 3 (“uncoupled” half of receptor 1) could offer insight into the nature of the guest-dependent binding conformations and possibly deconvolute the previously observed higher-ordered anion binding stoichiometry of 1. Receptor 2 is anticipated to interact with anions via either a U or W conformation (Fig. 2) where the U conformation is nearly identical to an anion-bound conformation observed for 1, and the W invokes coordination via an aryl C–H hydrogen bond, similar to the Z conformation proposed for 1.3 An S-type conformation is not possible for 2 due to the rigidity of the phenanthroline core. The U conformation is anticipated to be the dominant conformation observed for 2 given the preorganization of the receptor that directs the heteroaromatic phenanthroline nitrogens in a syn orientation, and places the urea groups on the same side of the heteroaromatic core promoting their simultaneous interaction with anionic guests.
Fig. 2. Representation of possible 1 : 1 host/guest binding conformations for receptors 1–3. Blue hexagons represent aromatic rings and the red wedges represent nitrogen atoms in the heteroarenes.

Pyridine-based receptor 3 presents a single urea binding unit capable of coordinating anions in two different L-shaped conformations, denoted L-N (pyridine N directed at the guest) or L-CH (pyridine CH directed at guest) differentiated by rotating freely around the pyridine-alkyne single bond (Fig. 2). This receptor permits analysis of anion binding restricted to a single urea moiety while also investigating the effect of aryl C–H hydrogen bonding (observed previously in the binding of 1 with halides) versus interaction with the basic pyridine nitrogen (favoured for protic oxoanions). Additionally, this mono-urea fragment of receptors 1 and 2 will help model the conformational and thermodynamic properties of the bisurea-based systems and help determine how important preorganization of the urea groups is to binding within the pocket of the receptor.
The synthesis of 2 and 3 (Scheme 1) is loosely based on strategies utilized for related aryl-acetylene receptor systems.5–10 Sonogashira cross-coupling of 2-ethynyl-4-t-butylaniline11 to either 2,9-diiodo-1,10-phenanthroline12 or 2-bromopyridine generates aniline precursors 4 and 5 in 74% and 89% yield, respectively. Subsequent reaction with 4-nitrophenylisocyanate produces receptors 2 and 3 in 40% and 72% yield, respectively. Full synthetic details are provided in the ESI.†
Scheme 1. Synthesis of receptors 2 and 3. Reagents and conditions: (a) 2-bromopyridine, Pd(PPh3)4, CuI, iPr2NH, THF, 25 °C, 4 h; (b) 1,10-diiodophenanthroline (0.5 equiv.), Pd(PPh3)4, CuI, iPr2NH, THF, 25 °C, 16 h; (c) 4-nitrophenylisocyante, toluene, 25 °C, 16 h.

Results and discussion
X-ray analysis
Single crystals of 2 and 3 were grown from slow evaporation of CHCl3–MeOH or CH2Cl2–MeOH solutions containing tetrabutyl-ammonium salts of the anionic guests.‡ Diffraction of the resultant crystals yielded representative free base structures of 2·(MeOH)2 (Fig. 3a) and 3·H2O (Fig. 3b). For 2, each phenanthroline nitrogen atom hydrogen bonds to the protic MeOH hydrogen atom. The MeOH oxygen atom in turn accepts hydrogen bonds from the urea NH units in either arm. A similar style of hydrogen bonding is portrayed by the adventitious water molecule in the structure of 3. Unfortunately, no anions co-crystallized with the receptor molecules under these conditions. The urea-bound solvent molecules do provide evidence for the hydrogen bond-accepting nature of the heteroaromatic nitrogens, and support the U and L-N binding conformations for 2 and 3 in the presence of protic guests.
Fig. 3. ORTEP representations of (a) 2·(MeOH)2 (left) top view and (right) side view, and (b) 3·H2O. Ellipsoids set at 50% probability. Non-H-bonding hydrogens were omitted for clarity, as was the disordered CH2Cl2 solvent molecule in 2. Gray = carbon, white = hydrogen, blue = nitrogen and red = oxygen.

