

Abstract
In cancer patients, visual identification of sentinel lymph nodes (LNs) is achieved by the injection of dyes that bind avidly to endogenous albumin, targeting these compounds to LNs where they are efficiently filtered by resident phagocytes1,2. Here we translate this “albumin hitchhiking” approach to molecular vaccines, via the synthesis of amphiphiles (amph-vaccines) comprised of an antigen or adjuvant cargo linked to a lipophilic albumin-binding tail by a solubility-promoting polar polymer chain. Structurally-optimized CpG-DNA/peptide amph-vaccines exhibited dramatic increases in LN accumulation and decreased systemic dissemination relative to their parent compounds, leading to 30-fold increases in T-cell priming and enhanced anti-tumor efficacy while greatly reducing systemic toxicity. Amph-vaccines provide a simple, broadly-applicable strategy to simultaneously increase the potency and safety of subunit vaccines.
A major challenge in the development of subunit vaccines is the efficient delivery of antigen/adjuvant to secondary lymphoid organs where immune responses are orchestrated3,4. Attempts to enhance vaccine delivery have included the use of depot-forming adjuvants5 or nanoparticulate carriers that are preferentially internalized by antigen presenting cells (APCs)4, 6–12, but approaches that could employ well-defined molecular conjugates would be attractive. Antigens conjugated to antibodies targeting dendritic cells (DCs) reach these cells in the draining LNs but also drain into the systemic circulation and access DCs in distal tissues13,14, which might promote tolerance unless inflammatory adjuvants are systemically co-administered. Lymph node targeting is also required for cancer staging in sentinel LN mapping procedures, where radioactive or colored dyes are injected at tumor resection sites1. Compounds which bind avidly to serum albumin are particularly effective LN tracers2; albumin binding targets these molecules to lymphatics and draining LNs, where they accumulate in APCs15,16. Inspired by this strategy, we set out to create LN-targeting molecular vaccines designed to similarly “hitchhike” on albumin to LNs. Exploiting albumin’s role as a fatty acid transporter, we hypothesized that antigens/adjuvants modified with a lipophilic albumin-binding domain would accumulate in lymphoid organs following injection via in situ complexation and transport with endogenous albumin. To develop this strategy, we studied model vaccines composed of peptide antigens and CpG DNAs, single-stranded oligonucleotides containing unmethylated cytosine-guanine motifs that bind Toll-like receptor-9 and serve as potent molecular adjuvants17,18.
To identify an optimal albumin-binding domain that could be appended to either CpG or peptide antigens, we constructed a series of amphiphilic 20-base phosphorothioate (PS)-stabilized CpG oligos 5′-linked to lipophilic tails (amph-CpGs, Fig. 1a). We first evaluated the interaction of fluorescein amidite (FAM)-labeled conjugates with serum proteins by size exclusion chromatography (SEC, Fig. 1b). Fetal bovine serum (FBS) exhibited a major protein fraction eluting at 5.3 min in SEC that coincided with albumin (Extended Data Fig. 1a). The vast majority of mono-acyl- (C18-CpG) or cholesterol-conjugated (Cho-CpG) oligos eluted as monomers at 5.8 min in the presence or absence of serum, indicating a lack of interaction with albumin (Fig. 1b and Extended Data Fig. 1a). In contrast, diacyl lipid-conjugated CpGs (lipo-CpGs) in aqueous solution eluted as micelles (3.7 min), but following incubation with serum, nearly 50% of the lipo-CpG co-migrated with albumin (Fig. 1b). Biotinylated lipo-CpG (but not CpG) incubated with FBS and then captured with magnetic beads was found to pull down albumin, and lipo-CpG was efficiently captured by albumin-conjugated agarose (Extended Data Figs. 1b–c). Biolayer interferometry and spectroscopy measurements of FRET between lipo-CpG and purified albumin further confirmed their molecular association in solution (Extended Data Figs. 1d–e).
Figure 1. Design of a lymph node-targeted molecular adjuvant.

a, Structure of amph-CpGs. b, SEC of FAM-lCpGs alone or following incubation with FBS for 2h (left), and %CpG co-migrating with albumin peaks (right). Vertical dashed line provides a guide to the eye. c–g, IVIS fluorescence imaging of excised draining LNs from C57Bl/6 mice (n=4 LNs/group) injected with FAM-CpGs (3.3 nmol) in soluble form, emulsified in IFA, entrapped in liposomes, or as amphiphile conjugates. c, IVIS images and quantification from inguinal and axillary nodes at 24h. d, CpG accumulation in draining LNs. e, IVIS quantification of CpG in LNs 24h after injection of G-quadruplex-forming Lipo-Gn-CpGs. f, Immunohistochemistry of inguinal LNs 24h post-injection (CD3, blue; B220, pink; CpG, green). g, LN CpG+ cells determined by flow cytometry at 24h. ***, p 0.001; **, p<0.01; *, p<0.05 compared to soluble CpG by one-way ANOVA with Bonferroni post-test. Data represent mean±SEM of 2–3 independent experiments.
Extended Data Figure 1. Interaction between albumin and amph-CpGs.
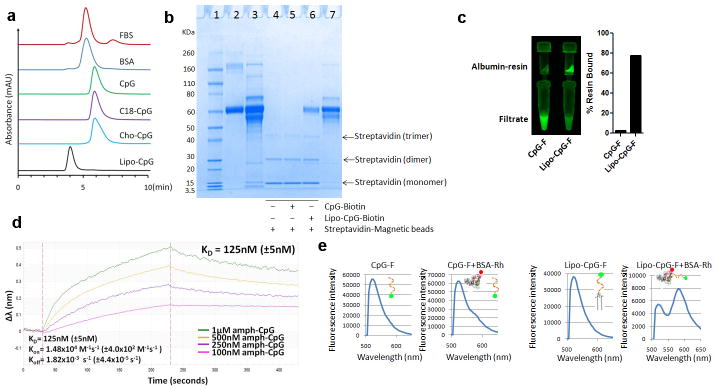
a, Size exclusion chromatography of fetal bovine serum (FBS), albumin, and fluorescein-labeled amph-CpGs. FBS and bovine serum albumin (BSA) were monitored using absorptions at 280 nm, while CpG oligos were monitored at 480 nm (Fluorescein peak). b, lipo-CpG, but not CpG, interact with serum albumin as shown by SDS-PAGE following protein pull-down assays: Fetal bovine serum (FBS) was incubated with 3′-biotin-labeled CpG (CpG-biotin), lipo-CpG (lipo-CpG-biotin), or PBS for 1 hr at 37°C. Streptavidin-conjugated magnetic beads were added to capture biotinylated CpGs and any associated proteins, separated by a magnet, boiled to release bound CpG/proteins, and subjected to SDS-PAGE analysis. Lane 1: protein MW ladder; lane 2: purified BSA; lane 3: FBS (100× dilution, 10 μL loading); lane 4: pull-down control, FBS incubated with streptavidin-magnetic beads; lane 5: pull-down with CpG-biotin, FBS was incubated with CpG-biotin and streptavidin-magetic beads; lane 6: pull-down with lipo-CpG-biotin, FBS incubated with lipo-CpG-biotin and streptavidin magnetic beads; lane 7: FBS (100× dilution, 5 μL loading). c, Fluorescein-labeled CpG or lipo-CpG was incubated with albumin-conjugated agarose resin for 1 hr at 37°C, and the resin was separated by filtration. The filtrate and recovered agarose were visualized by a gel imager and quantified by fluorescence measurements. d, Bio-layer interferometry measurements of lipo-CpG and CpG binding to immobilized bovine serum albumin. Albumin-conjugated BLI probes were immersed in solutions of lipo-CpG, and wavelength shifts (Δλ) of the interferometry pattern association and dissociation curves were followed over time to determine affinity constants of binding at 25°C. Shown are apparent kon, koff, and KD values from fits to the data. e, Fluorescence resonance energy transfer (FRET) between FAM-labeled lipo-CpG and rhodamine-conjugated albumin assessed by fluorescence spectroscopy. CpG-F or lipo-CpG-F (1.65 μM) alone or mixed with BSA-Rh (1.5 μM) in PBS were excited at 488 nm and emission was recorded from 500–650 nm.
