

Abstract
Increased activity of lung epithelial sodium channels (ENaCs) contributes to the pathophysiology of cystic fibrosis (CF) by increasing the rate of epithelial lining fluid reabsorption. Inter-α-inhibitor (IαI), a serum protease inhibitor, may decrease ENaC activity by preventing its cleavage by serine proteases. High concentrations of IαI were detected in the bronchoalveolar lavage fluid (BALF) of children with CF and lower airway diseases. IαI decreased amiloride-sensitive (IENaC) but not cAMP-activated Cl− currents across confluent monolayers of rat ATII, and mouse nasal epithelial cells grew in primary culture by 45 and 25%, respectively. Changes in IENaC by IαI in ATII cells were accompanied by increased levels of uncleaved (immature) surface α-ENaC. IαI increased airway surface liquid depth overlying murine nasal epithelial cells to the same extent as amiloride, consistent with ENaC inhibition. Incubation of lung slices from C57BL/6, those lacking phenylalanine at position 508 (∆F508), or CF transmembrane conductance regulator knockout mice with IαI for 3 hours decreased the open probability of their ENaC channels by 50%. ∆F508 mice had considerably higher levels the amiloride-sensitive fractions of ENaC nasal potential difference (ENaC-NPD) than wild-type littermates and only background levels of IαI in their BALF. A single intranasal instillation of IαI decreased their ENaC-NPD 24 hours later by 25%. In conclusion, we show that IαI is present in the BALF of children with CF, is an effective inhibitor of ENaC proteolysis, and decreases ENaC activity in lung epithelial cells of ∆F508 mice.
Keywords: alveolar type II cells, lung slices, human BAL, cystic fibrosis, biotinylation
Epithelial sodium (Na+) channels (ENaCs) are present at the apical membranes of epithelial cells and play a major role in the regulation of water movement, plasma osmolality, blood pressure, and Na+ homeostasis (1, 2). In the lung, ENaCs play an important role in maintaining the composition and the thickness of the airway surface liquid (3). ENaCs are formed by the assembly of at least three subunits, α, β, and γ (2), although lung epithelial cells are known to contain δENaCs as well (4). Because of its critical role, ENaC activity is tightly regulated at many levels (gene expression, synthesis, assembly, degradation, and trafficking to and from the plasma membrane) by complex mechanisms. At the plasma membrane, ENaC function is controlled by a finely tuned equilibrium between proteolytic activation (5, 6) and ubiquitination/internalization followed by degradation (7–9). Two membrane pools of ENaCs are known to be present in epithelial surfaces: an active, cleaved type, which transports Na+, and an uncleaved type, which does not transport Na+ but acts as a depot for the active ENaC.
Inter-α-inhibitor (IαI) is a serum protease inhibitor consisting of three polypeptides (10, 11). The two heavy chains of the IαI molecule (which can be H1, H2, or H3), sized between 75 and 90 kD, have similar primary structures. The third so-called light chain, also known as bikunin, is of smaller size and molecular weight (25 kD) and confers the protease inhibitory properties. The three polypeptides are covalently linked by ester bonds to a chondroitin sulfate backbone chain (11). This complex, with a molecular weight of 225 kD, is produced by hepatocytes and released into the blood stream, where it reaches concentrations as high as 0.5 mg/ml (12, 13). IαI covalently binds the glycosaminoglycan hyaluronan (HA) (14), and in the lung its presence is necessary for HA-mediated airway hyperresponsiveness after exposure to oxidants such as ozone (15). However, IαI is also present in the bronchoalveolar lavage fluid (BALF) after lung injury (16). Thus, its presence could confer benefit to patients with cystic fibrosis (CF) by inhibiting ENaC activity and consequently the Na+ hyperabsorption associated with a lack of functional CF transmembrane conductance regulator (CFTR) (17).
We designed a series of experiments to show the presence of IαI in BALF of children with CF. Subsequently, we measured the extent of IαI inhibition of amiloride-sensitive currents across Xenopus oocytes expressing human ENaCs and confluent monolayers of rodent ATII and nasal epithelial cells. We then incubated thinly cut lung slices from C57BL/6 mice and those lacking phenylalanine at position 508 (∆F508) with IαI and measured single-channel activity in ATII cells in situ. Finally, because of our previous studies demonstrating increased ENaC activity in ∆F508 mice (17), we instilled IαI intranasally in ∆F508 mice and measured the amiloride-sensitive components of nasal potential difference (ENaC-NPD) as an index of Na+ transport across the nasal airway epithelium. Taken together, our findings suggest that IαI plays a protective role by limiting the degree of Na+ hyperabsorption in wild-type and murine models of CF.
Materials and Methods
Additional details are provided in the online supplement.
Human Subjects and Bronchoalveolar Lavage
BALF was obtained from children undergoing clinically indicated bronchoscopy at the Children’s Hospital at the University of North Carolina, Chapel Hill, North Carolina. The Institutional Review Board approved collection of excess BALF. All parents signed consent, and assent was obtained in children old enough to read.
Animals
Cftr knockout mice (B6.129P2-Cftrtm1Unc/J; Cftr(−/−)) (18) and Cftr-∆F508 mice (B6.129S6-Cftrtm1Kth/J; ∆F508) (19) backcrossed extensively to C57BL/6 mice to make them congenic were provided by the University of Alabama at Birmingham Cystic Fibrosis Center Mouse Genetics Core Facility. All procedures involving animals were approved by the University of Alabama at Birmingham Institutional Animal Care and Use Committee.
Measurement of IαI and HA Concentrations in Human BALF
IαI in BALF were determined using a competitive ELISA as previously described (20). HA concentrations were determined using a competitive ELISA (Echelon, San Jose, CA).
Recording of Single-Channel Currents from ATII Cells in Lung Slices
ATII cells in lung slices were patched in the cell-attached mode, and Na+ single-channel activity was recorded after incubation of slices with vehicle or IαI (0.1 mg/ml) for 3 hours as previously described (17, 21).
Short-Circuit Currents across ATII Cells
Confluent monolayers of rat ATII cells (17) or murine (C57BL/6) nasal epithelial cells (22) were incubated with IαI (0.1 mg/ml) in 100 μl of Normal Ringers added on their apical side for 2 or 4 hours. Cells were mounted in Ussing chambers, and short-circuit currents (Isc) and transepithelial resistances were measured before and after addition of amiloride (10 μM), forskolin (10 μM), and glibenclamide (400 μM) in the apical compartments (21, 23, 24).
