

Abstract
Background
In bone, NADPH oxidase (NOX)-derived reactive oxygen species (ROS) superoxide and/or hydrogen peroxide are an important stimulus for osteoclast differentiation and activity. Previously, we have demonstrated that chronic ethanol (EtOH) consumption generates excess NOX-dependent ROS in osteoblasts, which functions to stimulate NFκB receptor ligand (RANKL)—RANK signaling, thus increasing osteoclastogenesis and activity. This activity can be blocked by co-administration of EtOH with the pan-NOX inhibitor diphenylene idonium (DPI).
Methods
To test if EtOH-induced bone loss is dependent on a functional NOX2 enzyme, six week old female C57BL/6J-Ncf1/p47phox-/- (p47phox KO) and wild-type (WT) mice were pair-fed EtOH diets for 40 days. Bone loss was assessed by 3-point bending, μCT and static histomorphometric analysis. Additionally, ST2 cultured cells were co-treated with EtOH and NOX inhibitors, DPI, gliotoxin and plumbagin, after which changes in ROS production, and in RANKL and NOX mRNA expression were analyzed.
Results
In WT mice, EtOH treatment significantly reduced bone density and mechanical strength, and increased total osteoclast number and activity. In EtOH-treated p47phoxKO mice, bone density and mechanical strength were completely preserved. EtOH p47phoxKO mice had no changes in osteoclast numbers or activity, and no elevations in serum CTX or RANKL gene expression (p<0.05). In both WT and p47phox KO mice, EtOH-feeding reduced biochemical markers of bone formation (P<0.05). In vitro EtOH exposure of ST2 cells increased ROS, which was blocked by pre-treating with DPI or the NOX2 inhibitor gliotoxin. EtOH induced RANKL and NOX2 gene expression which was inhibited by the NOX4-specific inhibitor plumbagin.
Conclusion
These data suggest that NOX2-derived ROS is necessary for EtOH-induced bone resorption. In osteoblasts NOX2 and NOX4 appear to work in tandem to increase RANKL expression whereas EtOH-mediated inhibition of bone formation occurs via a NOX2-independent mechanism.
Keywords: NADPH oxidase, bone loss, ethanol consumption, p47phox-/- KO mice
Introduction
Chronic alcohol abuse is a well known risk factor for osteoporotic bone loss. Numerous epidemiological studies have reported significant decreases in bone mineral density (BMD) and increased risk for osteoporotic fractures in heavy alcohol abusers compared to non-drinkers (Berg et al., 2008;Pasoto et al., 2011;Wuermser et al., 2011). The molecular mechanisms underlying alcohol’s action in bone are multifactorial, involving indirect disruption of normal hormonal function controlling bone growth and development (Badger et al., 1993;Mercer et al., 2012;Shankar et al., 2008b;Turner et al., 2010) and direct inhibition of bone formation and repair (Chakkalakal et al., 2005). In addition clinical studies and rodent models of chronic and binge drinking have also reported increased bone resorption following EtOH consumption (Alvisa-Negrin et al., 2009;Diez-Ruiz et al., 2010;Wezeman et al., 2007;Shankar et al., 2008). Previously, we have reported bone loss in female rats infused intragastrically with an isocaloric ethanol (EtOH)-liquid diet as part of a system of total enteral nutrition (TEN)(Chen et al., 2006;Shankar et al., 2006). In osteoblasts EtOH is metabolized to acetaldehyde via alcohol dehydrogenase, and this metabolism is required for elevating reactive oxygen species (ROS) in these cells (Chen et al., 2006;Chen et al., 2008). Increased ROS activates and sustains extracellular signal regulated kinases (ERK) which signals the phosphorylation of a downstream target, signal transducer and activator of transcription (STAT) 3. Phospho-STAT3 induces of receptor activator of nuclear factor kappa-β ligand (RANKL) expression in osteoblasts, thus promoting osteoclastogenesis and osteoclastic bone resorption (Chen et al., 2006). EtOH exposure increases ROS in osteoblasts through the induction and activation of three NADPH oxidase (NOX; NOX1, NOX2, NOX4) enzymes (Chen et al., 2006;Chen et al., 2008). Interestingly, co -administration of EtOH with diphenylene idonium (DPI), a pan-inhibitor of NOX activity, completely prevented induction of RANKL and bone resorption in female rats fed TEN diets, through inhibition of the ERK signaling pathway (Chen et al., 2011).
The NOX family consists of gp91phox (NOX2), NOX1, NOX3, NOX4, NOX5, DUOX-1 and DOUX-2, and are considered key producers of superoxide, which is required for normal cell function (Lambeth et al., 2007). NOX proteins are membrane bound catalytic subunits which require an additional membrane bound subunit, p22phox, for activity. Additionally, NOX 1/2, activation requires rac1, and two cytosolic subunits, p47phox and p67phox subunits for NOX2, and NOX organizer 1 (NOXO1) and NOX activator 1 (NOXA1) for NOX 1. (Bedard and Krause, 2007). Once assembled, these complexes catalyze the reduction of molecular oxygen to superoxide using NAPDH as an electron donor, which can be converted to hydrogen peroxide (H202) through the action of superoxide dismutase. (Bedard and Krause, 2007;Lambeth et al., 2007). In the bone microenvironment, hematopoietic stem cells commit to the osteoclastic lineage in response to RANKL produced by osteoblasts, osteocytes, and bone marrow stromal cells(O’Brien et al., 2012). Binding of RANKL to its receptor RANK, initiates a NOX-derived ROS-dependent signaling cascade considered essential for osteoclast differentiation (Roodman, 2006). More specifically, NOX1 is involved in osteoclast differentiation in response to RANKL signaling, and NOX2 and NOX4 have been shown to participate in the bone resorption activity of mature osteoclasts (Lee et al., 2005;Sasaki et al., 2009a;Sasaki et al., 2009b;Yang et al., 2001). Moreover in osteoclasts, increased expression of different NOX enzymes are able to compensate for the loss of particular isoforms. Support of this hypothesis comes from absence of published reports describing abnormal bone phenotypes in NOX1-/- or NOX2-/- knockout mice, and from in vitro studies using RNA interference to impair NOX function in osteoclastic cell lines (Gavazzi et al., 2006;Lee et al., 2005;Matsuno et al., 2005;Sasaki et al., 2009a).
Thus, there is accumulating evidence to suggest that Nox enzymes participate in a flexible compensatory mechanism to maintain ROS signaling post RANKL/RANK signaling in osteoclasts. By contrast, information regarding the role of NOX enzymes in osteoblast differentiation and function is somewhat limited (Mandal et al., 2011;Wittrant et al., 2009). Previous studies in our laboratory have shown that NOX enzymes participate in EtOH-mediated ROS production which results in bone loss (Chen et al, 2006, Chen et al, 2008). The co-administration of DPI or the anti-oxidant n-acetylcysteine (NAC) resulted in complete protection against EtOH-mediated bone resorption and EtOH inhibition of bone formation suggesting that NOX-derived ROS is involved in both signaling pathways (Chen et al., 2010). In the present study, we chronically fed EtOH liquid diets to C57BL/6J wild-type (WT) and C57BL/6J-Ncf1/p47phox-/- (p47phox KO) (Huang et al., 2000) to test if impairment of NOX2-derived ROS production results in the prevention of bone loss associated with EtOH exposure.