We next characterized the in vivo trafficking of CpG conjugates. Amph-CpGs were injected s.c. in C57Bl/6 mice, and 24h later, draining LNs were excised intact for IVIS fluorescence imaging. C18- and Cho-CpG showed marginally increased uptake in LNs relative to unmodified CpG, reaching levels similar to CpG delivered by two prototypical vaccine vehicles known to enhance vaccine accumulation in LNs, incomplete Freund’s adjuvant (IFA) or poly(ethylene glycol) (PEG)-coated liposomes6 (Fig. 1c). In contrast, lipo-CpG accumulated 8-fold more than soluble CpG (Fig. 1c). Over 7 days post-injection, soluble CpG exhibited no LN accumulation above 0.3% of the injected dose at any time, while lipo-CpG accumulated for 3 days, giving a 12-fold greater area-under-the-curve (AUC) for total LN exposure to CpG (Fig. 1d). LN accumulation was not dependent on TLR-9-recognized CpG motifs, and was not due to increased nuclease resistance of lipid-modified PS-backbone CpGs (data not shown).
Our in vitro analysis indicated that lipo-CpG molecules equilibrated between micellar and albumin-bound forms in the presence of serum, making it unclear which state was responsible for LN targeting. However, injection of lipo-CpG pre-incubated for 5 hr with freshly isolated 90 vol% mouse serum at 37°C, conditions where SEC showed the vast majority of the amphiphile co-migrated with albumin, led to essentially identical LN targeting as direct lipo-CpG injections (data not shown). To further address this question, we introduced poly-guanine repeats between the diacyl lipid and CpG sequence to lock the amphiphiles in the micellar state and block disassembly by albumin. G-quadruplex hydrogen bonding between adjacent oligo strands in lipo-Gn-CpG micelles containing 4 or more guanine repeats rendered the micelles stable in the presence of serum (Extended Data Figs. 2a–d). Labeled albumin did not co-migrate with stabilized micelles as assessed by SEC (Extended Data Fig. 2e), suggesting that the amphiphiles do not interact with albumin as intact micelles. Despite forming micelles with similar sizes (Extended Data Fig. 2f), G-quartet-stabilized lipo-G4-CpG or lipo-G6-CpG micelles exhibited poor LN accumulation following injection compared to albumin-binding lipo-CpG and lipo-G2-CpG (Fig. 1d, e). A possible explanation is that amplification of nonspecific matrix binding by the PS-DNA backbones19,20 in the multivalent micellar form irreversibly trapped the stabilized micelles at the injection site. Histological sections of draining LNs showed little detectable CpG or lipo-G4-CpG, while albumin-binding lipo-CpG and lipo-G2-CpG accumulated in the subcapsular sinus and interfollicular areas (Fig. 1f). LN-accumulating amphiphiles were associated with F4/80+ macrophages and CD11c+ dendritic cells, with only a minor contribution from skin-derived CD207+ DCs (Fig. 1g and Extended Data Fig. 3). If albumin “hitchhiking” mediates LN targeting, then covalent conjugation of oligos to albumin should also enhance LN accumulation. We found that injection of CpG conjugated to mouse serum albumin (MSA) gave slightly lower oligo uptake in LNs than lipo-CpG, but much greater accumulation than soluble CpG (Extended Data Fig. 4). Altogether, these data suggest that efficient LN delivery of CpG oligonucleotides conjugated to lipophilic tails is enhanced by partitioning from micelles into a serum albumin-bound state.
Extended Data Figure 2. Construction and characterization of G-quadruplex-stabilized CpG adjuvants.

a, G-quadruplex stabilized CpG micelles are self-assembled from amphiphiles composed of three distinct segments: an immunostimulatory CpG sequence, a central repeat block containing n=1–10 G-quartet-forming guanines followed by (10–n) non-interacting thymidines, and a diacyl lipid tail. In aqueous solutions, these amphiphiles self-assembled into three-dimensional spherical micelles with a CpG corona and a lipid core. In the presence of K+, neighboring guanine repeats in the oligo corona form G-quadruplex structures via Hoogsteen hydrogen bonds and stabilize the micelle structure. The oligo micelles’ stabilities in the presence of serum were programmed by altering the length of the guanine repeat. b, Parallel G-quartet formation among DNA strands within the micelles was detected by circular dichroism (CD) spectroscopy, as manifested by the shifting of positive peaks from 278 nm toward 262 nm and troughs at 245 nm as the number of guanines in the structure increased. c, Pyrene excimer fluorescence was used to assay the stabilities of G-quadruplex micelles in the presence of albumin: Pyrene dye incorporated in stabilized CpG micelles (n > 2) retained excimer fluorescence in the presence of high concentrations of albumin. In contrast, albumin binds to the lipids moiety of unstabilized micelles (n ≤ 2) and disrupts the micelle structures, leading to loss of excimer fluorescence in an albumin concentration-dependent manner as the protein disrupts the micelles into albumin-bound unimers. Shown below the schematic is the fraction of amphiphile-CpG remaining in the micellar state as a function of albumin concentration as reported by excimer fluorescence. Arrow indicates the plasma concentration of albumin. d, Stability profiles of G-quadruplex CpG micelles as measured by size-exclusion chromatography in the presence of fetal bovine serum (FBS). Fluorescein-labeled CpG micelles were incubated with 20% FBS in PBS in the presence of 10 mM Mg2+ and 20 mM K+ at 37°C for 2 hours, then analyzed by SEC. FBS and BSA were monitored using absorptions at 280 nm, while lipo-Gn-CpG amphiphiles were monitored at 480 nm (Fluorescein peak). Lipo-Gn-CpG with n=0 or 2 partitioned to co-migrate with albumin, while amphiphiles with n>2 showed increasing fractions of the amphiphiles migrating as intact micelles in the presence of serum with increasing n. e, Fluorescein-labeled lipo-G6-CpG (5 μM) and Alexa Fluor® 647-labeled BSA (5 μM) were incubated for 2 hrs at 37 °C in PBS+20 mM KCl, and then analyzed by size exclusion chromatography. Spectra were monitored at 480 nm (ODN channel, green line, fluorescein) and 640 nm (protein channel, red line, Alexa Fluor® 647). The majority of BSA and CpG micellar aggregates eluted separately. f, Amph-CpG micelles sizes as determined by dynamic light scattering. All data are mean±s.e.m. Statistical analysis was performed by one-way ANOVA with Bonferroni post-tests.