Binding studies
Anion binding studies of 2 and 3 were undertaken using various halides and oxoanions as the tetrabutylammonium salts in either 10% DMSO–CHCl3 or the perdeutero-equivalent. The resulting association constants (K a) were obtained by fitting either 1H NMR or UV/Vis titration curves using the Hyperquad 2006 suite13,14 of non-linear curve fitting software (Table 1). In all cases NMR titration data were obtained by following the downfield shift of the urea resonances upon addition of guests. All binding values are the result of fitting titration data to a 1 : 1 binding model, which is consistent with Job's method of continuous variation.15
Table 1. Association constants (K a, M–1) of 2 and 3 fit to a 1 : 1 binding model.
| Anion | 1 | 2 | 3 |
| Cl– | — | (2.60 ± 0.45) × 102 | (4.0 ± 0.2) × 101 |
| Br– | — | (6.0 ± 0.1) × 101 | (5.0 ± 0.6) × 101 |
| I– | — | — c | (6.0 ± 0.4) × 101 |
| H2PO4 – | (7.80 ± 2.0) × 104 a | (4.60 ± 0.0045) × 104 b | (3.10 ± 0.32) × 103 |
| HSO4 – | — | (3.30 ± 0.50) × 102 | (1.00 ± 0.09) × 102 |
| OAc– | — | (2.60 ± 0.23) × 103 d | (2.30 ± 0.34) × 103 |
aValue previously reported.
bAssociation constant obtained from UV/Vis titrations.
cEvidence exists for binding, but reliable association constants could not be determined.
dRepresents an apparent association constant obtained from fitting to a 1 : 1 model; however, a 1 : 2 model gave considerably better fits, although high errors and negative K a values prevented report of this binding data and indicates that neither model appropriately reflects the solution stoichiometry. Association constants represent an average of at least three titrations with various anions added as the tetrabutylammonium salts in 10% DMSO–CHCl3 (or the perdeutero-equivalent for NMR titrations) at 298 K, and were determined using HypNMR 2006 or Hyperquad 2006. Table 3 contains revised values for K a's with receptor 1 based on the refitting previous data using receptors 2 and 3 as model systems.
Overall the phenanthroline-based receptor 2 has a greater affinity for the tested anions compared to receptor 3 which is unsurprising given that 2 contains two ureas with the propensity to converge on the guest molecules. Several interesting trends become apparent upon a more in-depth comparison of binding trends for 2 and 3. For instance, there are stark differences in the relative affinity toward halides for the two receptors. Receptor 2 has the greatest affinity for Cl– and binding drops off considerably for Br–. Weak binding was observed for I–, but accurate association constants could not be determined for 2. Receptor 3 shows little discrimination between the halides, though there may be a slight preference for I– over Cl–.
Receptors 2 and 3 both show a preference for oxoanions over halides, and H2PO4 – yields the highest association constants though the preference exhibited by 3 is minimal. Dihydrogen phosphate induces significant downfield shifting of the urea resonances in 2, but broadening of the resonances (also observed previously with 1·H2PO4 –) necessitated UV/Vis titrations for determination of accurate association constants. The results of these studies indicate that receptor 2 binds H2PO4 – over an order of magnitude better than the more basic OAc– which is comparable to other flexible hydrogen bonding receptors.1g,h Given the comparable magnitude of binding and selectivity for H2PO4 – by the similarly structured receptors 1 and 2, it is likely they coordinate via similar conformations. The similar binding conformation observed in these receptors is a testament to the multiple complementary hydrogen bond donors and acceptors afforded by the U conformation. Despite the similarities, 2 still has 1.7-fold lower association constant for H2PO4 – than the previously reported 1·H2PO4 – complex. The preorganization of 2 was anticipated to provide a greater preference for H2PO4 –, but these studies suggest that conformational flexibility is more beneficial than preorganization in these systems. The extreme preference for H2PO4 – over OAc– is not shared by simplified receptor 3, and both anions are bound similarly. Curiously, the association constants of 2 and 3 for OAc– are within error of one another. A possible explanation is the presence of more complicated and higher order stoichiometry binding of 2 and OAc–, but fitting of the binding data to an alternate model (e.g. 1 : 2 host/guest model) produced unsatisfactory association constants with either negative values or larger errors than the 1 : 1 model preventing confirmation of this theory.