Materials and Methods
Animals and experimental design
All experimental procedures involving animals were approved by the Institutional Animal Care and Use Committee at the University of Arkansas for Medical Sciences. Mice were housed in an Association Assessment and Accreditation of Laboratory Animal Care approved animal facility. Fourteen, 6-week-old WT and p47phox KO female mice (Jackson Laboratories, Bar Harbor, ME) were randomly assigned to 3 weight-matched groups: a standard rodent chow diet (n=5/group); a 30% EtOH liquid diet (n=6); and a corresponding pair-fed (PF) control (n=3). All groups had access to water ad libitum. EtOH was added to the Lieber-DeCarli liquid diet (35% of energy from fat, 18% from protein, 47% from carbohydrates) by slowly substituting carbohydrate calories for EtOH calories (Dyets#710260) in a stepwise manner until 30% total calories were reached, which constitutes final EtOH concentration of 5.2% (v/v), respectively, and maintained until sacrifice (40 d) (Mercer et al., 2012). WT and p47phox KO mice fed the Lieber-DeCarli control diet (Dyets#710027) were isocalorically matched to their corresponding EtOH group based on the diet consumptions of the previous day (pair-fed, PF). At sacrifice, trunk blood was collected, and right femurs were harvested and frozen in saline soaked gauze at -80°C for mechanical strength testing. Right tibial bones were fixed in formalin for pQCT and μCT analyses, and left tibial bones were fixed in EtOH for immunohistochemistry. Blood EtOH concentrations (BEC) were analyzed using an Analox analyzer as previously described (Shankar et al., 2006).
Mechanical strength testing
Whole femur mechanical strength testing was accomplished by three-point bending using a MTS 858 Bionix test system load frame (MTS, Eden Prairie, MH) as described (Brown et al., 2002). Loading point was displaced at 0.1 mm/second until failure, and load displacement data was recorded at 100 Hz. Test curves were analyzed using TestWorks software (MTS, Eden Prairie, MH) to determine measures of whole-bone strength, which are peak load and stiffness. Load to failure was recorded as the load after a 2% drop from peak load.
Peripheral quantitative computerized tomography (pQCT) analyses
Ex vivo BMD, bone area, and bone mineral content (BMC) were measured in the tibias collected from PF and EtOH-treated WT and p47phoxKO mice using a STRATEC XCT Research SA+ pQCT machine (Orthometrix, White Plains, NY, USA) in a blinded fashion as previously described (Shankar et al., 2008a). Proximal tibias were analyzed using the manufacturer’s software version 5.40. Five contiguous sections, 1 mm apart, distal to the proximal end were measured for cortical BMD, area, and BMC with a spatial resolution of 100 μm. A threshold of 285 mg/cm3 was used to distinguish cortical bone. Average values for slices 3, 4, and 5 were calculated for statistical analysis.
Micro-computed tomography (μCT) analyses
All μCT analyses were consistent with current guidelines for the assessment of bone microstructure in rodents using micro-computed tomography (Bouxsein et al., 2010). Formalin-fixed tibiae were imaged using a MicroCT 40 (Scanco Medical AG, Bassersdorf, Switzerland) using a 12 μm isotropic voxel size in all dimensions. The region of interest selected for analysis comprised 240 transverse CT slices representing the entire medullary volume extending 1.24 mm distal to the end of the primary spongiosa with a border lying 100 um from the cortex. Three-dimensional reconstructions were created by stacking the regions of interest from each two-dimensional slice and then applying a gray-scale threshold and Gaussian noise filter as described (Suva et al., 2008) using a consistent and pre-determined threshold with all data acquired at 70 kVp, 114 mA, and 200-ms integration time. Bone was segmented from soft tissue using the same threshold, 247 mg HA/cm3 for trabecular bone. Fractional bone volume (bone volume/tissue volume; BV/TV) and architectural properties of trabecular bone (trabecular thickness (Tb.Th, μm), trabecular number (Tb.N., mm -1), and connectivity density (Conn. D, mm 3) were calculated using previously published methods (Suva et al., 2008).
Serum analysis of bone turnover markers
Serum osteocalcin (Biomedical Technologies, Inc., Stoughton, MA), and c-terminal telopeptides of type 1 (CTX) (Immunodiagnostic Systems, Fountain Hills, AZ) were detected in serum by commercially available ELISA kits. Serum levels of bone-specific alkaline phosphatase (BAP) was measured by a colorimetric assay as previously described (Chen et al., 2010).
Real-time RT PCR analyses
Total RNA was isolated from femur shaft using TRI reagent (MRC, Cincinnati, OH) as described previously (Chen et al., 2008). In cell culture, pre-osteoblastic ST2 cells were seeded (1 × 105 per well) in triplicate in 6 well plates and maintained in αMEM supplemented with 10% FBS, penicillin and streptomycin overnight, at which cells were treated with increasing concentrations of EtOH (0-100 mM). Additionally, ST2 cells (1 × 105 cells/ well) were pre-treated with an alcohol dehydrogenase inhibitor, 4-methylpyrazole (4-MP) or plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone), followed by EtOH treatment. The inhibitor stocks were diluted into CO2-conditioned media (αMEM supplemented with 10% FBS), which was added to the wells for a final concentration of 100 μM and 2.5 μM respectively. Following the pre-incubation step, 50-100 μl/ml of a 1M EtOH stock solution made in CO2-conditioned media is added to the appropriate plates for final EtOH concentration of 50-100 mM. To prevent EtOH evaporation in the media, all plates, including control plates without EtOH treatment, were wrapped in paraffin and maintained at 37°C at 37°C and 5% CO2 for 16 hours, at which total RNA was isolated using the RNeasy RNA isolation kit (Qiagen) as per manufacturer’s instructions. All RNA was reverse transcribed using IScript cDNA synthesis (Bio-Rad Laboratories, Hercules, CA) according to manufacturer’s instructions, and subsequent real-time PCR analysis was carried out using SYBR green and an ABI 7500 sequence detection system (Applied Biosystems, Foster City, CA). Results were quantified using deltaCT method relative to cyclophilin A and then to WT PF controls. In cultured cell experiments, results were quantified using the deltaCT method relative GAPDH, and then to the appropriate no EtOH controls. Comparisons of the raw CT values did not differ between groups (p=0.6), indicating that cyclophilin A and GAPDH were an appropriate normalizers. Gene specific primers were: osteocalcin F 5’ TTGTGCTGGAGTGGTCTCTATGAC 3’, R 5’CACCCTCTTCCCACACTGTACA 3’; collagen type 1a F 5’ AGGGTCATCGTCGCTTCTC 3’, R 5’ CTCCAGAGGGGGCTTGTT 3’; RANKL F 5’ GGGTTCGACACCTGAATGCT 3’, R 5’ AACTGGTCGGGCAATTCTGG3’; NOX2 F 5’ ACCGCCATCCACACAATTG 3’, R 5’ CCGATGTCAGAGAGAGCTATTGAA 3’; NOX4 F 5’ CTGCATCTGTCCTGAACCTCAA 3’, R 5’ TCTCCTGCTAGGGACCTTCTGT 3’; NOX1F 5’ ATGCCCCTGCTGCTCGAATA 3’, R 5’ AAATTGCCCCTCCATTTCCT 3’; cyclophilin A F 5’AAGGTGGAGAGCACCAAGACA 3’, R 5’ GCAATGGCGAAGGGTTTCT 3’; and GAPDH 5’ GTATGACTCGACTCACGGCAAA 3’, R 5’ GGTCTCGCTCCTGGAAGATG 3’.
Quantitative histomorphometry
For static histomorphometric analyses, 4-μm-thick central sagittal sections of undecalcified MMA-embedded tibiae were stained for tartrate-resistant acid phosphatase (TRAP) and counterstained with hematoxylin to determine osteoclast numbers and eroded surface per trabecular bone surface within the region of interest using Osteomeasure software (Osteometrics, Atlanta, GA) as previously described (Perrien et al., 2007).
Detection of EtOH-stimulated ROS in ST2 cells
EtOH-stimulated hydrogen peroxide was measured in cultured cells using the Amplex Red hydrogen peroxide /peroxidase assay (Invitrogen Molecular Probes, Eugene, OR) as per manufacturer’s instructions. Briefly, ST2 cells were washed twice in suspension with Hank’s balanced salt solution, and seeded in triplicate (2 × 104 per well) in 96-well clear bottom microplates. After a 30 minute pre-incubation period with different NADPH oxidase inhibitors: DPI (0.5 μM), gliotoxin (5 μM) and plumbagin (5 μM), the Amplex Red reaction buffer (50 μM Amplex red, 0.1 U.ml-1 HRP) was added to the cells, which was followed by EtOH treatment (50 mM) for 1 hour at 37°C before measuring absorbance at 560 nm. Data are expressed as the rate of H202 production per minute, which was corrected for non-specific H202 production by subtracting experimental values from values obtained from control wells; ST2 cells plus inhibitor without EtOH treatment.