Extended Data Figure 3. Lymph node localization of amph-CpGs with macrophages and dendritic cells.

a, Immunofluorescent images of inguinal LN section 24 h after injection of 3.3 nmol lipo-CpG or lipo-G2-CpG, showing dendritic cells (CD11c, blue), macrophages (F4/80, red), and CpG (green). b–d, Mice (n=3/group) were injected s.c. with 3.3 nmol of Fluorescein-labeled CpG formulations. After 24 h, lymph nodes were digested and lymph nodes cells stained with DAPI and antibodies against F4/80, CD11c and CD207. Shown are representative flow cytometry plots of F4/80 staining (b) and CD11c staining (c) versus CpG fluorescence in viable (DAPI−) cells. d, percentages of CpG+ cells in the LNs determined by flow cytometry at 24 hr. ***, p < 0.001; All data are mean±s.e.m. Statistical analysis was performed by unpaired student’s t-test.
Extended Data Figure 4. CpG-albumin conjugates accumulate in LNs.
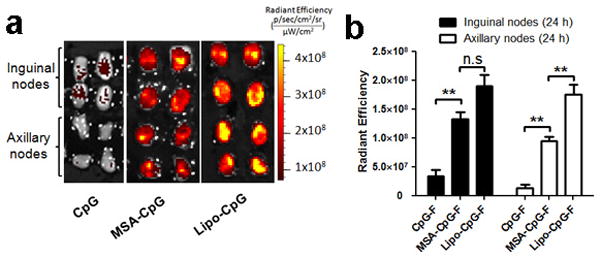
C57Bl/6 mice (n=4 LNs/group) were injected s.c. at the tail base with 3.3 nmol fluorescein-labeled free CpG, mouse albumin-CpG conjugates (MSA-CpG), or lipo-CpG. Inguinal LNs and axillary LNs were isolated 24 hours post injection and imaged (a) and quantified (b) by IVIS optical imaging. All data are mean±s.e.m. **, p < 0.01 by one-way ANOVA with Bonferroni post-test.
To identify potential differences in the function of CpG vs. optimally LN-targeted lipo-G2-CpG beyond altered biodistribution, we assessed several aspects of CpG bioactivity in vitro: lipo-G2-CpG was internalized by DCs into endolysosomes in a pattern indistinguishable from CpG in confocal microscopy, albeit to 2-fold higher levels (Extended Data Figs. 5a–b). Lipo-G2-CpG did not activate the lipid-binding receptor TLR2 in reporter cells, but both free CpG and amphiphile-CpG activated RAW macrophages bearing a NF-κB reporter in a CpG sequence-specific manner (Extended Data Figs. 5c–d). When DCs were activated with CpG or lipo-G2-CpG and pulsed with OVA protein to test cross-presentation of antigen to OT-I (OVA-specific) T-cells, DC activation by soluble or amphiphile-CpG led to similar T-cell proliferation (Extended Data Fig. 5e). Thus, lipid modification of CpG increased uptake in the presence of serum but did not otherwise greatly alter the bioactivity of CpG.
Extended Data Figure 5. In-vitro characterization of amph-CpG.
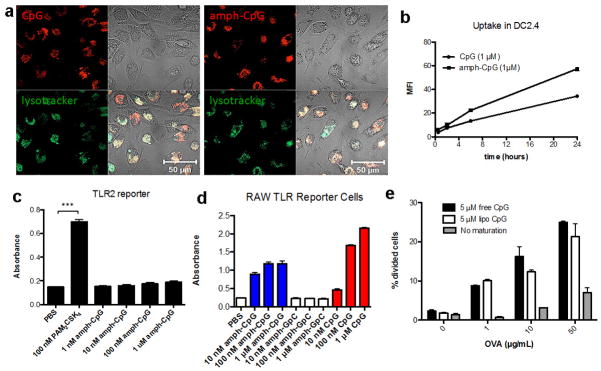
a, Rhodamine labeled CpG or amph-CpG (1 μM) was incubated with murine bone marrow-derived dendritic cells (BMDCs) at 37°C for 4 hours with LysoTracker® (Life Technologies) and imaged using a Zeiss LSM 510 confocal microscope (Oberkochen, Germany). b, Rhodamine labeled CpG or amph-CpG (1 μM) was incubated for 30 minutes, 2 hours, 6 hours, and 24 hours with the murine dendritic cell line DC2.4. Cells were stained with DAPI and uptake was quantified by flow cytometry using the mean fluorescence intensity (MFI) of viable (DAPI-) cells. c, Amph-CpG or PAM2CSK4 (a strong TLR2 agonist) was incubated for 24 hours with the InvivoGen HEK-Blue™ murine TLR2 reporter cell line, a secreted embryonic alkaline phosphatase (SEAP) reporter system. SEAP levels were quantified by incubating supernatant with QuantiBlue™ substrate for 1h and reading absorption at 620 nm. d, Amph-CpG, CpG, or control amph-GpC (1 μM) were incubated with InvivoGen RAW-Blue™ mouse macrophage reporter cells, which secrete SEAP upon TLR, NOD or Dectin-1 stimulation. SEAP levels were quantified by incubating supernatant with QuantiBlue™ substrate for 1h and reading absorption at 620 nm. e, Bone marrow-derived immature dendritic cells were incubated overnight with indicated concentrations of OVA and maturation stimuli (or medium alone). DCs were washed 3 times with PBS and 30,000 CFSE-labeled OT-I CD8+ T cells were then added to each well. Cells were collected after 2 days of co-culture, and stained and gated for DAPI- (viable) CD8+ T cells using Flowjo v.7.6.5 (Treestar). Extent of proliferation was quantified by determining the % of cells that had undergone division by determining % of viable CD8+ T cells that had diluted CFSE using T cells alone as a control for the no division/dilution peak. Shown are mean±s.e.m. in b–d; ***, p < 0.001 by one-way ANOVA with Bonferroni post-test. Bars in e represent medians and whiskers represent range (n=2 wells/condition).