Conformational analysis
The differences in the observed binding trends of 2 and 3 are likely attributed to the different modes of coordination that these receptors display toward the same guests. Inspection of the 1H NMR spectra of a titration of 3 with chloride (Fig. 4b) shows a significant downfield shift of the Hd′′ resonance (Δδ ≈ 0.6 ppm) indicating the formation of a weak aryl C–H hydrogen bond. This observation is analogous to what was observed for Cl– binding to 1. Analogous shifts were observed for Br– and I– but to a lesser extent. This demonstrates that 3 coordinates halides primarily in the L-CH conformation (akin to 1 binding halides in the Z conformation). Similar inspection of the 1H NMR spectra of a titration of 2 with Cl– shows a much smaller shift (Δδ ≈ 0.2 ppm) in the analogous hydrogen bonding aryl resonance (Hc′) which would suggest that the aryl C–H hydrogen bond is not being utilized to the same extent in this case, and the U conformation dominates (Fig. 4a). The 6.5-fold greater affinity for Cl– by 2 over 3 also supports coordination of multiple ureas to the anion in a U conformation. While the relative change in the chloride-induced downfield chemical shift (Δδ) of Hc′ is lower than for Hd′′, it is still evidence that a noticeable amount of the receptor is adopting the W conformation in binding Cl–. The resistance of Cl– to bind in the U conformation likely stems from a conflict between maximizing hydrogen bonding donors in the U conformation and minimizing repulsive interactions with the nitrogen lone pairs in the W conformation. This may also explain why halide binding for 2 is drastically reduced as the halide ionic radii increase.
Fig. 4. Stacked partial 1H NMR spectra and proposed binding conformations for (a) 2·Cl– and (b) 3·Cl–. Equiv. of guest at left of spectra.

The 1H NMR titration spectra for 2 and 3 with HSO4 – confirm this assertion (Fig. 5), showing little to no downfield shifting of Hc′ and Hd′′, respectively. The lack of shifting of the aryl C–H resonances indicates they are not involved in hydrogen bonding to HSO4 – in either case. Since the aryl C–H hydrogen bonding is not observed in any of the receptor oxoanion systems it is believed that the conformational preference exhibited by these receptors is a result of the ureas being directed toward the heteroaryl nitrogens, which allows for an additional advantageous hydrogen bond between the protic anions and the free base pyridyl lone pair in the U and L-N conformation for 2 and 3, respectively. The overall results of these analyses demonstrate that anion-dependent binding conformations are a trait represented by these receptors as a class even when the system is as simple as a single pyridine and urea unit (3). Additionally, preorganization of the receptor system (2) can influence the anion driven conformational outputs.
Fig. 5. Stacked partial 1H NMR spectra and proposed binding conformations for (a) 2·HSO4 – and (b) 3·HSO4 –. Equiv. of guest at left of spectra.

DFT calculations
DFT calculations were performed for each receptor:anion complex in each of the proposed binding conformations. Geometry optimizations were performed using the ωB97X-D method and 6-31G(d,p) basis set in the presence of a DMSO solvation model field.16 Host/guest complexes were placed in the approximate representative conformations and allowed to find a local minimum. Energy calculations were determined using the same level of theory (ωB97X-D/6-31G(d,p)) with the application of a DMSO solvation model field, and energy values (kcal mol–1) are displayed in Table 2.
Table 2. Calculated binding energies (kcal mol–1) of anion complexes of 2 and 3 in the likely binding conformations.
| Receptor | Conformation | Anion |
|||
| Cl– | H2PO4 – | HSO4 – | OAc– | ||
| 2 | W a | –13.9 | –25.6 | –18.7 | –32.7 |
| U a | –20.1 | –56.2 | –40.3 | –48.3 | |
| 3 | L-CH a | –13.7 | –27.1 | –20.5 | –31.1 |
| L-N a | –12.5 | –35.0 | –29.1 | –32.5 | |
aΔE = E(complex) – E(receptor) – E(anion).