Data and statistical analysis
Data are presented as means ± SEM. Comparisons between two groups were accomplished using Student’s T-test. Comparison between multiple groups was accomplished by one-way ANOVA, followed by Student Newman-Keuls post hoc analysis. The effect of the p47phox KO genotype, and EtOH and the interaction thereof were determined using two-way ANOVA, followed by Student Newman-Keuls post hoc analysis. Statistical significance was set at P<0.05. SigmaPlot software package 11.0 (Systat Software, Inc., San Jose, CA) was used to perform all statistical tests.
Results
Study observations
At sacrifice, the mean BECs were 197.52±44.2 mg/dL (range 10-386.2) and 138.14±40.8 mg/dL (range 10-266.1) for the WT EtOH group and the p47phox KO EtOH group, respectively and did not statistically differ from each other (p=0.367, Student’s T-test). These values are comparable to those observed in chronic alcoholics (Wadstein and Skude, 1979) and in mice fed Lieber-DeCarli EtOH diets (Mercer et al., 2012). Mean body weights were lower in both EtOH groups when compared to PF controls, 17.7 ± 0.45 and 22.0 ± 2.0, respectively (p<0.05) for WT mice and 20.1±0.3 and 22.0±1.1, respectively (p=0.06) for p47phox KO mice. We observed no differences in body weight between chow fed and PF controls from WT mice (p=0.160) and a slight increase in final weight between the chow fed and PF p47phox KO mice, 18.6±0.5 and 22±1.1, p<0.05).
P47phox KO mice are protected against EtOH-mediated bone loss
Following 40 d of chronic EtOH-feeding, cortical and trabecular bone was assessed by pQCT and μCT, respectively, in tibial bone taken from WT and p47phox KO chow-fed, PF and EtOH groups. In the chow-fed groups, there were no significant differences in any of the cortical bone parameters measured between the WT and p47phox KO mice (Table 1). In the trabecular compartment, feeding the high fat LieberDeCarli control diet decreased Tb.Th. in both WT and p47phox KO PF groups compared to chow-fed controls (p<0.05) (Table 2). In WT mice, chronic EtOH-feeding had a significant impact on cortical bone parameters, reducing BMC, BMD, area and thickness by 20%, 9%, and 13% and 11% respectively when compared to PF controls (Table 1). As shown in Table 2 EtOH-feeding also decreased trabecular BV/TV, Tb.N., and Conn.D., and increased Tb.Sp. in the WT EtOH mice compared to its PF control, (p<0.05). In p47phox KO mice, EtOH-feeding reduced BMD by 5.2% in comparison to its PF control (p<0.05), but had no effect on other cortical bone parameters measured, BMC, area, and thickness (Table 1). With respect to trabecular bone, we did not observe any significant changes in BV/TV, Tb.N, or Tb.Sp. between PF and EtOH-treated p47phox KO groups (Table 2). Consistent with these findings, chronic EtOH-feeding reduced the load-bearing strength and stiffness of femurs taken from EtOH-treated WT mice (p<0.05), but not in EtOH-treated p47phox KO mice (Figure 1).
Table 1.
pQCT analysis of tibial cortical bone in WT and p47phox KO chow, PF and EtOH-treated mice.
| WT | p47phox KO | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| Chow | PF | ETOH | Chow | PF | EtOH | |
| BMC, mg | 0.82 (0.02)a | 0.74 (0.03)a | 0.59 (0.02)b | 0.77 (0.025)a | 0.72 (0.03)a | 0.71 (0.02)a |
| BMD, mg/cm3 | 728.27 (6.87)a | 713.81 (9.72)a | 648.81 (7.55)b | 718.10 (6.87)a | 707.12 (9.72)a | 670.47 (8.15)b,c |
| Total area, mm2 | 1.13 (0.02)a | 1.05 (0.04)a | 0.91 (0.02)b | 1.08 (0.03)a | 1.03 (0.04)a | 1.07 (0.02)a |
| Thickness, mm | 0.27 (0.00)a | 0.26 (0.00)a | 0.23 (0.00)b | 0.27 (0.00)a | 0.261(0.00)a | 0.259 (0.00)a |
Data are means ±SEM; n=5animals for chow fed groups, n=3 for PF groups, n=6 for EtOH-treated groups. Statistical analysis was performed by two-way ANOVA followed by Student-Newman Keuls post hoc analysis. Groups within a row with different letters are significant from each other (p<0.05).
Table 2.
μCT analysis of tibial trabecular bone in WT and p47phox KO chow PF and EtOH-treated mice.
| WT | p47phox KO | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| Chow | PF | ETOH | Chow | PF | EtOH | |
| BV/TV (%) | 6.876 (0.814)a | 6.677 (1.051)a | 4.239 (0.913)b | 6.813 (0.688)a | 7.390 (1.051)a | 7.672 (0.646)a |
| Tb.N, 1/mm | 3.216 (0.254)a | 3.530 (0.288)a | 2.555 (0.157)b | 3.464 (0.189)a | 3.420 (0.288)a | 3.312 (0.249)a |
| Tb.Th, mm | 0.045 (0.001)a | 0.038 (0.001)b | 0.036 (0.001)b | 0.047 (0.001)a | 0.041 (0.001)b | 0.041 (0.001)b |
| Tb.Sp, mm | 0.329 (0.020)a | 0.297 (0.027)a | 0.405 (0.223)b | 0.304 (0.019)a | 0.293 (0.027)a | 0.313 (0.223)a |
| Conn.D, 1/mm3 | 34.7 (8.80)a | 50.0 (11.3)a | 18.87 (6.23)b | 42.4 (8.03)a | 43.3 (11.3)a | 54.6 (6.77)a |
Data are means ±SEM; n=5 animals for chow fed groups, n=3 for PF groups, n=6 for EtOH-treated groups. Statistical analysis was performed by two-way ANOVA followed by Student-Newman Keuls post hoc analysis. Groups within a row with different letters are significant from each other (p<0.05).
Figure 1.
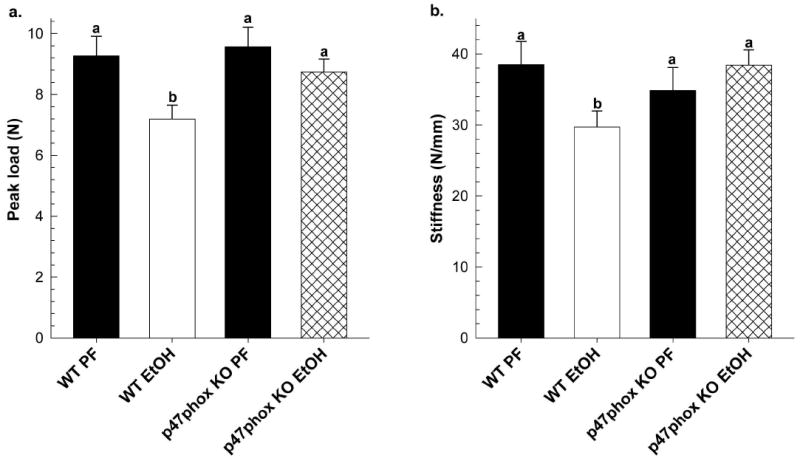
Mechanical strength testing of whole femurs from WT and p47phox KO EtOH-treated mice (n=6/group) and corresponding PF controls (n=3/group), (a) peak load and (b) stiffness. Statistical significance was determined by Two-way ANOVA followed by Student Newman-Keuls post hoc analysis. Values with different letter subscripts are significantly different from each other (P<0.05).