To determine the impact of LN targeting on the immune response, we immunized mice with ovalbumin protein (OVA) mixed with unmodified CpG, Cho-CpG, CpG emulsified in IFA, or lipo-Gn-CpGs (n=0, 2, 4, 6). OVA (which has only 13% sequence identity with albumin) showed minimal association with lipo-CpGs (data not shown) and thus these vaccinations assessed the impact of lymph node targeting of the adjuvant only, relying on normal lymphatic drainage of the OVA antigen. Lipo-Gn-CpG-adjuvanted vaccines primed significantly increased frequencies of antigen-specific, cytokine-producing CD8+ T-cells compared to unmodified CpG, Cho-CpG, or CpG in IFA, but the strongest responses (up to 32-fold greater than unmodified CpG) were elicited by lipo-CpG and lipo-G2-CpG (Figs. 2a–b; Extended Data Figs. 6a–b). Strikingly, the magnitude of the T-cell response was strongly correlated with LN accumulation of CpG (Fig. 2c). Lipo-CpG also modestly increased antibody responses by ~3-fold and enhanced CD8+ T-cell responses to the model HIV antigen SIV-gag (Extended Data Figs. 6c–d). Importantly, no antibodies against albumin were detected for any of the amphiphile-CpG vaccines (Extended Data Figs. 6e–f and data not shown). Control immunizations with non-TLR agonist lipo-GpC or diacyl-PEG conjugates (lipo-PEG) mixed with OVA were ineffective and amph-CpG responses were identical in TLR2−/− mice (Extended Data Figs. 6g–h), ruling out a direct adjuvant effect of the diacyl lipid tail. CpG that is not captured in local LNs drains to the systemic circulation, leading to systemic inflammatory toxicities21. Despite lymphadenopathy of draining LNs indicating local activity, repeated injections of lipo-CpGs showed greatly reduced systemic inflammation relative to free CpG (Figs. 2d–e and Extended Data Figs. 7a–b). While further work will be needed to determine any potential autoimmune toxicities related to LN targeting of CpG18, these results suggest that the LN targeting achieved by amph-CpGs greatly enhances the potency of this molecular adjuvant while simultaneously lowering acute systemic side effects.
Figure 2. Lymph node targeting enhances the potency while reducing systemic toxicity of CpG.
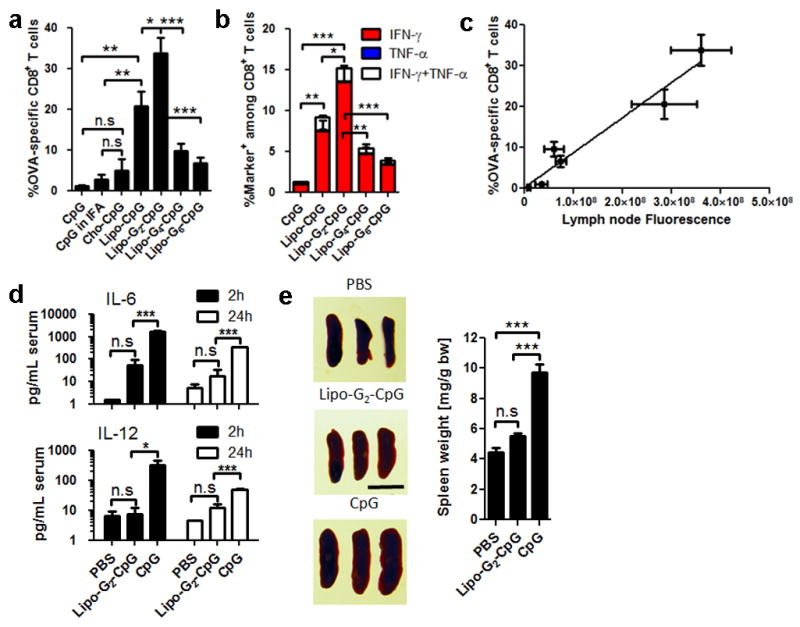
a–c, C57Bl/6 mice (n=4–8/group) were immunized with ovalbumin (10 μg) + CpG (1.24 nmol) on d0 and d14; shown are SIINFEKL tetramer (a) and intracellular cytokine staining (b) on peripheral blood at d20. c, LN CpG fluorescence correlation with T-cell response. d, Serum cytokines following injection (n=3/group) of 6.2 nmol CpG. e, Splenomegaly (n=3/group) assessed on d6 after 3 injections of CpG (scale bar: 1 cm). ***, p<0.001; **, p<0.01; *, p<0.05 by one-way ANOVA with Bonferroni post-test. Data show mean±SEM of 2–4 independent experiments.
Extended Data Figure 6. Albumin-binding lipo-CpGs elicit robust expansion of antigen-specific CD8+ T-cells when combined with soluble protein.
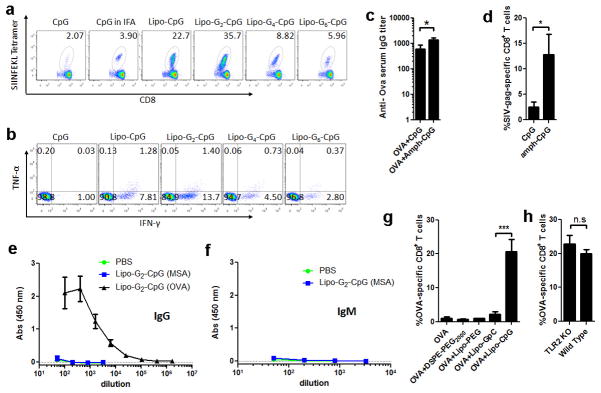
Groups of C57Bl/6 mice (n=4–8/group) were immunized s.c. on day 0 and day 14 with 10 μg OVA and 1.24 nmol CpG formulations as indicated. Six days after the final immunization, mice were bled and PBMCs were evaluated by SIINFEKL-tetramer staining and intracellular cytokine staining. a, Representative flow cytometric dot plots of H-2Kb/SIINFEKL tetramer staining of CD8+ cells. b, Representative flow dot plots of intracellular staining on CD8+ cells for IFN-γ and TNF-α after 6 h ex-vivo restimulation with SIINFEKL peptide. c, serum samples were collected and assayed by ELISA for anti-OVA IgG (day 34). d, mice were immunized on day 0 and day 14 with 1.24 nmol lipo-G2-CpG mixed with 10 μg SIV-gag protein, blood samples were collected and analyzed by peptide-MHC tetramer staining for CD8+ T-cells recognizing the immunodominant AL11 epitope of gag. e, f, anti-mouse serum albumin (MSA) and anti-OVA IgG (e, day 20) or IgM (f, day 20) were measured by ELISA. g, groups of C57Bl/6 mice (n=4/group) were immunized with 10 μg OVA alone or mixed with 1.24 nmol of a non-TLR agonist lipo-GpC, the same diacyl lipid tail conjugated to PEG (lipo-PEG, 48 EG units), or DSPE-PEG2000. Animals were boosted with the same formulation on day 14, and OVA tetramer+ CD8+ T-cells in peripheral blood were assayed by flow cytometry on day 20. ***, p < 0.001. h, TLR2 knockout or wild type mice were immunized as described in (a), and OVA tetramer+ CD8+ T-cells were assayed as previously. All data are mean±s.e.m. *, p < 0.05. Statistical analysis was performed by unpaired student’s t-test.