Surface Biotinylation and Western Blotting
Rat ATII cells were treated with IαI (100 μg/ml) or vehicle for 4 hours. They were rinsed with cold PBS and incubated with 1 mg/ml of EZ-Link Sulfo-NHS-SS-biotin (Pierce) in PBS (pH 8.0) for 30 minutes (25). The biotinylation reaction was quenched with 50 mM TRIS for 5 minutes, and the cells were washed again with PBS and lysed in RIPA buffer containing protease inhibitor cocktail (Roche). One milligram of lysate was incubated with 50 μl of packed streptavidin–agarose (Novagen) overnight at 4°C. After incubation, the streptavidin–agarose beads were extensively washed with lysis buffer. Bound proteins were eluted with Laemmle 2× sample buffer and separated by 10% SDS-PAGE gels (Bio-Rad). Proteins were then transferred to polyvinylidene fluoride membranes and immunoblotted with an antibody against α-ENaCs (Thermo Scientific, Rockford, IL). Proteins were visualized using chemiluminescence. Specific binding was assessed by immunostaining with the antibody against α-ENaCs in the presence of the immunizing peptide (21).
NPD
NPDs were measured following the method of Knowles and colleagues (26) and as previously described (24) 4 or 24 hours after intranasal instillation of 25 μl of lactated Ringer’s solution with IαI (100 μg/ml) or alone in each nostril.
Statistics
One-way ANOVA and the Bonferroni t test, adjusted for multiple comparisons, were used to determine differences among group means. Differences at P < 0.05 were considered significant.
Results
IαI and HA Concentrations Increased in BALF of Pediatric Patients with CF and in Patients with Pneumonia
BALF from 36 children with CF and 17 children with lower airway diseases (recurrent pneumonia, asthma, cough, and two patients with primary ciliary dyskinesia) were included. Indications for bronchoscopy in CF were mainly for exacerbations failing oral or intravenous antibiotics in nonexpectorating children, new detection of Pseudomonas aeruginosa infection (n = 6), and surveillance bronchoscopy at the time of another surgery (n = 3). As such, a majority of subjects were on antibiotics before bronchoscopy, but these were held at least 48 hours prior to the procedure in all but two subjects. Further clinical and BALF data are shown in Table E1 in the online supplement. The percentage of polymorphonuclear leukocytes in BALF was significantly higher in patients with CF versus patients without CF (67 ± 3.6 vs. 34 ± 8.1; X ± 1 SE; P = 0.002 with Mann-Whitney test; n = 36 and 17, respectively)
Because IαI accumulates in sites of inflammation (27), we investigated whether IαI concentrations were increased in the BALF of these patients. IαI values in patients with CF (41 ± 4.2 μg/ml; n = 36) were significantly higher than in patients withut CF (11.3 ± 51 μg/ml [n = 17 samples]; X ± 1 SE; P < 0.0001; Mann Whitney test) (Figure 1A). There was a linear correlation between IαI and number of inflammatory cells in the BALF (R2 = 0.49) for patients with CF (Figure 1B). Patients with CF also had significantly higher levels of HA in their BALF as compared with those without CF (15.6 ± 1.7 [n = 36 samples] vs. 4.1 ± 2.3 [n = 17 samples]; X ± 1 SEM;P = 0.0002 with Mann-Whitney test). There was a significant correlation among IαI and HA concentrations in the BALF (Figure 1C).
Figure 1.

Children with cystic fibrosis (CF) have increased concentrations of inter-α-inhibitor (IαI) in their bronchoalveolar lavage fluid (BALF). (A) Samples were stratified according to the primary diagnosis (CF = cystic fibrosis; Non-CF = lower airway disease). IαI was measured in the cell-free lavage as described in Materials and Methods. Box whisker plots showing individual data points, boxes, whiskers (2 SD from the mean), means, and standard errors (*P = 0.0.0026 with Mann-Whitney test; n = 16 for Non-CF and n = 36 for CF). (B) IαI concentrations versus log10 (number of inflammatory cells) in the BALF for patients with CF and non-CF subjects. The straight line represents best line of fit for the CF samples (IαI = 30.43 × log (cells) of cells −150; R = 0.7; P < 0.001). (C) Hyaluronan (HA) versus IαI in the BALF. The straight line represents best line of fit for all patients (R2 = 0.6; P < 0.05).
IαI Inhibits ENaCs Heterologously Expressed in Xenopus Oocytes
Perfusion of Xenopus oocytes expressing human α-, β-, and γ-ENaCs with ND96 containing 0.1 mg/ml IαI under voltage clamp conditions led to a time-dependent inhibition of whole-cell Na+ current (INa) (Figure 2A). The mean rate constant (t) of decay, calculated by fitting the INa with a mono-exponential decay equation [y = A × e(−INa/t) + B] (28), was 18.0 ± 0.85 minutes (X ± SEM; n = 17). Addition of trypsin (2 μM) in the perfusion medium reversed the inhibitory effects of IαI on INa by most likely activating silent Na+ channels at their plasma membranes (Figure 2A). Subsequently, oocytes were incubated with IαI (0.1 mg/ml) or medium (ND96) for 60 minutes, and current–voltage (I–V) relationships were obtained in the absence and presence of amiloride (10 μM) in the perfusion medium. Amiloride-sensitive currents (IENaC,; indicative of the component of Na+ currents due to ENaC) (Figure 2B) exhibited inwardly rectified currents, which were inhibited largely by IαI. On the other hand, IαI did not alter the reversal potential, suggesting that the ENaC permeability ratio of Na+/K+ was not affected. No change of IENaC currents was seen when ENaC-expressing oocytes were incubated with IαI and an antibody against IαI for 60 minutes (Figure 2C).
Figure 2.

IαI inhibits human epithelial sodium channel (ENaC) expressed in Xenopus oocytes. (A) Time course of inward Na+ currents (INa+) across Xenopus oocytes injected with cRNAs encoding wild-type α-, β-, and γ- human-ENaC (8.4 ng each). Addition of amiloride (10 μM) in the perfusion medium decreased INa+ from −40 to −5 μA. Perfusion with amiloride-free medium (ND96) restored INa+ to −40 μA. Perfusion with ND96 containing IαI (0.1 mg/ml) resulted in a time-dependent decrease of INa+. Addition of trypsin (2 μM) restored INa+ to its initial value. Results are from a typical experiment, which was repeated at least five times. (B) Oocytes were incubated with 0.1 mg/ml IαI or vehicle (ND96) for 60 minutes. Current voltage curves for amiloride-sensitive currents (IENaC) were calculated as described in Materials and Methods. Values are means ± SE; n (number of oocytes from 3 or 4 experiments) = 21 for vehicle and 19 for IαI. (C) ENaC-expressing oocytes were incubated with vehicle (ND96) or with equal concentrations of IαI and a specific blocking antibody (mAB) against IαI (0.1 mg/ml). Values means ± SE; n (number of oocytes from three or four experiments) = 8 for vehicle and 7 for IαI+mAB. Veh, vehicle.