The p47phox KO genotype protects against EtOH-mediated bone resorption
In response to EtOH-feeding, a 55% increase in circulating CTX, a marker for increased bone resorption, was observed in WT mice (p<0.05 vs. PF control). However, no significant increase was observed in CTX in EtOH-treated p47phox KO mice (Figure 2a). In Figure 3, static histomorphometric analysis of formalin-fixed tibia taken from EtOH-treated WT mice revealed significant increases in total osteoclast number (2-fold) and in osteoclast activity, as determined by the 3-fold increase in number of osteoclasts associated with the bone perimeter, p<0.05. In contrast, no significant increases in osteoclast number or osteoclast activity was observed in response to EtOH-feeding in p47phox KO EtOH-treated mice, p=0.145 and p=0.165, respectively (Figure 3). In WT mice, EtOH exposure increased the mRNA expression of RANKL and NOX enzymes in femur bone (Figure 4). A 3-fold increase in NOX2 mRNA expression was observed in the WT EtOH group compared to its PF control (p<0.05). Likewise, NOX4 expression in the WT EtOH group increased 2-fold, but this did not achieve statistical significance relative to PF controls (p=0.100). In the p47phox KO mice, EtOH exposure did not significantly increase RANKL, NOX2 or NOX4 mRNA expression compared to its PF control.
Figure 2.
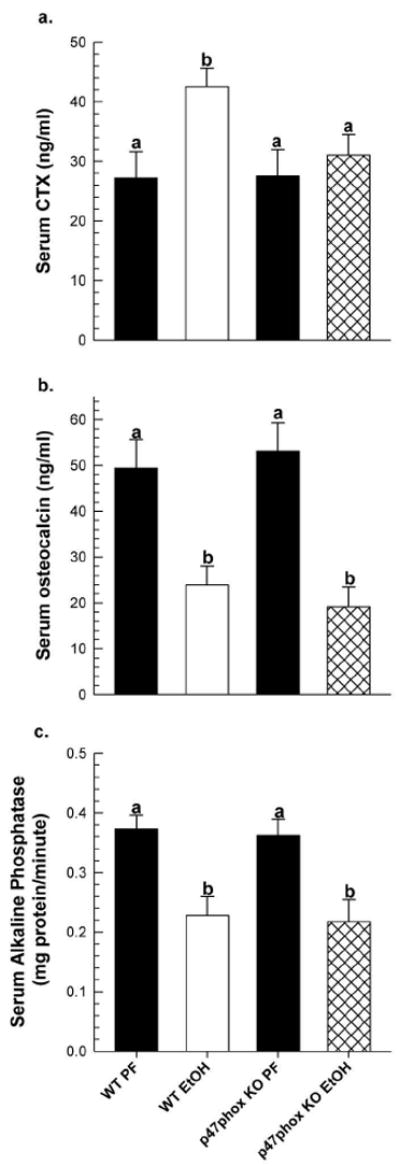
Changes in serum bone turnover markers in WT and p47phox KO EtOH-treated mice (n=6/group) and corresponding PF controls (n=3/group); bone resorption marker (a) CTX, and bone formation markers (b) osteocalcin, and (c) bone-specific alkaline phosphatase. All data is expressed as mean ± SEM. Statistical significance was determined by Two-way ANOVA followed by Student Newman-Keuls post hoc analysis. Groups with different letter subscripts are significantly different from each other (P<0.05).
Figure 3.
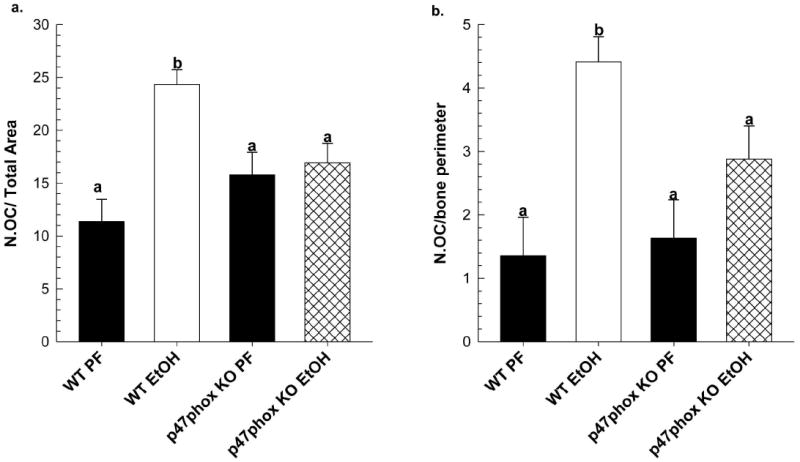
Quantitative histomorphometry analysis of TRAP-stained osteoclasts; (a) osteoclast number (N.OC) per total area, and (b) N.OC per bone perimeter in undecalcified MMA-embedded tibias from WT and p47phox KO EtOH (n=6/group) and PF controls (n=3/group) as described in Materials and Methods. Data are expressed as mean ± SEM. Statistical significance was determined by Two-way ANOVA followed by Student-Newman Keuls post hoc analysis. Groups with different letters are significantly different from each other (p<0.05).
Figure 4.
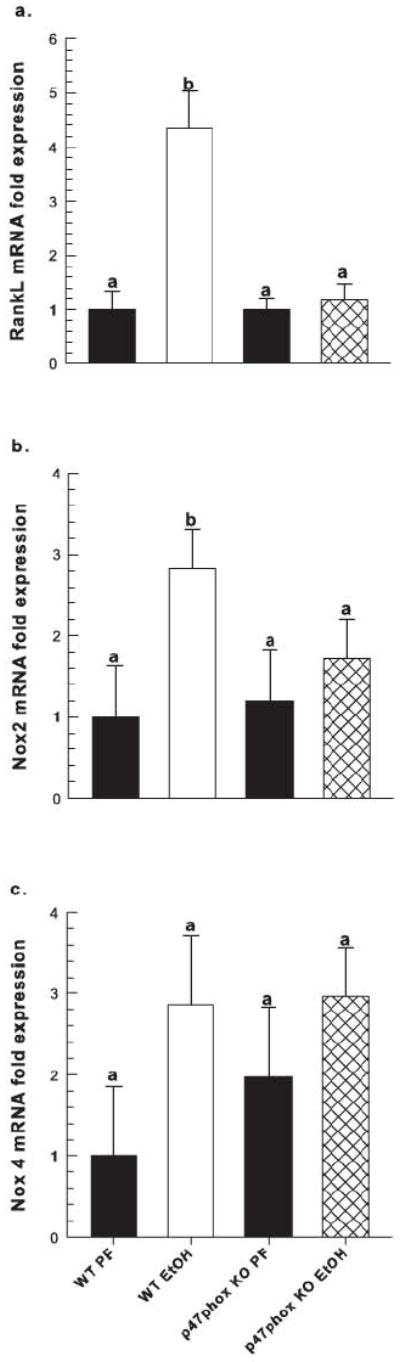
Gene expression of (a) RANKL, (b) NOX2, and (c) NOX4, (d) osteocalcin and (e) collagen a type 1(COLA1) in femur shaft of EtOH-treated WT and p47phox KO mice (n=6/group) and their corresponding PF controls (n=3/group) as measured by real-time PCR, normalized to cyclophilinA mRNA. Fold expression is reported as mean ± SEM. Statistical significance was determined by Two-way ANOVA followed by Student Newman-Keuls post hoc analysis. Groups with different letter subscripts are significantly different from each other (P<0.05). For osteocalcin a vs. ab, p=0.154, for COLA1 a vs. ab, p=0.105.
The p47phox KO genotype does not protect against EtOH-mediated inhibition of bone formation
Interestingly, EtOH-treated p47phox KO mice were not protected from reduced bone formation. In both WT and p47phox KO EtOH-treated mice, serum osteocalcin concentrations in both EtOH groups decreased by 52% and 64% respectively, in comparison to PF controls, p,0.05 (Figure 2b). Likewise serum alkaline phosphatase activity, another biochemical marker for bone formation, was significantly decreased in WT EtOH- and p47phox KO-treated groups in comparison to PF controls (Figure 2c). Additionally, real-time RT PCR analysis of gene expression in EtOH-treated WT and p47phox KO femurs showed 2-fold reductions in osteocalcin mRNA expression, p=0.154 and p=0.035 respectively and 2-3 fold reductions in collagen a type 1 expression, p=0.046 and p=0.022, respectively, as compared to their appropriate PF controls (data not shown).