Extended Data Figure 7. Albumin-binding CpG induces local lymphadenopathy but reduces systemic toxicity compared to soluble CpG adjuvant.
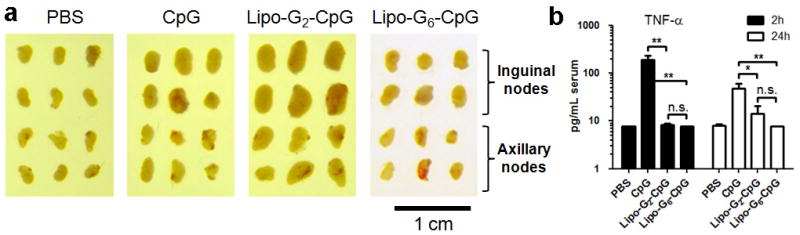
a, C57Bl/6 mice (n=3/group) were injected with 1.24 nmol CpGs subcutaneously on day 0 and 2.48 nmol CpGs on days 2 and 4. On day 6 animals were sacrificed and lymph nodes were isolated and photographed with a digital camera. b, Bead-based flow analysis of proinflammatory cytokines elicited in peripheral blood of mice injected with a single dose (6.2 nmol) of different CpG formulations, blood samples were collect at different time interval and analyzed for TNF-α per manufacturer’s instructions. All data are mean±s.e.m. **, p < 0.01; *, p < 0.05. Statistical analysis was performed by one-way ANOVA with Bonferroni post-test.
Synthesis of lipo-CpG is straightforward due to the solubility imparted by the long polar oligonucleotide block, but depending on the amino acid sequence, lipidated polypeptides can be essentially insoluble22. To generalize this LN targeting strategy to antigens and other potential vaccine components, we synthesized peptides linked to a diacyl lipid tail via a PEG block chosen to promote the conjugate solubility (amph-peptides, Fig. 3a). Amph-peptides and lipo-PEGs in water form micelles, but these amphiphiles can also insert their diacyl tails into cell membranes. We found that lipo-PEG amphiphiles with short PEG blocks exhibited preferential plasma membrane insertion when incubated with cells in the presence of albumin in vitro (Figs. 3b and Extended Data Figs. 8a–b), which might limit transit to LNs on albumin in vivo. However, increasing the polar block to 48 ethylene glycol units gave amphiphiles that partitioned preferentially into solution while retaining albumin binding (Figs. 3b and Extended Data Fig. 8c), consistent with prior studies23. This in vitro partitioning directly predicted in vivo draining patterns, as lipo-PEG-FAM amphiphiles injected s.c. showed increasing LN accumulation with increasing PEG block length (Fig. 3c). Although optimal immunostimulatory CpGs are ~20 bases, an analogous trend was observed for DNA amphiphiles as a function of oligo length (Extended Data Figs. 8d–e). Like amph-CpGs, the structure of the hydrophobic block was also important; while lipo-PEG amphiphiles with long diacyl tails (≥16 carbons, which exhibit a high affinity for albumin24) showed intense LN accumulation, shorter lipid tails with low affinity for albumin showed low LN accumulation (Fig. 3d). Based on these findings establishing design rules for efficient targeting of lipo-PEG amphiphiles to LNs, we conjugated peptide antigens to DSPE-PEG(2KDa) to generate amph-peptides for vaccination studies (Extended Data Fig. 9a).
Figure 3. Design of lymph node-targeted amphiphile-peptides.
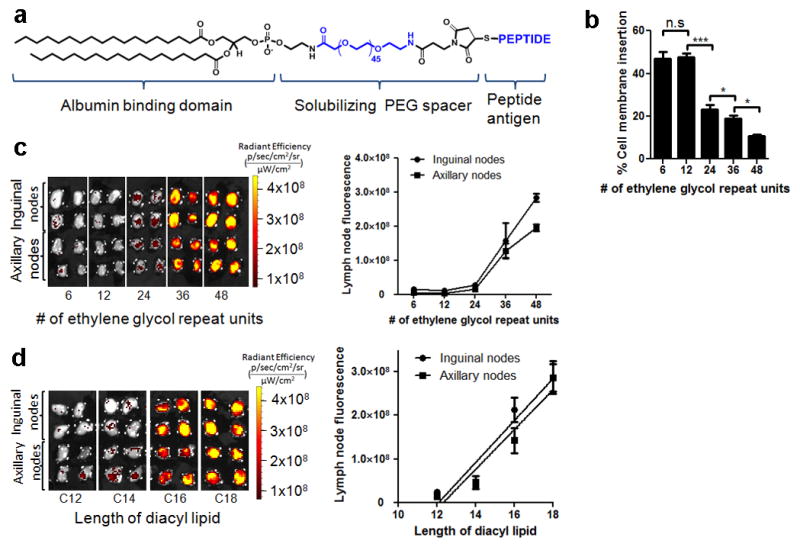
a, Structure of amph-peptides. b, Amph-PEG-fluorescein insertion into cell membranes quantified by fluorescence spectroscopy following 1h incubation with splenocytes in the presence of 100 μM albumin. c–d, C57Bl/6 mice (n=4 LNs/group) were injected with fluorescent amph-PEGs having varying PEG length (fixed C18 diacyl lipid tails, c) or lipid tail length (fixed PEG length 48 EG units, d); draining LNs were excised and imaged by IVIS after 24h. ***, p < 0.001; **, p < 0.01; *, p < 0.05 by one-way ANOVA with Bonferroni post-test. Data represent mean±SEM of two independent experiments.
Extended Data Figure 8. Hydrophilic block length of amphiphiles determines cell membrane insertion and lymph node accumulation.

a, Amphiphiles with varying hydrophilic PEG lengths were prepared by solid phase synthesis of diacyl tails coupled to 1–8 hexa-ethylene glycol phosphorothioate units. Fluorescein was incorporated either at the 3′ terminal (for membrane insertion analyses) or adjacent to the lipid moiety (for albumin-binding analyses). b, Splenocytes from C57Bl/6 mice (5×107cells/mL) were incubated with lipo-(PEG)n-Fluorescein (1.67 μM) and albumin (100 μM) at 37°C for 1 hour. Shown is a representative image of membrane insertion observed by confocal microcopy for lipo-(PEG)1-fluorescein. c, Equilibrium partitioning measurements shown as a function of albumin concentration at 37°C. Lipo-fluorescein-(PEG)n (5 μM) was incubated with varying concentrations of BSA and fluorescence intensities were monitored by fluorescence spectroscopy. BSA binding disrupted the micellar structure and decreased the self-quenching of fluorescein. All samples reached maximum fluorescence intensities at around 10 μM BSA, indicating 100% micelle breakup at this concentration (at higher BSA concentrations, fluorescence decreases due to solution turbidity). Arrow indicates plasma concentration of albumin. d, e, Lipo-Tn-FAM amphiphiles (n=5, 10, 15, 20) were injected s.c. in C57Bl/6 mice (n=4 LNs/group) and excised LNs were imaged after 24 hr (d). Mean LN fluorescence from groups of mice are plotted in (e). All data are mean±s.e.m.