IαI Inhibits Na+ but Not Cl− Currents across Rat ATII Monolayers
To test the effects of IαI on native ENaC, IαI (0.1 mg/ml) or vehicle were added to the apical surfaces of confluent monolayers of rat ATII cells (21, 29) for 2 or 4 hours. ATII cells were then mounted in Ussing chambers, and Isc was recorded continuously (Figures 3A and 3B). No changes were seen at 2 hours after IαI addition. However, IαI decreased the amiloride-sensitive component of Isc (IENaC) at 4 hours after incubation from 4.3 ± 0.4 μA to 2.34 ± 0.25 μA (X ± 1 SEM; vehicle = 19; IαI = 21; **P < 0.01). The forskolin-activated and glibenclamide-inhibited component of Isc (Figures 3A and 3B) and the transepithelial resistances remained at baseline levels (data not shown). Similarly, incubation of murine nasal cells with IαI (0.1 mg) for 4 hours decreased their IENaC from 3.8 ± 0.23 to 3.0 ± 0.08 (X ± 1 SEM; n = 6; P = 0.0023). These data indicate that IαI decreased native murine ENaCs and human ENaCs expressed in Xenopus oocytes.
Figure 3.

IαI inhibits ENaC but not CF transmembrane conductance regulator (CFTR) short-circuit current (Isc) across ATII cell monolayers. (A) ATII cell monolayers were incubated with IαI (0.1 mg/ml added to their apical surface) for 2 or 4 hours at 37°C and mounted in Ussing chambers for the measurement of Isc. Amiloride (A; 10 μM), forskolin (F; 10 μM), and glibenclamide (G; 400 μM) were added sequentially in the apical chambers. (A, B) Typical experiments after incubation of ATII cell monolayers with IαI for 2 or 4 hours.
To obtain additional insight into the mechanisms by which IαI decreased ENaC activity, we isolated total and surface proteins from ATII cells incubated with IαI for 4 hours and immunostained them using an α-ENaC antibody. Bands around 95 kD, indicative of uncleaved αENaCs, were seen in total and surface proteins (Figure 4). These bands increased significantly after incubation of ATII cells with IαI for 4 hours. The specificity of the anti–α-ENaC antibody was verified by the disappearance of these bands when Western blotting studies were repeated in the presence of the immunizing peptide. In previous studies we have shown that α-antitrypsin decreased the 65 kD band in Xenopus oocytes injected with human α-, β-, and γ-ENaCs (28). Planes and colleagues (30) detected a 65-kD band in surface but not total proteins from ATII cells, which they attributed to cleaved αENaCs. However, these authors were unable to detect the high-molecular weight-bands as we did in our experiments. Differences in culture conditions and protein loading and the use of antibodies from different sources may account for these differences.
Figure 4.

IαI increase uncleaved ENaC. Western blots of total and surface proteins from rat ATII cells with an antibody against α-ENaC in the absence and presence of the immunizing peptide are shown. ATII cells were isolated from rat lungs, seeded on permeable supports, and cultured under liquid–liquid (3 d) and air–liquid (1–2 d) interfaces. IαI (0.1 mg/ml) was added on their apical surface. Four hours later, cells were lysed, and total and surface (biotinylated) proteins were isolated. Representative total (all proteins except those in the apical plasma membranes) and surface (biotinylated; proteins in apical surface) αENaC Western blots in the absence and presence of the immunizing peptide are shown. For surface labeling, 250 μg of protein were loaded with neutravidin beads as described in Materials and Methods. A total of 40 μg of protein were used. This experiment was repeated three times with similar results. Equal amounts of protein were loaded in each lane as verified by amido-black staining at the end of each experiment (data not shown). Notice the disappearance of all bands in the surface pool in the absence of biotin. The specificity of the anti-αENaC antibody was verified by the absence of staining in the presence of the immunizing peptide. IαI increased the intensity of the 95-kD band (uncleaved ENaC) in total and surface proteins. MW, molecular weight.
IαI Inhibits ENaCs In Situ
To more definitively document the effects of IαI on ATII cell ENaCs, we incubated freshly prepared lung slices from C57BL/6 mice with IαI (0.1 mg/ml) for 4 hours, patched ATII cells in situ using the cell-attached mode of patch clamp configuration, and recorded ENaC activity in situ as reported earlier (17, 21). Two distinct Na+ channels with conductances of 4 and 18 pS were observed (Figure 5A). Both channels were inhibited by amiloride (21). The 4-pS channel is thought to represent ENaCs, whereas the 18-pS channel is a cation channel with equal permeability to Na+ and K+ ions. Incubation of lung slices with IαI inhibited the activity of both channels (Figures 5B and 5C) and decreased their open probabilities (Po) (Figures 5D and 5E). This effect was abolished when lung slices were incubated simultaneously with IαI and an antibody against IαI (Figures 5D and 5E).
Figure 5.
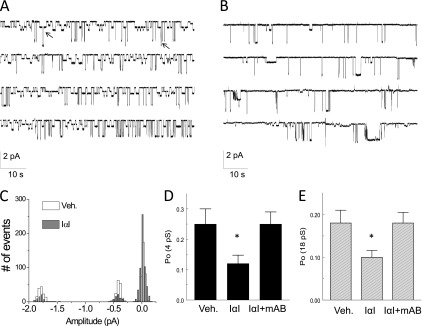
IαI inhibits ENaC single-channel activity in AT2 cells in mouse lung slices. (A) Cell-attached patches of AT2 cells in lung slices at a holding potential of −100 mV . Note the presence of two Na+ channels with conductances of 4 and 18 pS. Both channels were totally inhibited by the addition of amiloride (10 μM) in the pipette solution (data not shown; see Reference 21). Arrows indicate the simultaneous openings of the 4- and 18-pS channels. (B) A similar record showing decreased channel activity after incubation of the slice with IαI (0.1 mg/ml) for 3 hours. (C) Histogram showing the number of events versus current amplitude (pA) for the records shown in A and B. The corresponding conductances for the three peaks are 4 and 18 pS. Typical records of experiments, which were repeated at least five times. (D, E) Mean values (± 1 SE) (n = 20 for each group) of the open probabilities (Po) of the 4-pS (left panel) and 18-pS (right panel) Na+ channels obtained by patching ATII cells in lung slices incubated with medium (DMEM; Veh), IαI (0.1 mg/ml), or IαI and an equal concentration of an antibody against IαI (mAB IαI; 0.1 mg/ml for ∼ 3 h; *P < 0.0035, as compared with the corresponding control values by one-way ANOVA followed by the Bonferroni modification of the t test for multiple comparisons).