Inhibition of NOX2 activity by chemical inhibitors reduces transient EtOH-mediated ROS production in cultured cells
Previously we have shown that EtOH exposure generates excess NOX-derived ROS production in osteoblasts, which in turn increases RANKL-RANK signaling to pre-osteoclastic cells (Chen et al., 2008). In the ST2 cells, a stromal cell line derived from mouse bone marrow, three NOX isoforms, NOX4, NOX2, and NOX1, are expressed as determined by real-time RT-PCR. As shown in Figure 5a, NOX4 is highly expressed, followed by NOX2, and minimal expression of NOX1. To show that NOX2 generates significant ROS following EtOH exposure, we measured H202 production in EtOH-treated ST2 cells using a stable fluorogenic reagent, Amplex Red. As expected acute EtOH exposure (1hr) increased H202 production in the ST2 cells when compared to their no EtOH control cells, 0.179 mM/minute versus 0.102 mM/minute, respectively, p<0.05. DPI is a broad-spectrum inhibitor of electron transporters including NOX2, NOX4, mitochondrial oxidase and xanthine oxidase (Drummond et al., 2011). As expected, pre-treating ST2 cells with 0.5 μM DPI prior EtOH exposure significantly reduced H202 production (60%). Pre-treating cells with 5 μM gliotoxin, a known NOX2 inhibitor (Serrendar et al., 2007), also inhibited EtOH-generated H202 production by 72%, p<0.05. Interestingly, plumbagin, a plant derived napthoquinone, which has been used to specifically inhibit NOX4 activity in cell lines (Ding et al., 2005), did not significantly decrease H202 production compared to either DPI or gliotoxin.
Figure 5.
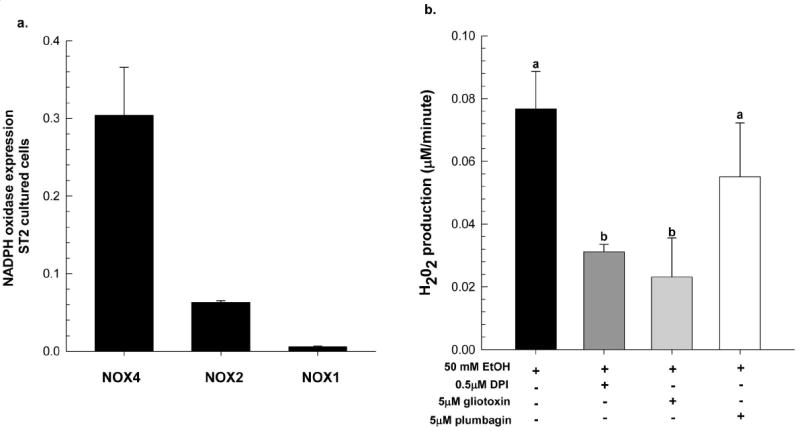
Gene expression of (a) NADPH oxidase enzymes in cultured ST2 cells as measured by real-time PCR, normalized to GAPDH mRNA; fold expression is reported as mean ± SEM. In a separate experiment, (b) ST2 cells were pre-incubated with different NADPH oxidase inhibitors for 30 minutes prior to EtOH-treatment (50 mM) for 1 hr at 37°C, at which H202 production was measured using the Amplex Red hydrogen peroxide/peroxidase assay kit as described in Materials and Methods. Data presented here are representative of two independent experiments performed in triplicate and expressed as the rate of H202 production per minute which were corrected for background absorbance by subtracting experimental values from those obtained in control wells; ST2 cells +/- inhibitor without EtOH treatment. Statistical significance was determined by One-way ANOVA followed by Student-Newman Keuls post hoc analysis, a<b, p<0.05.
NOX2- and NOX4-derived ROS are important in increasing RANKL expression in osteoblastic cells
In ST2 cells, prolonged exposure to EtOH (16hrs) resulted in significant increases in RANKL and NOX2 mRNA, p<0.05 (Figure 6a). Treating ST2 cells with an alcohol dehydrogenase inhibitor, 4-methylpyrozole (4MP) 30 minutes prior to EtOH exposure reduced both RANKL and NOX2 expression by 30 to 40% (p<0.05) (Figure 6b). As shown in Figure 6d, pre-treatment with plumbagin (2.5 μM) also decreased RANKL mRNA expression following EtOH exposure, p<0.05.
Figure 6.
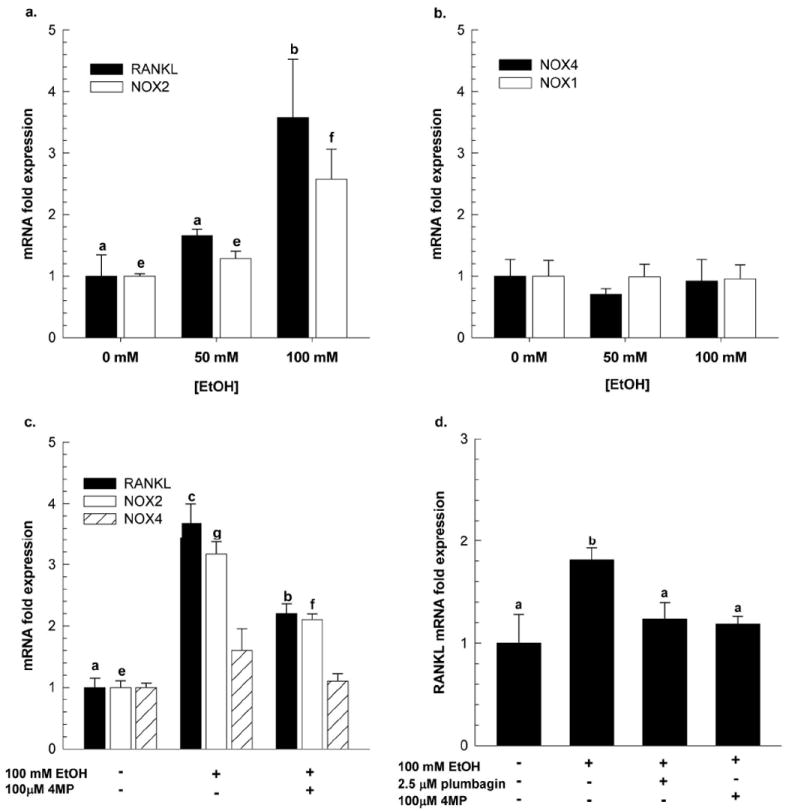
ST2 cells were treated with increasing concentrations of EtOH for 16 hrs and gene expression of (a) RANKL and NOX2, and (b) NOX4 and NOX1 were measured by real-time PCR, which was normalized to GAPDH mRNA. Fold expression is reported as mean ± SEM. In separate experiments, ST2 cells were treated with either (c) 100 mM EtOH +/- 100μM 4 methylpyrazole (4MP) or (f) 100mM EtOH +/- 2.5 μM plumbagin for 16 hours and gene expression of RankL, NOX2 and NOX4 were measured by real-time PCR, normalized to GAPDH mRNA. Data is expressed as the fold increase in RANKL expression relative to no EtOH controls plus the appropriate inhibitor. Data presented here are representative of two independent experiments performed in triplicate. Statistical significance was determined by One-way ANOVA followed by Student-Newman Keuls post hoc analysis. For RANKL expression, p<0.05 a<b<c; for Nox2 expression, p<0.05 e<f<g.
Discussion
Previously, we have reported that EtOH feeding in female rats increases ROS in bone which in return up-regulates RANKL expression and stimulates osteoclastogenesis and bone resorption through increased RANKL/RANK signaling. In osteoblasts, EtOH is metabolized to acetaldehyde by alcohol dehydrogenase (ADH) instead of through cytochrome P450 2E1 which is not expressed. Therefore the EtOH-generated ROS necessary for increased RANKL expression may be dependent on increased NOX expression and activity or changes in cellular redox status associated with ADH or ALDH catalyzed formation of NADH in response to EtOH and acetaldehyde metabolism (Chen et al., 2006). Interestingly, co-treatments of EtOH with DPI or the antioxidant N-acetyl cysteine prevented EtOH-dependent ROS formation in bone through inhibition of NOX enzymes, specifically NOX1, NOX2 and NOX4 (Chen et al., 2008;Chen et al., 2011). Similar NOX-dependent signaling pathways may regulate RANKL expression in osteoblasts, osteocytes and other bone marrow cell types.