Extended Data Figure 9. Lipo-PEG-peptide amphiphiles exhibit greatly enhanced lymph node accumulation compared to unmodified peptides.
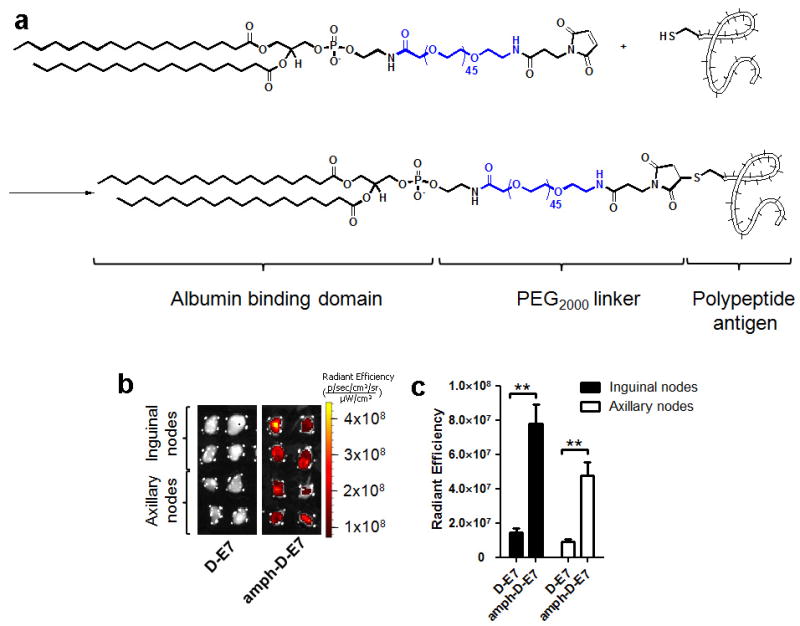
a, Peptides with N-terminal cysteines were conjugated to maleimide-PEG2000-DSPE.b–c, FAM-labeled immunodominant peptide derived from the HPV-16 E7 protein (FAM-FTVINYHARC, synthesized in reverse sequence order using D-amino acids to obtain the same chiral organization of side chains as the typical L-amino acid sequence in a protease-resistant peptide) was injected s.c. at the tail base as a free peptide (D-E7) or as a PEG-DSPE conjugate (amph-D-E7). Shown are IVIS images of draining lymph nodes 24 hours post injection (b) and fluorescence quantifications (c). All data are mean±s.e.m. **, p < 0.01. Statistical analysis was performed by unpaired student’s t-test.
To test the potency of combined antigen and adjuvant targeting to LNs, we prepared amph-peptide (DSPE-PEG-peptide) conjugates of a model HIV antigen (AL11 epitope from Simian Immunodeficiency Virus (SIV) gag25), the tumor-associated self-antigen Trp2 from melanoma26, and a peptide derived from the human papillomavirus (HPV)-derived cervical cancer antigen E727. Amph-peptides accumulated efficiently in LNs (Extended Data Figs. 9b–c). C57Bl/6 mice immunized with amph-peptides and amph-CpG (lipo-G2-CpG) showed dramatically increased expansion of antigen-specific, cytokine-producing CD8+ T-cells and enhanced cytolytic activity relative to unmodified peptide/CpG immunizations (Figs. 4a–c). To test whether amph-vaccine delivery enhances the protective efficacy of peptide vaccines, animals bearing established TC-1 tumors (expressing the E7 oncoprotein from HPV) or B16F10 melanomas were vaccinated. Amph-vaccines triggered sustained regression of large TC-1 tumors that were only modestly impacted by soluble vaccines (Fig. 4d) and slowed the growth of melanoma tumors, where a traditional soluble vaccine had no effect (Fig. 4e). In addition to enhancing the effectiveness of optimal T-cell epitopes, synthetic “long peptide” antigens28 also exhibited enhanced immunogenicity when delivered as amph-peptides (Extended Data Figs. 10a–e). This result is of particular interest since a finite pool of long sequences permit peptide vaccines to provide effective coverage of epitopes across the diverse haplotypes of a given target patient population. Amphiphile-long peptides were also ~10-fold more potent than soluble peptides when combined with non-CpG, non-LN-targeted alternative TLR adjuvants (Extended Data Fig. 10f), showing that CpG is not required to see an enhanced response to LN-targeted peptides. Altogether, the results presented here define design rules for amphiphile conjugates as a general strategy to enhance the potency and safety of LN-active compounds, an approach that may be applicable to a broad range of immunomodulatory therapeutics and imaging agents. These findings also have implications for how the immune system may survey lipophilic antigens. Further work will be needed to determine whether albumin binding is functionally critical, or alternatively whether other rare serum components may have a role in the observed lymph node targeting.
Figure 4. Amph-vaccines maximize immunogenicity and therapeutic efficacy of polypeptide vaccines.
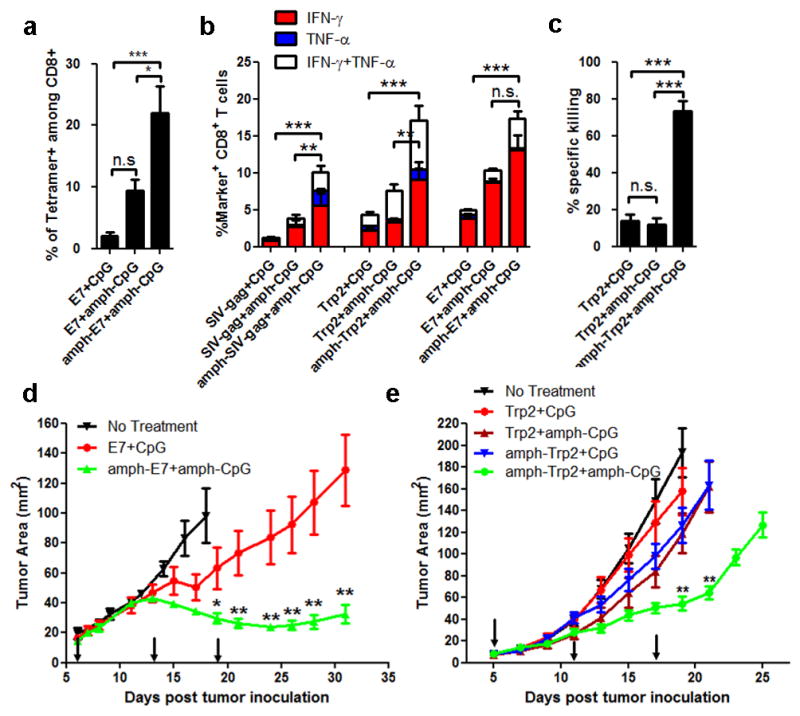
a–c, C57Bl/6 mice (n=3–4/group) were immunized with SIV-gag, Trp2, or E7 peptides (10 μg) + CpG (1.24 nmol) on d0 and d14; shown are tetramer-positive CD8+ T-cells (a) and intracellular cytokine production (b) in peripheral blood on d20. c, Trp2-specific cytotoxicity measured using an in vivo killing assay on d21. d–e, Tumor growth in C57BL/6 mice (n=8/group) inoculated with 3×105 TC-1 (d) or B16F10 (e) tumor cells and vaccinated with CpG + E7 peptide or Trp2 peptide (10 μg prime, 20 μg boost), respectively, on days indicated by arrows. Statistically-significant differences between soluble and amph-vaccines indicated by asterisks: ***, p<0.001; **, p<0.01; *, p<0.05 by one-way ANOVA with Bonferroni post-test. Shown are mean±SEM of 2–4 independent experiments.