IαI Partially Reverses Increased ENaC Activity in ATII Cells from CFTR−/− and ∆F-508 ATII Mice
∆F-508 and CFTR−/− mice have higher levels of lung ENaC activity (17, 31) but only background levels of IαI in their BALS (∆F-508: 0.1 ± 0.02 [n = 8 mice]; C57BL/6: 0.075 ± 0.045 [n = 7 mice]; X ± SEM; P = 0.66). We questioned whether incubation of lung slices from ∆F-508 and CFTR−/− mice with IαI would reduce ENaC activity. The results of these studies are shown in Figure 6. As reported previously (17), ATII cells from ∆F-508 and CFTR(−/−) mice exhibited significantly higher Po values for the 4-pS (Figure 6) and the 18-pS channels (Figure E1). Incubation of slices with 0.1 mg/ml IαI for 4 hours reduced Po values of both channels close to normal levels.
Figure 6.

IαI decreases ENaC activity of lung epithelial cells from wild-type (Wt) and CFTR-deficient mice in vitro and in situ. (A) Lung slices from Wt (Control), ∆F508 (∆F), and CFTR(−/−) mice were incubated with vehicle (V) or IαI (0.1 mg/ml) for 4 hours. Open probability (Po) for the 4- pS channel. Values are means ± SE (n = 20 patches per group; *,#P < 0.001) compared with the corresponding value to the immediate right and the control (Wt) value, respectively; one-way ANOVA followed the Bonferroni modification of the t test adjusted for multiple comparisons. (B, C) Measurements of airway surface liquid volume on murine nasal epithelial cells after addition of IαI (0.1 mg/ml) or vehicle (PBS) for 4 hours. (B) Confocal microscopy figures with the cells labeled with Cell Tracker Green CMFDA and the apical surface fluid with Texas Red (further details are provided in the online supplement). Notice the significant increase of the apical surface liquid on monolayers treated with IαI. Mean values ± 1 SE are shown in C. For comparison, some monolayers were treated with amiloride (10 μM), which completely inhibits ENaC. Number of monolayers used were vehicle (control) = 11; IαI = 11; amiloride = 14 (*P < 0.05 compared with Veh.; ANOVA (P = 0.001) followed by the Bonferroni modification of the t test for multiple comparisons). (D) Measurements of the amiloride-sensitive component of nasal potential difference (NPD) in Wt and ∆F508 mice 4 or 24 hours after intranasal instillation of IαI (0.1 mg/ml; 25 μl in each nostril). Values are means ± 1 SE; number of mice are as follows: Wt (vehicle = 10); Wt IαI = 10; ∆F508 = 20 for each group. Statistical significance is indicated in the graph and was determined as described above.
IαI Increased the Airway Surface Liquid Depth Overlying Primary Murine Nasal Epithelial Cells
The volume of fluid on apical surface of murine nasal epithelial cells depends on the balance between Na+ reabsorption (mainly through ENaCs) and Cl− secretion (mainly through CFTR) (22). Addition of IαI (0.1 mg) to the apical surface of confluent monolayers of primary murine nasal epithelial cells increased the airway surface liquid depth to the same extent as amiloride (vehicle, 5.6 ± 0.1 [n = 11]; IαI, 7.2 ± 0.3 [n = 11]; amiloride, 8 ± 0.44 [n = 14]) (Figures 6B and 6C). These data indicated that IαI decreased ENaCs but not CFTR (or at least it decreased ENaCs to a much higher extent than CFTR), in agreement with our Isc measurements across ATII cell monolayers (Figure 3).
IαI Reduces Amiloride-Sensitive NPD Values of ∆F-508 Mice
NPD values are dependent on the rate of Na+ absorption and Cl− secretion by nasal epithelial cells. The amiloride-sensitive NPD (ENaC-NPD) values in wild-type and ∆F508 mice were −6.95 ± 0.37 mV (mean ± SE; n = 10) and −23 ± 0.31 mV (mean ± SE; n = 20), respectively. Four hours after intranasal instillation of IαI, the absolute values of these variables were reduced by 23% (Figure 6D). The magnitude of this decrease is similar to the decrease of the amiloride-sensitive Isc across nasal epithelial cells in primary culture. This effect was still present at 24 hours after instillation.
Discussion
The mechanisms involved in the activation and removal of ENaCs from the plasma membranes of epithelial cells are not well understood. Activation involves membrane-bound proteases, such as furin, prostasin, and matriptase, which activate ENaCs by cleaving critical amino acids in α- and γ-ENaC subunits or by activating signaling pathways (6, 32–35). Kunitz-type protease inhibitors, such as aprotinin, inactivate serine proteases, including trypsin, plasmin, and kallikreins (36); α1-antitrypsin, an acute phase glycoprotein and a member of the SERPIN (serine protease inhibitors) superfamily (37), also inhibit ENaC activity (6, 28, 38). Herein, we demonstrate for the first time that IαI, a plasma protein that is also expressed by airway epithelia, is present in the BALF of children with CF and lower airway disease. In addition, our data show that IαI inhibits ENaC activity across Xenopus oocytes expressing human ENaCs, decreases amiloride-sensitive currents across confluent monolayers of rat ATII and mouse nasal epithelial cells, and decreases the open probabilities of two different types of ATII cell amiloride-sensitive currents in situ. In agreement with our previous findings (17), data shown here indicate that ATII cells from ∆F508 and CFTR(−/−) mice have higher ENaC activity than wild-type mice. Furthermore, ∆F508 mice showed more negative amiloride-sensitive NPDs, consistent with higher ENaC activity in the lungs of these mice. Intranasal instillation of IαI decreased ENaC-NPD by 25%, and this effect persisted for up to 24 hours after instillation. Western blotting studies in surface proteins of ATII cells show that IαI increases uncleaved (inactive) αENaCs, thus providing insight into the mechanisms involved. Our findings contribute to the understanding of the role played by ENaCs in lung fluid regulation in health and disease and suggest that IαI contributes to the homeostatic regulation of ENaCs in wild-type mice and in those with the most common CFTR mutation and in mice lacking the CFTR genes.
IαI consists of two heavy chains and a light chain, bikunin (39), which is responsible for its antiprotease activity. IαI is a fairly weak and nonspecific protease inhibitor and makes up only approximately 5% of the total protease inhibitory activity in plasma, even though it is present at a fairly high concentrations (0.1–0.5 mg/ml) (40). IαI is present in low concentrations in the lung lining epithelial fluid of naive mice, but its concentration increases significantly when the epithelial permeability is compromised, such as in lung injury or inflammation. For example, we show that IαI levels are very high in the BALF of children with CF (Figure 1). There is a linear correlation between the number of inflammatory cells and levels of IαI in the BALF of children with CF (Figure 1). This suggests that IαI may be especially up-regulated during inflammation. Our current set of patients was not large enough to stratify by other parameters (e.g., type of infection). In previous studies, Garantziotis and colleagues (27) showed colocalization of IαI and HA in fibroblastic foci in patients with interstitial pneumonitis. In contrast to the human data, ∆F508 mice (with no evidence of lung disease) have normal levels of IαI in their BALF (Figure 1).