To test the hypothesis that NOX2 specifically is involved in EtOH up-regulation of RANKL expression in bone and in EtOH-induced bone resorption, we chronically fed EtOH to WT and p47phox KO female mice to see if EtOH-induced bone loss is dependent on a functional NOX2 enzyme. As expected and consistent with our previous studies, chronic EtOH-feeding in WT mice produced significant reductions in cortical BMD and trabecular bone architecture resulting in a loss in mechanical strength (Mercer et al., 2012). Bone loss was associated with significant reductions in serum concentrations of osteocalcin and alkaline phosphatase activity, suggestive of decreased bone formation, and increased serum concentrations of CTX, a marker for bone resorption and increased expression RANKL mRNA in femur bone. In addition, static histomorphometric analysis of tibial bone demonstrated that bone loss in EtOH-treated WT mice was associated with increased osteoclast numbers and enhanced osteoclastic activity. In contrast, p47phox KO mice were protected against EtOH-induced bone loss. We observed no reductions in cortical BMC, area or thickness or adverse changes in trabecular %BV/TV, Tb.N or Tb.Sp. to suggest a loss of bone. Likewise, mechanical strength was also preserved in the EtOH-treated p47phox KO femurs in comparison to the EtOH-treated WT femurs. Mechanistically, RANKL expression was not increased in EtOH-treated p47phox KO femurs, and there were no observable increases in osteoclast numbers or bone resorbing activity in these mice. Given the relatively small number of WT and p47phox KO animals used in this study, it was surprising to observe such statistically robust differences between the genotypes in response to EtOH feeding. Several laboratories have reported that in osteoclastic cells NOX isoforms participate in ROS signaling pathway downstream of RANKL/RANK binding to mediate osteoclast differentiation and activity (Lee et al., 2005;Sasaki et al., 2009a;Yang et al., 2001). Thus loss of NOX2 activity in osteoclast precursors and thus reduced signaling post RANKL/RANK binding may explain in part our in vivo findings in the p47phox KO mice receiving EtOH. However, in WT and p47phox KO chow fed and PF controls, we observed no appreciable differences in bone density, strength, or turnover that would support a phenotype of deficiency in osteoclastogenesis or bone resorption at this age associated iwht the p47phox KO genotype itself.
Surprisingly the p47phox KO mice were not protected against EtOH-mediated reductions in bone formation serum markers. Moreover, we observed decreases in osteocalcin and collagen a type 1 mRNA expression in femurs from EtOH-treated WT and p47phox KO mice in comparison to their PF controls. These findings suggest that in response to EtOH, NOX enzymes have distinct roles and that NOX1 or NOX4 may be involved in regulating EtOH effects on osteoblast differentiation. Other studies have reported that NOX isoforms may regulate separate signaling pathways (Anilkumar et al., 2008;Mandal et al., 2011;Piccoli et al., 2007). For example, Anilkumar et al. has reported increased signaling of mitogen-activated protein kinase (MAPK) pathways, JNK, Akt, and GSK3β in response to insulin stimulation in HEK293 NOX4-overxpressing cells, but not in NOX2 over-expressing cells. Moreover, tumor necrosis factor-α stimulation increased MAPK signaling through ERK1/2 in NOX2-overexpressing cells, but had no stimulatory effect on MAPK pathways in NOX4-overexpressing cells (Anilkumar et al., 2008). Of the two enzymes, NOX4 is the more likely candidate to mediate effects of EtOH on bone formation. We and others have reported that NOX4 is the predominate NOX isoform expressed in primary osteoblasts and cell lines (Chen et al., 2011;Mandal et al., 2011;Wittrant et al., 2009). Again in the ST2 cell line used in this study, NOX4 mRNA is more abundant compared NOX1 or NOX2 transcripts. It is worth mentioning that EtOH and acetaldehyde metabolism can stimulate NOX-independentsources of ROS for example as a result of mitochondrial respiratory uncoupling, and DPI is also known to inhibit these NOX-independent sources of ROS. Thus it is possible that these additional ROS sources may be involved in EtOH effects on osteoblast differentiations (Lieber, 1996). Future studies will include in vitro experiments using RNA interference to knock down NOX4 or key EtOH metabolizing enzymes to determine if NOX4- and/or mitochondrial-derived ROS are responsible for EtOH-mediated inhibition of osteoblastogenesis.
In this study, we demonstrated that p47phox KO mice are protected against EtOH-mediated bone resorption through the loss of a functional NOX2 enzyme. Still, this finding does not necessarily exclude the involvement of other NOX isoforms in increasing RANKL expression. Several NOX isoforms are up-regulated in rat femurs receiving TEN EtOH diets, and in osteoblastic cell lines treated with physiological doses of EtOH (Chen et al., 2006; Chen et al., 2008; Chen et al., 2011). In the present study, NOX2, but not NOX4, was significantly up-regulated in femurs taken from EtOH-treated WT mice. In the literature, authors have suggested that in some cell types NOX isoforms work in tandem (Piccoli et al., 2007;Yeligar et al., 2012). One hypothesis is that the constitutively active NOX4 is on the top of the cascade, acting as an oxygen sensor producing ROS, which signals to activate other NOX isoforms (Piccoli et al., 2007). In ST2 cells, transient EtOH-derived ROS (1hr), as measured by H202 production was inhibited by DPI at a dose specific for NOX inhibition (Serrander et al., 2007) and the NOX2 inhibitor gliotoxin, but not plumbagin, a recently described NOX4 inhibitor. In longer exposures to EtOH, real-time RT-PCR analysis showed a significant increase in NOX2 expression coinciding with the up-regulation of RANKL expression. Pre-treatment of cells with 4MP or plumbagin prior to EtOH exposure decreased RANKL expression. Taken together, we propose that in response to EtOH, NOX isoforms work in tandem in regulating the signaling cascade for increased RANKL expression in osteoblasts. NOX2 activation appears to be the major initial source of ROS in response to EtOH. However, amplification of the ROS signal and subsequent signaling through the ERK-STAT3 pathway appears to involve NOX4-dependent induction of additional NOX2 expression (Figure 7). Consistent with this model are our previous data demonstrating that the induction of ERK-STAT3 signaling by EtOH in osteoblast cells lines in vitro requires a minimum of 6 h exposure and can be blocked by the protein synthesis inhibitor cycloheximide (Chen et al. 2008). It is worth noting that some authors speculate that plumbagin’s structure may impart ROS-scavenging effects and limit its usefulness as a true NOX4 inhibitor (Drummond et al., 2011). Therefore, additional studies using RNAi technology to knockdown NOX2, NOX4 or both, are ongoing to delineate the role of NOX4 in increasing RANKL expression in EtOH-treated osteoblasts.
Figure 7.