Extended Data Figure 10. Long peptide amphiphiles, when combined with amph-CpG, elicit potent antigen specific CD8+ T cells response with therapeutic benefits, as compared to soluble formulation.
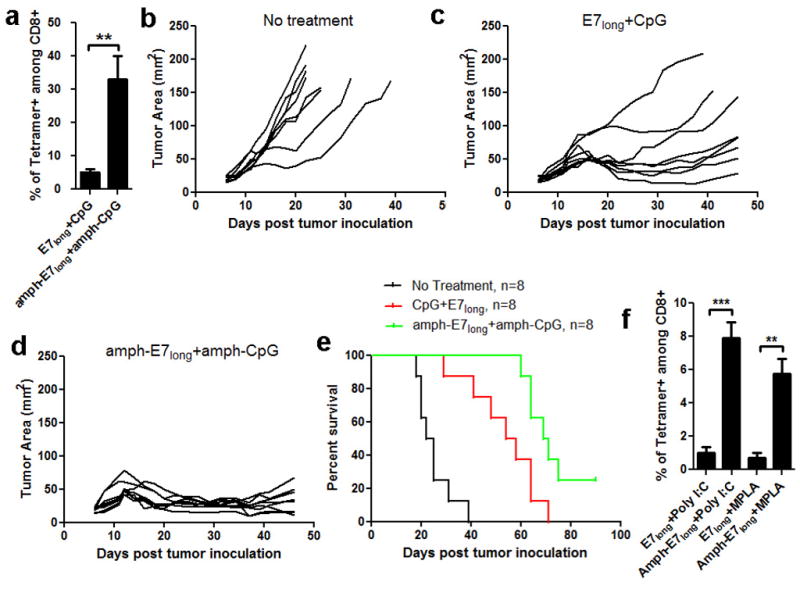
a, C57Bl/6 mice were primed on day 0 and boosted on day 14 with amph-E7long (HPV-16 E743–62, 10 μg peptide) and amph-CpG (Lipo-G2-CpG, 1.24 nmol) or equivalent soluble peptide/CpG vaccines. Six days post boost, mice were bled and analyzed for tetramer positive CD8+ T-cells in peripheral blood. b–e, C57BL/6 mice (n=8/group) were inoculated with 3×105 TC-1 tumor cells s.c. in the flank and left untreated or immunized with soluble or amphiphile long peptide vaccines on days 6 (10 μg peptide, 1.24 nmol CpG), 13 (20 μg peptide, 1.24 nmol CpG), and 19 (20 μg peptide, 1.24 nmol CpG). Shown are individual tumor growth curves for no treatment (b), immunization with soluble E7long and CpG (c), or immunization with amph-E7long+amph-CpG (d). Kaplan-Meier survival curves of eight mice per group are shown in (e). f, long peptide amphiphiles also elicit potent immune responses when combined with non-CpG, non-lymph node targeting alternative adjuvants. C57Bl/6 mice (n=4/group) were immunized as before, using amph-E7long peptide (10 μg) combined with monophosphoryl lipid A (MPLA, 10 μg) or Polyinosinic:polycytidylic acid (Poly I:C, 50 μg). The frequencies of E7 tetramer+ CD8+ T-cells in peripheral blood were assayed on day 20. All data are mean±s.e.m. ***, p < 0.001; **, p < 0.01; *, p < 0.05 by unpaired student’s t-test.
METHODS SUMMARY
Synthesis of vaccine amphiphiles
CpG/ODN amphiphiles were synthesized using an ABI 394 synthesizer on a 1.0 micromole scale. All lipophilic phosphoramidites were conjugated as a final ‘base’ on the 5′ end of oligos19. Antigen amphiphiles were synthesized by reacting N-terminal cysteine-modified peptides with maleimide-PEG2000-DSPE in DMF.
Immunizations
Six-to-eight week old C57BL/6 mice (female, Jackson Laboratory) were immunized with 10 μg of antigen mixed with 1.24 nmol CpG adjuvant in 100 μl of PBS subcutaneously at the base of the tail. All procedures were performed in accordance with the guidelines for animal care in USDA-inspected MIT animal facility.
Full Methods and any associated references are available in the online version of the paper.
Supplementary Material
Acknowledgments
This work was supported in part by the Koch Institute Support (core) Grant P30-CA14051 from the National Cancer Institute, the NIH (grants AI091693, AI104715, and AI095109), the Dept. of Defense (W911NF-13-D-0001 and W911NF-07-D-0004, T.O. 8) and the Ragon Institute of MGH, MIT, and Harvard. D.J.I. is an investigator of the Howard Hughes Medical Institute. We thank Dr. T.C. Wu for kindly providing the TC-1 tumor cells. We thank the Koch Institute Swanson Biotechnology Center for technical support, specifically the applied therapeutics & whole animal imaging core facility, histology and flow cytometry core facility. The authors wish to dedicate this paper to the memory of Officer Sean Collier, for his caring service and sacrifice in protecting the MIT community.
Footnotes
Supplementary Information is available in the online version of the paper.
Author Contributions H.L. designed and performed most experiments and analyzed the data, and wrote the manuscript; Y.Z. carried out tumor therapy experiments and analyzed the data. K.D.M. carried out in vitro bioactivity studies of CpG, bio-layer interforometry binding studies and in vivo immunizations of SIV-gag and analysed the data. A.V.L. and B.H. assisted in tetramer/in vivo cytotoxicity assays and contributed experimental suggestions. G.L.S. assisted optimization of proinflamatory cytokines assays. G.L.S., C.P., and D.S.V. contributed to in vitro T-cell proliferation assays. D.J.I. supervised all experiments and wrote the manuscript.
Author Information Reprints and permissions information is available at www.nature.com/reprints. The authors declare competing financial interests: a patent application for amphiphile vaccines has been filed, with D.J.I. and H.L. as inventors. Readers are welcome to comment on the online version of the paper. Correspondence and requests for materials should be addressed to D.J.I (djirvine@mit.edu).