The binding energies for all of the anionic complexes with 2 indicate that guest coordination is preferred in a U conformation where both urea groups can simultaneously bind guests (Fig. 6a and b), and the anion stability trend in this conformation is H2PO4 – > OAc– > HSO4 – > Cl– which is consistent with 1H NMR data. The calculated structures of 3 demonstrate that the protic anions also prefer interaction with the free base pyridyl nitrogen (L-N conformation) versus the aryl C–H (L-CH conformation) (Fig. 6d). Chloride binding is stabilized most by the L-CH conformation, which avoids a repulsive interaction with the pyridyl lone pair and allows for formation of an opportunistic C–H aryl hydrogen bond. Again, the binding energies of the most stable conformer of 3 are identical to those from the solution binding studies. Overall, the computational studies mirror the findings from NMR experiments: the heteroaryl nitrogen directs oxoanions toward the binding pocket while diverting halides to settle with the weak C–H hydrogen bond.
Fig. 6. Calculated structures (ωB97X-D/6-31G(d,p)) of preferred binding conformations for (a) 2·Cl–, (b) 2·HSO4 –, (c) 3·Cl– and (d) 3·HSO4 –.

Conformational intermediates
The conformationally flexible receptor 1 demonstrates interesting anion-dependent switching, but this effect has also complicated accurate assessment of the stepwise association constants for this system.3 This makes determination of K 11 and K 11⇌12 involved in the overall association constant, K 12, of the likely formed 1 : 2 host/guest complexes difficult. The binding studies of 2 and 3 offer the potential to shed light on some of the steps in the many possible equilibria in this complex system (Scheme 2). The various equilibria for such a ditopic system are related to one another by K m (microscopic association constant), α (cooperativity factor), and statistical correction factors and/or EM (effective molarity).17,18
Scheme 2. Some of the representative equilibria involved in the formation of the 1·(anion)2 complex described by the overall association constant (K 12) and stepwise association constants (K 11 and K 11⇌12). These macroscopic association constants are related to each other by K m (microscopic association constant), α (cooperativity factor), statistical correction factors and/or EM (effective molarity).

The microscopic association constant (K m) represents the binding of a guest with a single binding site. Since 3 showed a similar differential coordination motif for halides versus oxoanions compared to 1 this allows the binding constants for complexes of 3 to be representative of K m in systems of 1 believed to form 1 : 2 host/guest complexes (e.g. Cl–, Br–, HSO4 – and OAc–).19 If it is assumed that there is no cooperativity between the two structurally equivalent binding sites of 1 (e.g. α = 1), the overall association constant (K 12) and stepwise constant K 11( Z / S ) of binding in either a Z or S conformation can be calculated (Table 3). Using this calculated K 12 value as a constant, a more realistic approximation of the K 11 value was determined by re-fitting the binding data for 1 (Table 3). One thing that stands out is that the values from the re-fitting process (K 11) are noticeably larger than the K 11( Z / S ) values calculated from the statistical relationship (2K m). In the case of HSO4 – and OAc– the larger K 11 values could be explained by the formation of a weak intramolecular chelate complex with the receptor which would be anticipated for binding these anions in a more U-like conformation for 1 : 1 complexes. Assuming that the energetic differences between 1 and 2 in adopting the U conformation are negligible, association constants for 2 may serve as reasonable estimates for the formation of 1 : 1 complexes of 1·anion in the U conformation (K 11( 2 ), Scheme 2). Comparison of K 11 for HSO4 – with the K a for 2·HSO4 – (a representative of binding in the U conformation) produces very similar values which suggests the 1·HSO4 – complex is more U-like in its binding conformation.