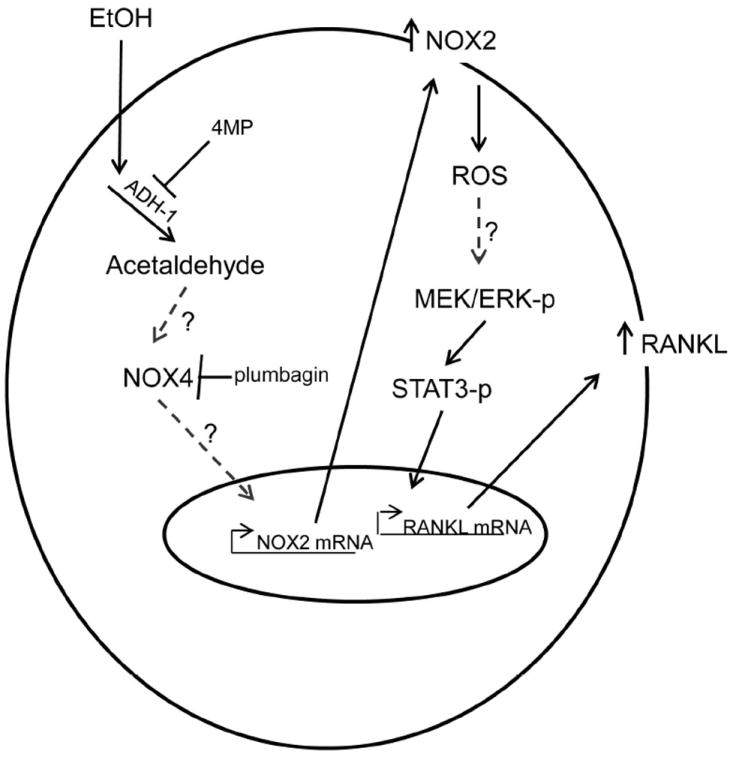
Schematic diagram describing the role of NOX/ROS in the EtOH-mediated signaling cascade for increased RankL expression in osteoblasts. EtOH metabolism via alcohol ADH produces ROS which induces/activates NOX4 and NOX2 in tandem, followed by chronic activation of the ERK1/2/STAT3 phosphorylation cascade to increase RANKL expression. The ADH-1 inhibitor 4MP and NOX4-specific inhibitor plumbagin, both inhibit EtOH-induced increases in RANKL expression. Dashed lines represent unknown mechanisms of action to be determined in future studies.
In summary, we have demonstrated that NOX2-derived ROS is necessary for increased RANKL/RANK signaling between osteoblasts and osteoclasts, and enhanced bone resorption in response to chronic EtOH administration. In the p47phox KO mice, NOX1 and NOX4 activity were unable to compensate for the loss in NOX2 function in osteoblasts resulting in a lack of RANKL induction and subsequent EtOH-mediated bone resorption in these mice. However, loss of NOX2 function did not prevent EtOH-mediated inhibition of bone formation. Other NOX enzymes, particularly NOX4 may regulate osteoblastogenesis independently from NOX2, as suggested from our previous studies showing complete protection against both decreased bone formation and increased bone resorption in rats co-administered with DPI or N-acetyl cysteine (Chen et al., 2011). Our findings suggests that NOX-derived ROS signaling in osteoblasts is complex with NOX enzymes working separately or in tandem of each other, to regulate bone turnover pathways in the cell.
Acknowledgments
Support: This work was supported by the National Institute of Health [RO1 AA18282], the and the Carl L. Nelson Chair in Orthopaedic Creativity, University of Arkansas for Medical Sciences.
Reference List
- Alvisa-Negrin J, Gonzalez-Reimers E, Santolaria-Fernandez F, Garcia-Valdecasas-Campelo E, Valls MR, Pelazas-Gonzalez R, Duran-Castellon MC, de Los Angeles Gomez-Rodriguez Osteopenia in alcoholics: effect of alcohol abstinence. Alcohol Alcoholism. 2009;44:468–475. doi: 10.1093/alcalc/agp038. [DOI] [PubMed] [Google Scholar]
- Anilkumar N, Weber R, Zhang M, Brewer A, Shah AM. Nox4 and nox2 NADPH oxidases mediate distinct cellular redox signaling responses to agonist stimulation. Arterioscler Thromb Vasc Biol. 2008;28:1347–1354. doi: 10.1161/ATVBAHA.108.164277. [DOI] [PubMed] [Google Scholar]
- Badger TM, Ronis MJ, Lumpkin CK, Valentine CR, Shahare M, Irby D, Huang J, Mercado C, Thomas P, Ingelman-Sundberg M. Effects of chronic ethanol on growth hormone secretion and hepatic cytochrome P450 isozymes of the rat. J Pharmacol Exp Ther. 1993;264:438–447. [PubMed] [Google Scholar]
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- Berg KM, Kunins HV, Jackson JL, Nahvi S, Chaudhry A, Harris KA, Jr, Malik R, Arnsten JH. Association between alcohol consumption and both osteoporotic fracture and bone density. Am J Med. 2008;121:406–418. doi: 10.1016/j.amjmed.2007.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Muller R. Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J Bone Miner Res. 2010;25:1468–1486. doi: 10.1002/jbmr.141. [DOI] [PubMed] [Google Scholar]
- Brown EC, Perrien DS, Fletcher TW, Irby DJ, Aronson J, Gao GG, Hogue WJ, Skinner RA, Suva LJ, Ronis MJ, Hakkak R, Badger TM, Lumpkin CK., Jr Skeletal toxicity associated with chronic ethanol exposure in a rat model using total enteral nutrition. J Pharmacol Exp Ther. 2002;301:1132–1138. doi: 10.1124/jpet.301.3.1132. [DOI] [PubMed] [Google Scholar]
- Chakkalakal DA. Alcohol-induced bone loss and deficient bone repair. Alcohol Clin Exp Res. 2005;29:2077–2090. doi: 10.1097/01.alc.0000192039.21305.55. [DOI] [PubMed] [Google Scholar]
- Chen JR, Haley RL, Hidestrand M, Shankar K, Liu X, Lumpkin CK, Simpson PM, Badger TM, Ronis MJ. Estradiol protects against ethanol-induced bone loss by inhibiting up-regulation of receptor activator of nuclear factor-kappaB ligand in osteoblasts. J Pharmacol Exp Ther. 2006;319:1182–1190. doi: 10.1124/jpet.106.109454. [DOI] [PubMed] [Google Scholar]
- Chen JR, Lazarenko OP, Shankar K, Blackburn ML, Badger TM, Ronis MJ. A role for ethanol-induced oxidative stress in controlling lineage commitment of mesenchymal stromal cells through inhibition of Wnt/beta-catenin signaling. J Bone Miner Res. 2010;25:1117–1127. doi: 10.1002/jbmr.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JR, Lazarenko OP, Shankar K, Blackburn ML, Lumpkin CK, Badger TM, Ronis MJ. Inhibition of NADPH oxidases prevents chronic ethanol-induced bone loss in female rats. J Pharmacol Exp Ther. 2011;336:734–742. doi: 10.1124/jpet.110.175091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JR, Shankar K, Nagarajan S, Badger TM, Ronis MJ. Protective effects of estradiol on ethanol-induced bone loss involve inhibition of reactive oxygen species generation in osteoblasts and downstream activation of the extracellular signal-regulated kinase/signal transducer and activator of transcription 3/receptor activator of nuclear factor-kappaB ligand signaling cascade. J Pharmacol Exp Ther. 2008;324:50–59. doi: 10.1124/jpet.107.130351. [DOI] [PubMed] [Google Scholar]
- Diez-Ruiz A, Garcia-Saura PL, Garcia-Ruiz P, Gonzalez-Calvin JL, Gallego-Rojo F, Fuchs D. Bone mineral density, bone turnover markers and cytokines in alcohol-induced cirrhosis. Alcohol Alcoholism. 2010;45:427–430. doi: 10.1093/alcalc/agq037. [DOI] [PubMed] [Google Scholar]
- Ding Y, Chen ZJ, Liu S, Che D, Vetter M, Chang CH. Inhibition of Nox-4 activity by plumbagin, a plant-derived bioactive naphthoquinone. J Pharm Pharmacol. 2005;57:111–116. doi: 10.1211/0022357055119. [DOI] [PubMed] [Google Scholar]
- Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov. 2011;10:453–471. doi: 10.1038/nrd3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavazzi G, Banfi B, Deffert C, Fiette L, Schappi M, Herrmann F, Krause KH. Decreased blood pressure in NOX1-deficient mice. FEBS Lett. 2006;580:497–504. doi: 10.1016/j.febslet.2005.12.049. [DOI] [PubMed] [Google Scholar]
- Huang CK, Zhan L, Hannigan MO, Ai Y, Leto TL. P47(phox)-deficient NADPH oxidase defect in neutrophils of diabetic mouse strains C57BL/6J-m db/db and db/+ J Leukoc Biol. 2000;67:210–215. doi: 10.1002/jlb.67.2.210. [DOI] [PubMed] [Google Scholar]