IαI is a known HA binding protein (27). We show that the BALF concentration of HA and IαI increased concomitantly in the BALF of patients with CF (Figure 1C), suggesting that these two extracellular matrix components may be bound to each other. Luminal HA is bound on the apical surface of bronchial epithelia (41) and may thus be the linking molecule that brings IαI in apposition to the cell membrane. Bikunin inhibits its protease substrates on the cell surface much more efficiently than in solution (42). Because ENaC activation requires membrane-bound serine proteases, IαI may be able to regulate ENaC activity via increased concentration and proximity to the cell surface. IαI is expressed by lung epithelia as well (16), where it promotes epithelial repair after injury. Thus, IαI may also contribute to homeostatic ENaC regulation in health and disease.
There is considerable controversy whether increased ENaC activity contributes to the pathophysiology of CF disease (31, 43). Although it is known that CF results from a defect in CFTR, a cAMP-activated Cl− channel, increased Na+ reabsorption in the airways of patients with CF leading to decreased mucous hydration, stasis, and increased susceptibility to infections has been demonstrated (44, 45). Furthermore, mice overexpressing β-ENaCs develop CF-like symptoms with mucus plugging (46). Our previous studies in ∆F508, CFTR(+/−), and CFTR(−/−) mice have shown an inverse correlation between putative levels of CFTR and ENaC activity in ATII cells (17). Another study reported that increased functional CFTR decreases ENaC activity by raising intracellular Cl− (47). However, studies in newborn pigs with CF disease found no evidence for increased Na+ reabsorption across airway epithelia (48, 49). Herein we report that ∆F508 mice have significantly more negative amiloride-sensitive NPD values (−23 vs. −7 mV for wild-type mice), which is in agreement with previous reports in mice (50) and humans with CF (51). A single intranasal instillation of IαI decreased NPD by approximately 25% for 24 hours.
Previously, we reported that α1-antitrypsin (a member of the SERPIN family that lacks the Kunitz-type domain) down-regulates both amiloride-sensitive currents in vitro and Na+-dependent alveolar fluid clearance across the distal lung epithelia of anesthetized mice in vivo (28). Protease inhibitor nexin 1, another member of the SERPIN family that inhibits matriptase (52, 53), also inhibited Na+ currents across human airway epithelial cells isolated from CF lungs (52). Based on Western blotting studies of surface proteins of ATII cells in culture and in situ, we conclude that IαI decreased ENaC activity by preventing membran- bound proteases from cleaving portions of the extracellular loops of α- and γ-ENaC subunits, thereby inhibiting the activation of ENaC at the cell membrane of lung epithelial cells and oocytes. To the best of our knowledge, this is the first demonstration of IαI inhibiting membrane proteases involved in the activation of ENaC at the surface of epithelial cells in vitro, in situ, and in vivo and involving oocytes expressing ENaC.
Our data show that IαI did not alter CFTR activity because it does not require proteolytic cleavage for its activation as ENaCs do. The specific target proteases of IαI have not been identified. Prostasin, trypsin, and matriptase are possible targets because they have been shown to activate Na+ currents across human airway epithelial cells isolated from CF lungs (6, 32, 52, 53). Prostasin is synthesized as an inactive zymogen that requires a site-specific endoproteolytic cleavage to be converted to an active protease. It has been recently reported that matriptase is necessary for prostasin activation (54). Protease inhibitor nexin-1, like A1AT, inhibits matriptase, which inactivates ENaCs (52, 53). Another important protease involved in ENaC activation is furin (55). In fact, IαI has been shown to inhibit furin activation of anthrax lethal toxin and to protect animals from anthrax intoxication (56). Furin is mainly found intracellularly and cleaves ENaC during trafficking to the cell surface (55). However, furin-like activity has been shown at the cell surface as well (57), suggesting that extracellular IαI may inhibit furin activity without having to enter the cytoplasm.
There is significant evidence to show that there is a large increase of protease to antiprotease balance in the lungs of patients with CF. There is excess secretion of serine proteases (such as elastase, by activated neutrophils), cysteine proteases (by alveolar macrophages), matrix metalloproteinases (airway cells), and bacterial proteases (by P. aeruginosa). Unless deactivated by antiproteases, these proteases increase lung inflammation, degrade structural proteins, and activate ENaCs, leading to compromised respiratory function (58). For this reason, administration of α-antitrypsin to patients with CF has been shown to decrease inflammation (59). However, the antiprotease activity of α-antitrypsin may be abrogated by oxidation of methionine in its active site by a number of oxidants (60), including peroxynitrite (28), the concentration of which is known to increase in the lungs of patients with acute lung injury and CF (61, 62). Whether or not IαI is subject to oxidative posttranslational modifications has not been determined. The overall increase of antiprotease balance will benefit the lungs by decreasing inflammation and ENaC activity. Very little IαI was present in the lung lavage fluid of ∆F508 mice, which is in sharp contrast to the large amounts of IαI and HA seen in the BALF of patients with CF and in patients with lower airway disease. This is consistent with the lack of pulmonary disease in ∆F508 mice and with the absence of lung inflammation in these animals. It is possible that the presence of significant amounts of IαI in patients will down-regulate increased levels of airway ENaCs, thus limiting mucous stasis, airway obstruction, and compromised pulmonary function.
Acknowledgments
Acknowledgments
The authors thank Dr. Lan Chen for technical assistance with the isolation and culture of rat ATII cells and for the measurements of short circuit currents and Dr. David M Bedwell and the Gregory Fleming James Cystic Fibrosis Center for providing us with ∆F508 and CFTR(−/−) mice.
Footnotes
This work was supported by the CounterACT Program; by National Institutes of Health Office of the Director and the National Institute of Neurological Disorders and Stroke grants 5U01ES015676 and 1R21ES02402701; by National Heart, Lung and Blood Institute grant 5R01HL031197 (S.M.); by the Flight Attendant’s Medical Research Institute Young Clinical Scientist Award (072218) and National Institutes of Health/National Heart, Lung and Blood Institute grant 1K08HL107142-02 (B.A.W.); and by the Division of Intramural Research, National Institute of Environmental Health Sciences, National Institutes of Health (S.G.).