Table 3. Revised association constants for 1 from previously obtained data using receptors 2 and 3 as model systems.
| Anions | Macroscopic association constants |
||
| K 11 (M–1) a | K 11( Z / S ) (M–1) b , d | K 12 (M–1) c , d | |
| Cl– | (2.30 ± 0.06) × 102 | 8.0 × 101 | 1.59 × 103 |
| Br– | (1.50 ± 0.06) × 102 | 1.00 × 102 | 1.59 × 103 |
| HSO4 – | (3.70 ± 0.06) × 102 | 2.00 × 102 | 1.00 × 104 |
| OAc– | (2.11 ± 0.002) × 104 | 4.60 × 103 | 5.25 × 106 |
aValues determined from re-fitting of titration data to a stepwise 1 : 2 host/guest model with the K 12 value held constant.
bValues calculated from expected statistically corrected equilibrium relationship (K ( Z / S ) = 2K m).
cValues calculated from expected statistically corrected equilibrium relationship (K 12 = αK m 2) and assumed α = 1.
dValues for K m were assumed to be equal to the determined association constant for 3 with the corresponding anion.
The K 11 for 1 toward OAc– is much higher than 2·OAc– (Table 1), but these differences might be a result of the unreliable stoichiometry for 2·OAc–. These assertions cannot be definitively confirmed without the determination of the EM value which is not presently viable. The calculated and re-fit 1 : 1 values, K 11 and K 11( Z / S ) respectively, for Br– on the other hand are quite similar, and seem to confirm that binding in the Z conformation is the most dominant form. Gratifyingly, this was previously indicated by 1H NMR shifting of the aryl C–H resonance. The re-fit K 11 value for Cl– is significantly higher than the anticipated binding constant for chloride binding strictly in the Z conformation (K 11( Z / S )), and is similar to the value for 2·Cl–. The different shifting profiles of 2·Cl– and 1·Cl– (Fig. S38†) would indicate that 2·Cl– is in a U conformation and 1·Cl– is in a Z conformation. The previously reported 1H NMR spectra for the titration of 1 with Cl– indicated similar involvement of the urea and aryl C–H hydrogens in the coordination of Cl– throughout the titration. These observations make it difficult to imagine an intramolecular chelate complex forming that involves the aryl C–H hydrogen bond. The larger K 11 value may instead indicate some form of intermolecular complex though no direct evidence yet exists for this in solution. While using binding data from 2 and 3 have yielded more acceptable binding values for 1 and a reasonable assessment of the 1·HSO4 – binding conformation, discrepancies with other complexes indicate that either more complicated equilibria are present or that cooperativity cannot be neglected in these systems.
Conclusions
We have demonstrated through binding studies of pyridyl- and phenanthroline-containing, urea-based anion-binding receptors, that the same anion influenced conformational binding that was observed for previously reported receptor 1 also exists for the presented receptors as well. This seems to represent a common coordination motif for this class of receptor when compared to the previously reported bipyridine-containing receptor. In all cases the ureas represent the dominant anion binding unit, but the conformational flexibility is dependent on supporting intramolecular interactions. The presence of halides induces a heteroaryl C–H hydrogen bond to form between the binding halide and the pyridyl proton ortho to the alkyne group. This conformation seems to be driven by the repulsive interaction with the heteroaromatic nitrogen lone pair and suggests a possible new “streamlined” design for receptors targeting oxoanions. Even when the Z conformation is eliminated (as in 2) and a conformation exists with the two ureas convergent and in close proximity to the nitrogen lone pair, as in 2·Cl–, a clear resistance to the U conformation still exists. Protic oxoanions on the other hand neglect the assistance of any heteroaryl C–H hydrogen bonding interactions presumably as a result of favorable hydrogen bond donation to the nitrogen lone pair and adopt a U conformation. The dichotomous conformational binding observed demonstrates the potential of this receptor motif to direct conformational changes via anionic stimuli.
Additionally, in comparison of the binding studies of 2 and 3 to 1, more reliable binding data for the stepwise formation of 1 : 2 host/guest complexes of 1 for several anions were able to be determined. These comparisons also suggest that oxoanions, particularly HSO4 –, seem to prefer a 1 : 1 host/guest complex in a U conformation en route to higher ordered complexes. This comparison unfortunately does not clarify the binding conformation of 1·Cl–, and in fact contradicts previously acquired solid state and solution phase binding data. Ultimately, these studies have shed light on the anion-induced conformational preferences of an interesting class of receptors, and the insights gained from these systems can be extended toward the improved design of anion affected supramolecular switches and foldamer systems.