- Lambeth JD, Kawahara T, Diebold B. Regulation of Nox and Duox enzymatic activity and expression. Free Radic Biol Med. 2007;43:319–331. doi: 10.1016/j.freeradbiomed.2007.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee NK, Choi YG, Baik JY, Han SY, Jeong DW, Bae YS, Kim N, Lee SY. A crucial role for reactive oxygen species in RANKL-induced osteoclast differentiation. Blood. 2005;106:852–859. doi: 10.1182/blood-2004-09-3662. [DOI] [PubMed] [Google Scholar]
- Mandal CC, Ganapathy S, Gorin Y, Mahadev K, Block K, Abboud HE, Harris SE, Ghosh-Choudhury G, Ghosh-Choudhury N. Reactive oxygen species derived from Nox4 mediate BMP2 gene transcription and osteoblast differentiation. Biochem J. 2011;433:393–402. doi: 10.1042/BJ20100357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuno K, Yamada H, Iwata K, Jin D, Katsuyama M, Matsuki M, Takai S, Yamanishi K, Miyazaki M, Matsubara H, Yabe-Nishimura C. Nox1 is involved in angiotensin II-mediated hypertension: a study in Nox1-deficient mice. Circulation. 2005;112:2677–2685. doi: 10.1161/CIRCULATIONAHA.105.573709. [DOI] [PubMed] [Google Scholar]
- Mercer KE, Wynne RA, Lazarenko OP, Lumpkin CK, Hogue WR, Suva LJ, Chen JR, Mason AZ, Badger TM, Ronis MJ. Vitamin D supplementation protects against bone loss associated with chronic alcohol administration in female mice. J Pharmacol Exp Ther. 2012;343:401–412. doi: 10.1124/jpet.112.197038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien CA, Nakashima T, Takayanagi H. Osteocyte control of osteoclastogenesis. Bone. 2012;54:258–263. doi: 10.1016/j.bone.2012.08.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasoto SG, Yoshihara LA, Maeda LC, Bernik MM, Lotufo PA, Bonfa E, Pereira RM. Osteoporotic hip fractures in non-elderly patients: relevance of associated co-morbidities. Rheumatol Int. 2011;32:3149–3153. doi: 10.1007/s00296-011-2154-x. [DOI] [PubMed] [Google Scholar]
- Perrien DS, Akel NS, Edwards PK, Carver AA, Bendre MS, Swain FL, Skinner RA, Hogue WR, Nicks KM, Pierson TM, Suva LJ, Gaddy D. Inhibin A is an endocrine stimulator of bone mass and strength. Endocrinology. 2007;148:1654–1665. doi: 10.1210/en.2006-0848. [DOI] [PubMed] [Google Scholar]
- Piccoli C, D’Aprile A, Ripoli M, Scrima R, Lecce L, Boffoli D, Tabilio A, Capitanio N. Bone-marrow derived hematopoietic stem/progenitor cells express multiple isoforms of NADPH oxidase and produce constitutively reactive oxygen species. Biochem Biophys Res Commun. 2007;353:965–972. doi: 10.1016/j.bbrc.2006.12.148. [DOI] [PubMed] [Google Scholar]
- Roodman GD. Regulation of osteoclast differentiation. Ann N Y Acad Sci. 2006;1068:100–109. doi: 10.1196/annals.1346.013. [DOI] [PubMed] [Google Scholar]
- Sasaki H, Yamamoto H, Tominaga K, Masuda K, Kawai T, Teshima-Kondo S, Matsuno K, Yabe-Nishimura C, Rokutan K. Receptor activator of nuclear factor-kappaB ligand-induced mouse osteoclast differentiation is associated with switching between NADPH oxidase homologues. Free Radic Biol Med. 2009a;47:189–199. doi: 10.1016/j.freeradbiomed.2009.04.025. [DOI] [PubMed] [Google Scholar]
- Sasaki H, Yamamoto H, Tominaga K, Masuda K, Kawai T, Teshima-Kondo S, Rokutan K. NADPH oxidase-derived reactive oxygen species are essential for differentiation of a mouse macrophage cell line (RAW264.7) into osteoclasts. J Med Invest. 2009b;56:33–41. doi: 10.2152/jmi.56.33. [DOI] [PubMed] [Google Scholar]
- Serrander L, Cartier L, Bedard K, Banfi B, Lardy B, Plastre O, Sienkiewicz A, Forro L, Schlegel W, Krause KH. NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem J. 2007;406:105–114. doi: 10.1042/BJ20061903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar K, Hidestrand M, Haley R, Skinner RA, Hogue W, Jo CH, Simpson P, Lumpkin CK, Jr, Aronson J, Badger TM, Ronis MJ. Different molecular mechanisms underlie ethanol-induced bone loss in cycling and pregnant rats. Endocrinology. 2006;147:166–178. doi: 10.1210/en.2005-0529. [DOI] [PubMed] [Google Scholar]
- Shankar K, Hidestrand M, Liu X, Chen JR, Haley R, Perrien DS, Skinner RA, Lumpkin CK, Jr, Badger TM, Ronis MJ. Chronic ethanol consumption inhibits postlactational anabolic bone rebuilding in female rats. J Bone Miner Res. 2008a;23:338–349. doi: 10.1359/jbmr.071023. [DOI] [PubMed] [Google Scholar]
- Shankar K, Liu X, Singhal R, Chen JR, Nagarajan S, Badger TM, Ronis MJ. Chronic ethanol consumption leads to disruption of vitamin D3 homeostasis associated with induction of renal 1,25 dihydroxyvitamin D3-24-hydroxylase (CYP24A1) Endocrinology. 2008b;149:1748–1756. doi: 10.1210/en.2007-0903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suva LJ, Hartman E, Dilley JD, Russell S, Akel NS, Skinner RA, Hogue WR, Budde U, Varughese KI, Kanaji T, Ware J. Platelet dysfunction and a high bone mass phenotype in a murine model of platelet-type von Willebrand disease. Am J Pathol. 2008;172:430–439. doi: 10.2353/ajpath.2008.070417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner RT, Rosen CJ, Iwaniec UT. Effects of alcohol on skeletal response to growth hormone in hypophysectomized rats. Bone. 2010;46:806–812. doi: 10.1016/j.bone.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadstein J, Skude G. Serum ethanol, hepatic enzymes and length of debauch in chronic alcoholics. Acta Med Scand. 1979;205:317–318. doi: 10.1111/j.0954-6820.1979.tb06055.x. [DOI] [PubMed] [Google Scholar]
- Wezeman FH, Juknelis D, Himes R, Callaci JJ. Vitamin D and ibandronate prevent cancellous bone loss associated with binge alcohol treatment in male rats. Bone. 2007;41:639–645. doi: 10.1016/j.bone.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittrant Y, Gorin Y, Mohan S, Wagner B, Abboud-Werner SL. Colony-stimulating factor-1 (CSF-1) directly inhibits receptor activator of nuclear factor-{kappa}B ligand (RANKL) expression by osteoblasts. Endocrinology. 2009;150:4977–4988. doi: 10.1210/en.2009-0248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuermser LA, Achenbach SJ, Amin S, Khosla S, Melton LJ., III What accounts for rib fractures in older adults? J Osteoporos. 2011;2011 doi: 10.4061/2011/457591. 457591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Madyastha P, Bingel S, Ries W, Key L. A new superoxide-generating oxidase in murine osteoclasts. J Biol Chem. 2001;276:5452–5458. doi: 10.1074/jbc.M001004200. [DOI] [PubMed] [Google Scholar]
- Yeligar SM, Harris FL, Hart CM, Brown LA. Ethanol induces oxidative stress in alveolar macrophages via upregulation of NADPH oxidases. J Immunol. 2012;188:3648–3657. doi: 10.4049/jimmunol.1101278. [DOI] [PMC free article] [PubMed] [Google Scholar]