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1165/rcmb.2013-0215OC on December 4, 2013
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Canessa CM, Horisberger JD, Rossier BC.Epithelial sodium channel related to proteins involved in neurodegeneration Nature 1993361467–470.(see Comments [DOI] [PubMed] [Google Scholar]
- 2.Rossier BC, Canessa CM, Schild L, Horisberger JD. Epithelial sodium channels. Curr Opin Nephrol Hypertens. 1994;3:487–496. doi: 10.1097/00041552-199409000-00003. [DOI] [PubMed] [Google Scholar]
- 3.Matalon S, O'Brodovich H. Sodium channels in alveolar epithelial cells: molecular characterization, biophysical properties, and physiological significance. Annu Rev Physiol. 1999;61:627–661. doi: 10.1146/annurev.physiol.61.1.627. [DOI] [PubMed] [Google Scholar]
- 4.Ji HL, Su XF, Kedar S, Li J, Barbry P, Smith PR, Matalon S, Benos DJ. Delta-subunit confers novel biophysical features to alpha beta gamma-human epithelial sodium channel (ENaC) via a physical interaction. J Biol Chem. 2006;281:8233–8241. doi: 10.1074/jbc.M512293200. [DOI] [PubMed] [Google Scholar]
- 5.Vuagniaux G, Vallet V, Jaeger NF, Pfister C, Bens M, Farman N, Courtois-Coutry N, Vandewalle A, Rossier BC, Hummler E. Activation of the amiloride-sensitive epithelial sodium channel by the serine protease MCAP1 expressed in a mouse cortical collecting duct cell line. J Am Soc Nephrol. 2000;11:828–834. doi: 10.1681/ASN.V115828. [DOI] [PubMed] [Google Scholar]
- 6.Planes C, Leyvraz C, Uchida T, Angelova MA, Vuagniaux G, Hummler E, Matthay M, Clerici C, Rossier B. In vitro and in vivo regulation of transepithelial lung alveolar sodium transport by serine proteases. Am J Physiol Lung Cell Mol Physiol. 2005;288:L1099–L1109. doi: 10.1152/ajplung.00332.2004. [DOI] [PubMed] [Google Scholar]
- 7.Staub O, Gautschi I, Ishikawa T, Breitschopf K, Ciechanover A, Schild L, Rotin D. Regulation of stability and function of the epithelial Na+ channel (ENaC) by ubiquitination. EMBO J. 1997;16:6325–6336. doi: 10.1093/emboj/16.21.6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Valentijn JA, Fyfe GK, Canessa CM. Biosynthesis and processing of epithelial sodium channels in xenopus oocytes. J Biol Chem. 1998;273:30344–30351. doi: 10.1074/jbc.273.46.30344. [DOI] [PubMed] [Google Scholar]
- 9.Malik B, Schlanger L, Al Khalili O, Bao HF, Yue G, Price SR, Mitch WE, Eaton DC. Enac degradation in A6 Cells by the ubiquitin-proteosome proteolytic pathway. J Biol Chem. 2001;276:12903–12910. doi: 10.1074/jbc.M010626200. [DOI] [PubMed] [Google Scholar]
- 10.Bost F, Diarra-Mehrpour M, Martin JP. Inter-alpha-trypsin inhibitor proteoglycan family: a group of proteins binding and stabilizing the extracellular matrix. Eur J Biochem. 1998;252:339–346. doi: 10.1046/j.1432-1327.1998.2520339.x. [DOI] [PubMed] [Google Scholar]
- 11.Malki N, Balduyck M, Maes P, Capon C, Mizon C, Han KK, Tartar A, Fournet B, Mizon J. The heavy chains of human plasma inter-alpha-trypsin inhibitor: their isolation, their identification by electrophoresis and partial sequencing: differential reactivity with concanavalin A. Biol Chem Hoppe Seyler. 1992;373:1009–1018. doi: 10.1515/bchm3.1992.373.2.1009. [DOI] [PubMed] [Google Scholar]
- 12.Choi-Miura NH, Otsuyama K, Sano Y, Saito K, Takahashi K, Tomita M. Hepatic injury-specific conversion of mouse plasma hyaluronan binding protein to the active hetero-dimer form. Biol Pharm Bull. 2001;24:892–896. doi: 10.1248/bpb.24.892. [DOI] [PubMed] [Google Scholar]
- 13.Choi-Miura NH, Takahashi K, Yoda M, Saito K, Mazda T, Tomita M. Proteolytic activation and inactivation of the serine protease activity of plasma hyaluronan binding protein. Biol Pharm Bull. 2001;24:448–452. doi: 10.1248/bpb.24.448. [DOI] [PubMed] [Google Scholar]
- 14.Fries E, Kaczmarczyk A. Inter-alpha-inhibitor, hyaluronan and inflammation. Acta Biochim Pol. 2003;50:735–742. [PubMed] [Google Scholar]
- 15.Garantziotis S, Li Z, Potts EN, Kimata K, Zhuo L, Morgan DL, Savani RC, Noble PW, Foster WM, Schwartz DA, et al. Hyaluronan mediates ozone-induced airway hyperresponsiveness in mice. J Biol Chem. 2009;284:11309–11317. doi: 10.1074/jbc.M802400200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 16.Adair JE, Stober V, Sobhany M, Zhuo L, Roberts JD, Negishi M, Kimata K, Garantziotis S. Inter-alpha-trypsin inhibitor promotes bronchial epithelial repair after injury through vitronectin binding. J Biol Chem. 2009;284:16922–16930. doi: 10.1074/jbc.M808560200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lazrak A, Jurkuvenaite A, Chen L, Keeling KM, Collawn JF, Bedwell DM, Matalon S. Enhancement of alveolar epithelial sodium channel activity with decreased cystic fibrosis transmembrane conductance regulator expression in mouse lung. Am J Physiol Lung Cell Mol Physiol. 2011;301:L557–L567. doi: 10.1152/ajplung.00094.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Snouwaert JN, Brigman KK, Latour AM, Malouf NN, Boucher RC, Smithies O, Koller BH. An animal model for cystic fibrosis made by gene targeting. Science. 1992;257:1083–1088. doi: 10.1126/science.257.5073.1083. [DOI] [PubMed] [Google Scholar]
- 19.Zeiher BG, Eichwald E, Zabner J, Smith JJ, Puga AP, McCray PB, Jr, Capecchi MR, Welsh MJ, Thomas KR. A mouse model for the Delta F508 allele of cystic fibrosis. J Clin Invest. 1995;96:2051–2064. doi: 10.1172/JCI118253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lim YP, Bendelja K, Opal SM, Siryaporn E, Hixson DC, Palardy JE. Correlation between mortality and the levels of inter-alpha inhibitors in the plasma of patients with severe sepsis. J Infect Dis. 2003;188:919–926. doi: 10.1086/377642. [DOI] [PubMed] [Google Scholar]