Acknowledgments
This research was supported by the NIH (GM087398), which also funded early stage intellectual property that was licensed by SupraSensor Technologies, a company co-founded by the PIs. We gratefully acknowledge the use of UO CAMCOR facilities, which have been purchased with a combination of federal and state funding.
Footnotes
‡Crystallographic data for: 2·(MeOH)2: (C50H50N8O6)·2(CH3OH)·0.69(CH2Cl2), M = 972.99, 0.16 × 0.11 × 0.03 mm, T = 100(2) K, CuKα radiation λ = 1.54178 Å, triclinic, space group P1, a = 13.5061(5) Å, b = 13.6726(5) Å, c = 15.5153(5) Å, α = 112.007(2)°, β = 103.362(2)°, γ = 102.907(2)°, V = 2427.06(15) Å3, Z = 2, D c = 1.331 Mg m–3, μ = 1.409 mm–1, F(000) = 1021, 2θ max = 135.82°, 23032 reflections, 8453 independent reflections [R int = 0.0381], R 1 = 0.0589, wR 2 = 0.1677 and GOF = 1.048 for 8453 reflections (849 parameters) with I > 2σ(I), R 1 = 0.0751, wR 2 = 0.1833 and GOF = 1.048 for all reflections, max/min residual electron density +1.252/–0.481 eÅ3. 3·H2O: C24H24N4O4, M = 432.47, 0.38 × 0.21 × 0.10 mm, T = 193(2) K, MoKα radiation λ = 0.71073 Å, monoclinic, space group P21/n, a = 12.6483(16) Å, b = 6.9763(9) Å, c = 25.125(3) Å, α = γ = 90, β = 101.115(2)°, V = 2175.4(5) Å3, Z = 4, D c = 1.320 Mg m–3, μ = 0.092 mm–1, F(000) = 912, 2θ max = 54.00°, 23515 reflections, 4739 independent reflections [R int = 0.0282], R 1 = 0.0487, wR 2 = 0.1339 and GOF = 1.011 for 4739 reflections (385 parameters) with I > 2σ(I), R 1 = 0.0635, wR 2 = 0.1503 and GOF = 1.011 for all reflections, max/min residual electron density +0.383/–0.170 eÅ3.
References
- (a) Zhang M., Zhu K. and Huang F., in Supramolecular Chemistry: From Molecules to Nanomaterials, ed. P. A. Gale and J. W. Steed, John Wiley and Sons, 2012, pp. 2425–2496. [Google Scholar]; (b) Hua Y., Liu Y., Chen C.-H., Flood A. H. J. Am. Chem. Soc. 2013;135:14401–14412. doi: 10.1021/ja4074744. [DOI] [PubMed] [Google Scholar]; (c) Jeong K.-S., Chae M. K., Suk J.-m. Pure Appl. Chem. 2012;84:953–964. [Google Scholar]; (d) Juwarker H., Suk J.-m., Jeong K.-S. Chem. Soc. Rev. 2009;38:3316–3325. doi: 10.1039/b909034g. [DOI] [PubMed] [Google Scholar]; (e) Wu B., Jia C., Wang X., Li S., Huang X., Yang X.-J. Org. Lett. 2012;14:684–687. doi: 10.1021/ol2031153. [DOI] [PubMed] [Google Scholar]; (f) Juwarker H., Jeong K.-S. Chem. Soc. Rev. 2010;39:3664–3674. doi: 10.1039/b926162c. [DOI] [PubMed] [Google Scholar]; (g) Suk J.-m., Kim J.-i., Jeong K.-S. Chem. – Asian J. 2011;6:1992–1995. doi: 10.1002/asia.201100087. [DOI] [PubMed] [Google Scholar]; (h) Cao L., Jiang R., Zhu Y., Wang X., Li Y., Li Y. Eur. J. Org. Chem. 2014:2687–2693. [Google Scholar]
- (a) Steed J. W. and Atwood J. L., Supramolecular Chemistry, John Wiley and Sons, 2nd edn, 2009. [Google Scholar]; (b) Clegg J. K., Cremers J., Hogben A. J., Breiner B., Smulders M. M. J., Thoburn J. D., Nitschke J. R. Chem. Sci. 2013;4:68–76. [Google Scholar]