References
- 1.Salhab M, Patani N, Mokbel K. Sentinel lymph node micrometastasis in human breast cancer: an update. Surg Oncol. 2011;20:E195–E206. doi: 10.1016/j.suronc.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 2.Tsopelas C, Sutton R. Why certain dyes are useful for localizing the sentinel lymph node. J Nucl Med. 2002;43:1377–1382. [PubMed] [Google Scholar]
- 3.Johansen P, Mohanan D, Martínez-Gómez JM, Kündig TM, Gander B. Lympho-geographical concepts in vaccine delivery. J Controlled Release. 2010;148:56–62. doi: 10.1016/j.jconrel.2010.05.019. [DOI] [PubMed] [Google Scholar]
- 4.Moon JJ, Huang B, Irvine DJ. Engineering nano- and microparticles to tune immunity. Adv Mater. 2012;24:3724–3746. doi: 10.1002/adma.201200446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gupta RK. Aluminum compounds as vaccine adjuvants. Adv Drug Deliv Rev. 1998;32:155–172. doi: 10.1016/s0169-409x(98)00008-8. [DOI] [PubMed] [Google Scholar]
- 6.Bachmann MF, Jennings GT. Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns. Nat Rev immunol. 2010;10:787–796. doi: 10.1038/nri2868. [DOI] [PubMed] [Google Scholar]
- 7.Hubbel JA, Thomas SN, Swartz MA. Materials engineering for immunomodulation. Nature. 2009;462:449–460. doi: 10.1038/nature08604. [DOI] [PubMed] [Google Scholar]
- 8.Temmerman MD, et al. Particulate vaccines: on the quest for optimal delivery and immune response. Drug discovery today. 2011;16:569–582. doi: 10.1016/j.drudis.2011.04.006. [DOI] [PubMed] [Google Scholar]
- 9.Pal I, Ramsey JD. The role of the lymphatic system in vaccine trafficking and immune response. Adv Drug Deliv Rev. 2011;63:909–922. doi: 10.1016/j.addr.2011.05.018. [DOI] [PubMed] [Google Scholar]
- 10.Reddy ST, et al. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat Biotechnol. 2007;25:1159–1164. doi: 10.1038/nbt1332. [DOI] [PubMed] [Google Scholar]
- 11.St John AL, Chan CY, Staats HF, Leong KW, Abraham NA. Synthetic mast-cell granules as adjuvants to promote and polarize immunity in lymph nodes. Nat Mater. 2012;11:250–257. doi: 10.1038/nmat3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trubetskoy VS, Torchilin VP. Use of polyoxyethylene-lipid conjugates as long-circulating carriers for delivery of therapeutic and diagnostic agents. Adv Drug Deliv Rev. 1995;16:311–320. [Google Scholar]
- 13.Keler T, He L, Ramakrishna V, Champion B. Antibody-targeted vaccines. Oncogene. 2007;26:3758–3767. doi: 10.1038/sj.onc.1210375. [DOI] [PubMed] [Google Scholar]
- 14.Tenbusch M, et al. Immunogenicity of DNA vaccines encoding simian immunodeficiency virus antigen targeted to dendritic cells in rhesus macaques. PLoS ONE. 2012;7:e39038. doi: 10.1371/journal.pone.0039038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Faries MB, et al. Active macromolecule uptake by lymph node antigen-presenting cells: a novel mechanism in determining sentinel lymph node status. Ann Surg Oncol. 2000;7:98–105. doi: 10.1007/s10434-000-0098-6. [DOI] [PubMed] [Google Scholar]
- 16.Schaafsma BE, et al. The clinical use of indocyanine green as a near-infrared fluorescent contrast agent for image-guided oncologic surgery. J Surg Oncol. 2011;104:323–332. doi: 10.1002/jso.21943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vollmer J, Krieg AM. Immunotherapeutic applications of CpG oligodeoxynucleotide TLR9 agonists. Adv Drug Deliv Rev. 2009;61:195–204. doi: 10.1016/j.addr.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 18.Bode C, Zhao G, Steinhagen F, Kinjo T, Klinman DM. CpG DNA as a vaccine adjuvant. Expert Rev Vaccines. 2011;10:499–511. doi: 10.1586/erv.10.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu H, Kwong B, Irvine DJ. Membrane anchored immunostimulatory oligonucleotides for in vivo cell modification and localized immunotherapy. Angew Chem, Int Ed. 2011;50:7252–7255. doi: 10.1002/anie.201101266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krieg AM, Stein CA. Phosphorothioate oligodeoxynucleotides: antisense or anti-protein? Antisense Res Dev. 1995;5:241–241. doi: 10.1089/ard.1995.5.241. [DOI] [PubMed] [Google Scholar]
- 21.Bourquin C, et al. Targeting CpG oligonucleotides to the lymph node by nanoparticles elicits efficient antitumoral immunity. J Immunol. 2008;181:2990–2998. doi: 10.4049/jimmunol.181.5.2990. [DOI] [PubMed] [Google Scholar]
- 22.Zeng M, Ghosh S, Lau YF, Brown LE, Jackson DC. Highly immunogenic and totally synthetic lipopeptides as self-adjuvanting immunocontraceptive vaccines. J Immunol. 2002;169:4905–4912. doi: 10.4049/jimmunol.169.9.4905. [DOI] [PubMed] [Google Scholar]
- 23.Kastantin M, Missirlis D, Black M, Ananthanarayanan B, Peters D, Tirrell M. Thermodynamic and kinetic stability of DSPE-PEG(2000) micelles in the presence of bovine serum albumin. J Phys Chem B. 2010;114:12632–12640. doi: 10.1021/jp1001786. [DOI] [PubMed] [Google Scholar]
- 24.Peters T. All About Albumin: Biochemistry, Genetics, and Medical Applications. Academic Press; San Diego: 1995. [Google Scholar]
- 25.Barouch DH, et al. Immunogenicity of recombinant adenovirus serotype 35 vaccine in the presence of pre-existing anti-Ad5 immunity. J Immunol. 2004;172:6290–6297. doi: 10.4049/jimmunol.172.10.6290. [DOI] [PubMed] [Google Scholar]
- 26.Schreurs MW, et al. Dendritic cells break tolerance and induce protective immunity against a melanocyte differentiation antigen in an autologous melanoma model. Cancer Res. 2000;60:6995–7001. [PubMed] [Google Scholar]
- 27.Feltkamp MC, et al. Vaccination with cytotoxic T lymphocyte epitope-containing peptide protects against a tumor induced by human papillomavirus type 16-transformed cells. Eur J Immunol. 1993;23:2242–2249. doi: 10.1002/eji.1830230929. [DOI] [PubMed] [Google Scholar]
- 28.Kenter GG, et al. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N Engl J Med. 2009;361:1838–1847. doi: 10.1056/NEJMoa0810097. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.