- 21.Lazrak A, Chen L, Jurkuvenaite A, Doran SF, Liu G, Li Q, Lancaster JR, Jr, Matalon S. Regulation of alveolar epithelial Na+ channels by ERK1/2 in chlorine-breathing mice. Am J Respir Cell Mol Biol. 2012;46:342–354. doi: 10.1165/rcmb.2011-0309OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Woodworth BA, Antunes MB, Bhargave G, Palmer JN, Cohen NA. Murine tracheal and nasal septal epithelium for air-liquid interface cultures: a comparative study. Am J Rhinol. 2007;21:533–537. doi: 10.2500/ajr.2007.21.3068. [DOI] [PubMed] [Google Scholar]
- 23.Chen L, Song W, Davis IC, Shrestha K, Schwiebert E, Sullender WM, Matalon S. Inhibition of Na+ transport in lung epithelial cells by respiratory syncytial virus infection. Am J Respir Cell Mol Biol. 2009;40:588–600. doi: 10.1165/rcmb.2008-0034OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen L, Bosworth CA, Pico T, Collawn JF, Varga K, Gao Z, Clancy JP, Fortenberry JA, Lancaster JR, Jr, Matalon S. DETANO and nitrated lipids increase chloride secretion across lung airway cells. Am J Respir Cell Mol Biol. 2008;39:150–162. doi: 10.1165/rcmb.2008-0005OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tucker TA, Schwiebert LM. CD40 ligation decreases its protein half-life at the cell surface. Eur J Immunol. 2008;38:864–869. doi: 10.1002/eji.200737828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Knowles MR, Paradiso AM, Boucher RC. In vivo nasal potential difference: techniques and protocols for assessing efficacy of gene transfer in cystic fibrosis. Hum Gene Ther. 1995;6:445–455. doi: 10.1089/hum.1995.6.4-445. [DOI] [PubMed] [Google Scholar]
- 27.Garantziotis S, Zudaire E, Trempus CS, Hollingsworth JW, Jiang D, Lancaster LH, Richardson E, Zhuo L, Cuttitta F, Brown KK, et al. Serum inter-alpha-trypsin inhibitor and matrix hyaluronan promote angiogenesis in fibrotic lung injury. Am J Respir Crit Care Med. 2008;178:939–947. doi: 10.1164/rccm.200803-386OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lazrak A, Nita I, Subramaniyam D, Wei S, Song W, Ji HL, Janciauskiene S, Matalon S. Alpha(1)-antitrypsin inhibits epithelial Na+ transport in vitro and in vivo. Am J Respir Cell Mol Biol. 2009;41:261–270. doi: 10.1165/rcmb.2008-0384OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Factor P, Mutlu GM, Chen L, Mohameed J, Akhmedov AT, Meng FJ, Jilling T, Lewis ER, Johnson MD, Xu A, et al. Adenosine regulation of alveolar fluid clearance. Proc Natl Acad Sci USA. 2007;104:4083–4088. doi: 10.1073/pnas.0601117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Planes C, Blot-Chabaud M, Matthay MA, Couette S, Uchida T, Clerici C. Hypoxia and beta 2-agonists regulate cell surface expression of the epithelial sodium channel in native alveolar epithelial cells. J Biol Chem. 2002;277:47318–47324. doi: 10.1074/jbc.M209158200. [DOI] [PubMed] [Google Scholar]
- 31.Collawn JF, Lazrak A, Bebok Z, Matalon S. The CFTR and ENaC debate: how important is ENaC in CF lung disease? Am J Physiol Lung Cell Mol Physiol. 2012;302:L1141–L1146. doi: 10.1152/ajplung.00036.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rossier BC. The epithelial sodium channel: activation by membrane-bound serine proteases. Proc Am Thorac Soc. 2004;1:4–9. doi: 10.1513/pats.2306007. [DOI] [PubMed] [Google Scholar]
- 33.Snyder PM. Minireview: regulation of epithelial Na+ channel trafficking. Endocrinology. 2005;146:5079–5085. doi: 10.1210/en.2005-0894. [DOI] [PubMed] [Google Scholar]
- 34.Hughey RP, Mueller GM, Bruns JB, Kinlough CL, Poland PA, Harkleroad KL, Carattino MD, Kleyman TR. Maturation of the epithelial Na+ channel involves proteolytic processing of the alpha- and gamma-subunits. J Biol Chem. 2003;278:37073–37082. doi: 10.1074/jbc.M307003200. [DOI] [PubMed] [Google Scholar]
- 35.Bengrine A, Li J, Hamm LL, Awayda MS. Indirect activation of the epithelial Na+ channel by trypsin. J Biol Chem. 2007;282:26884–26896. doi: 10.1074/jbc.M611829200. [DOI] [PubMed] [Google Scholar]
- 36.Vogel R, Werle E, Zickgraf-Rudel G. Current aspects of kinin research: I. Potentiation and blocking of biological kinin activity. Z Klin Chem Klin Biochem. 1970;8:177–185. [PubMed] [Google Scholar]
- 37.Carrell RW, Lomas DA. Alpha1-antitrypsin deficiency: a model for conformational diseases. N Engl J Med. 2002;346:45–53. doi: 10.1056/NEJMra010772. [DOI] [PubMed] [Google Scholar]
- 38.Fan B, Wu TD, Li W, Kirchhofer D. Identification of hepatocyte growth factor activator inhibitor-1B as a potential physiological inhibitor of prostasin. J Biol Chem. 2005;280:34513–34520. doi: 10.1074/jbc.M502119200. [DOI] [PubMed] [Google Scholar]
- 39.Zhuo L, Kimata K. Structure and function of inter-alpha-trypsin inhibitor heavy chains. Connect Tissue Res. 2008;49:311–320. doi: 10.1080/03008200802325458. [DOI] [PubMed] [Google Scholar]
- 40.Potempa J, Korzus E, Travis J. The serpin superfamily of proteinase inhibitors: structure, function, and regulation. J Biol Chem. 1994;269:15957–15960. [PubMed] [Google Scholar]
- 41.Forteza R, Lieb T, Aoki T, Savani RC, Conner GE, Salathe M. Hyaluronan serves a novel role in airway mucosal host defense. FASEB J. 2001;15:2179–2186. doi: 10.1096/fj.01-0036com. [DOI] [PubMed] [Google Scholar]
- 42.Kobayashi H, Shinohara H, Takeuchi K, Itoh M, Fujie M, Saitoh M, Terao T. Inhibition of the soluble and the tumor cell receptor-bound plasmin by urinary trypsin inhibitor and subsequent effects on tumor cell invasion and metastasis. Cancer Res. 1994;54:844–849. [PubMed] [Google Scholar]