- Gavette J. V., Mills N. S., Zakharov L. N., Johnson C. A., Johnson D. W., Haley M. M. Angew. Chem., Int. Ed. 2013;52:10270–10274. doi: 10.1002/anie.201302929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tresca B. W., Zakharov L. N., Carroll C. N., Johnson D. W., Haley M. M. Chem. Commun. 2013;49:7240–7242. doi: 10.1039/c3cc44574g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson C. A., Berryman O. B., Sather A. C., Zakharov L. N., Haley M. M., Johnson D. W. Cryst. Growth Des. 2009;9:4247–4249. [Google Scholar]
- Carroll C. N., Coombs B. A., McClintock S. P., Johnson II C. A., Berryman O. B., Johnson D. W., Haley M. M. Chem. Commun. 2011;47:5539–5541. doi: 10.1039/c1cc10947b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll C. N., Berryman O. B., Johnson C. A., Zakharov L. N., Haley M. M., Johnson D. W. Chem. Commun. 2009:2520–2522. doi: 10.1039/b901643k. [DOI] [PubMed] [Google Scholar]
- Watt M. M., Zakharov L. N., Haley M. M., Johnson D. W. Angew. Chem., Int. Ed. 2013;52:10275–10280. doi: 10.1002/anie.201303881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berryman O. B., Johnson C. A., Zakharov L. N., Haley M. M., Johnson D. W. Angew. Chem., Int. Ed. 2008;47:117–120. doi: 10.1002/anie.200703971. [DOI] [PubMed] [Google Scholar]
- Engle J. M., Carroll C. N., Johnson D. W., Haley M. M. Chem. Sci. 2012;3:1105–1110. doi: 10.1039/C2SC00975G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan W. B., Haley M. M. J. Org. Chem. 2001;66:3893–3901. doi: 10.1021/jo010183n. [DOI] [PubMed] [Google Scholar]
- Guo H. C., Zheng R. H., Jiang H. J. Org. Prep. Proced. Int. 2012;44:392–396. [Google Scholar]
- Frassineti C., Ghelli S., Gans P., Sabatini A., Moruzzi M. S., Vacca A. Anal. Biochem. 1995;231:374–382. doi: 10.1006/abio.1995.9984. [DOI] [PubMed] [Google Scholar]
- Gans P., Sabatini A., Vacca A. Talanta. 1996;43:1739–1753. doi: 10.1016/0039-9140(96)01958-3. [DOI] [PubMed] [Google Scholar]
- Hirose K. J. Inclusion Phenom. Macrocyclic Chem. 2001;39:193–209. [Google Scholar]
- Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Mennucci B., Petersson G. A., Nakatsuji H., Caricato M., Li X., Hratchian H. P., Izmaylov A. F., Bloino J., Zheng G., Sonnenberg J. L., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Mongomery Jr J. A., Peralta J. E., Ogliaro F., Bearpark M., Heyd J. J., Brothers E., Kudin N., Staroverov V. N., Keith T., Kobayashi R., Normand J., Raghavachari K., Rendell A., Burant J. C., Iyengar S. S., Tomasi J., Cossi M., Rega N., Millam J. M., Klene M., Knox J. E., Cross J. B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R. E., Yazyev O., Austin A. J., Cammi R., Pomelli C., Ochterski J. W., Martin R. L., Morokuma K., Zakrzewski V. G., Voth G. A., Salvador P., Dannenberg J. J., Dapprich S., Daniels A. D., Farkas O., Foresman J. B., Ortiz J. V., Cioslowski J. and Fox D. J., Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2010.
- Ballester P., Costa A., Castilla A. M., Deya P. M., Frontera A., Gomila R. M., Hunter C. A. Chem.–Eur. J. 2005;11:2196–2206. doi: 10.1002/chem.200400772. [DOI] [PubMed] [Google Scholar]
- Sun H., Hunter C. A., Navarro C., Turega S. J. Am. Chem. Soc. 2013;135:13129–13141. doi: 10.1021/ja406235d. [DOI] [PubMed] [Google Scholar]
- Only 1 : 1 complexes of 1·H2PO4 – were observed
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.