- 43.Collawn JF, Matalon S. The role of CFTR in transepithelial liquid transport in pig alveolar epithelia. Am J Physiol Lung Cell Mol Physiol. 2012;303:L489–L491. doi: 10.1152/ajplung.00216.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boucher RC, Stutts MJ, Knowles MR, Cantley L, Gatzy JT. Na+ transport in cystic fibrosis respiratory epithelia: abnormal basal rate and response to adenylate cyclase activation. J Clin Invest. 1986;78:1245–1252. doi: 10.1172/JCI112708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Knowles MR, Stutts MJ, Spock A, Fischer N, Gatzy JT, Boucher RC. Abnormal ion permeation through cystic fibrosis respiratory epithelium. Science. 1983;221:1067–1070. doi: 10.1126/science.6308769. [DOI] [PubMed] [Google Scholar]
- 46.Mall M, Grubb BR, Harkema JR, O'Neal WK, Boucher RC. Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat Med. 2004;10:487–493. doi: 10.1038/nm1028. [DOI] [PubMed] [Google Scholar]
- 47.Konig J, Schreiber R, Voelcker T, Mall M, Kunzelmann K. The cystic fibrosis transmembrane conductance regulator (CFTR) inhibits ENaC through an increase in the intracellular Cl- concentration. EMBO Rep. 2001;2:1047–1051. doi: 10.1093/embo-reports/kve232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Itani OA, Chen JH, Karp PH, Ernst S, Keshavjee S, Parekh K, Klesney-Tait J, Zabner J, Welsh MJ. Human cystic fibrosis airway epithelia have reduced Cl- conductance but not increased Na+ conductance. Proc Natl Acad Sci USA. 2011;108:10260–10265. doi: 10.1073/pnas.1106695108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li X, Comellas AP, Karp PH, Ernst SE, Moninger TO, Gansemer ND, Taft PJ, Pezzulo AA, Rector MV, Rossen N, et al. CFTR is required for maximal transepithelial liquid transport in pig alveolar epithelia. Am J Physiol Lung Cell Mol Physiol. 2012;303:L152–L160. doi: 10.1152/ajplung.00116.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Salinas DB, Pedemonte N, Muanprasat C, Finkbeiner WF, Nielson DW, Verkman AS. CFTR involvement in nasal potential differences in mice and pigs studied using a thiazolidinone CFTR inhibitor. Am J Physiol Lung Cell Mol Physiol. 2004;287:L936–L943. doi: 10.1152/ajplung.00354.2003. [DOI] [PubMed] [Google Scholar]
- 51.Knowles M, Gatzy J, Boucher R. Increased bioelectric potential difference across respiratory epithelia in cystic fibrosis. N Engl J Med. 1981;305:1489–1495. doi: 10.1056/NEJM198112173052502. [DOI] [PubMed] [Google Scholar]
- 52.Myerburg MM, McKenna EE, Luke CJ, Frizzell RA, Kleyman TR, Pilewski JM. Prostasin expression is regulated by airway surface liquid volume and is increased in cystic fibrosis. Am J Physiol Lung Cell Mol Physiol. 2008;294:L932–L941. doi: 10.1152/ajplung.00437.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Janciauskiene S, Nita I, Subramaniyam D, Li Q, Lancaster JR, Jr, Matalon S. Alpha1-antitrypsin inhibits the activity of the matriptase catalytic domain in vitro. Am J Respir Cell Mol Biol. 2008;39:631–637. doi: 10.1165/rcmb.2008-0015RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Netzel-Arnett S, Currie BM, Szabo R, Lin CY, Chen LM, Chai KX, Antalis TM, Bugge TH, List K. Evidence for a matriptase-prostasin proteolytic cascade regulating terminal epidermal differentiation. J Biol Chem. 2006;281:32941–32945. doi: 10.1074/jbc.C600208200. [DOI] [PubMed] [Google Scholar]
- 55.Hughey RP, Bruns JB, Kinlough CL, Harkleroad KL, Tong Q, Carattino MD, Johnson JP, Stockand JD, Kleyman TR. Epithelial sodium channels are activated by furin-dependent proteolysis. J Biol Chem. 2004;279:18111–18114. doi: 10.1074/jbc.C400080200. [DOI] [PubMed] [Google Scholar]
- 56.Opal SM, Artenstein AW, Cristofaro PA, Jhung JW, Palardy JE, Parejo NA, Lim YP. Inter-alpha-inhibitor proteins are endogenous furin inhibitors and provide protection against experimental anthrax intoxication. Infect Immun. 2005;73:5101–5105. doi: 10.1128/IAI.73.8.5101-5105.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Koo BH, Longpre JM, Somerville RP, Alexander JP, LeDuc R, Apte SS. Cell-surface processing of Pro-ADAMTS9 by furin. J Biol Chem. 2006;281:12485–12494. doi: 10.1074/jbc.M511083200. [DOI] [PubMed] [Google Scholar]
- 58.Quinn DJ, Weldon S, Taggart CC. Antiproteases as therapeutics to target inflammation in cystic fibrosis. Open Respir Med J. 2010;4:20–31. doi: 10.2174/1874306401004010020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Griese M, Latzin P, Kappler M, Weckerle K, Heinzlmaier T, Bernhardt T, Hartl D. Alpha1-antitrypsin inhalation reduces airway inflammation in cystic fibrosis patients. Eur Respir J. 2007;29:240–250. doi: 10.1183/09031936.00047306. [DOI] [PubMed] [Google Scholar]
- 60.Taggart C, Cervantes-Laurean D, Kim G, McElvaney NG, Wehr N, Moss J, Levine RL. Oxidation of either methionine 351 or methionine 358 in alpha 1-antitrypsin causes loss of anti-neutrophil elastase activity. J Biol Chem. 2000;275:27258–27265. doi: 10.1074/jbc.M004850200. [DOI] [PubMed] [Google Scholar]
- 61.Haddad IY, Pataki G, Hu P, Galliani C, Beckman JS, Matalon S. Quantitation of nitrotyrosine levels in lung sections of patients and animals with acute lung injury. J Clin Invest. 1994;94:2407–2413. doi: 10.1172/JCI117607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Balint B, Kharitonov SA, Hanazawa T, Donnelly LE, Shah PL, Hodson ME, Barnes PJ. Increased nitrotyrosine in exhaled breath condensate in cystic fibrosis. Eur Respir J. 2001;17:1201–1207. doi: 10.1183/09031936.01.00072501. [DOI] [PubMed] [Google Scholar]